

Abstract
Human cytomegalovirus is a ubiquitous infectious agent that affects mainly immunosuppressed, fetuses, and newborns. The virus has several polymorphic regions, in particular in the envelope glycoproteins. The UL55 gene encodes the glycoprotein B that has a variable region, containing a furin cleavage site and according to the variability different genotypes are characterized. Here we investigated variability and existence of selective pressure on the UL55 variable region containing the furin cleavage site in 213 clinical sequences from patients worldwide. We showed the occurrence of positive selective pressure on gB codons 461 and 462, near the furin cleavage site. Cleavage analysis of synthesized peptides demonstrated that most mutations confer better cleavage by furin, suggesting that evolution is acting in order to increase the efficiency cleavage and supporting the hypothesis that gB processing is important in the host. We also demonstrated that peptides containing sequences, that characterize genotypes gB2 and 3, are differentially cleaved by furin. Our data demonstrate for the first time that variability in the cleavage site is related to degree of gB processing by furin.
Keywords: human cytomegalovirus, glycoprotein B, selective pressure, furin cleavage, gB genotypes
Introduction
Human Cytomegalovirus (HCMV) is a ubiquitous human pathogen, present in 40–100% of the global population. In most cases, viral replication is controlled by the host immune system and infection is asymptomatic. HCMV-associated pathogenesis, therefore, predominantly affects immunocompromised individuals such as transplant recipients and AIDS patients (Soderberg-Naucler, 2006; Steininger, 2007), while congenital infection by HCMV is a major cause of birth defects (Gaytant et al., 2002; Cannon, 2009). Additionally, evidence suggests a linkage between HCMV and pathologies such as cancer (Soroceanu and Cobbs, 2011; Dziurzynski et al., 2012; Cobbs et al., 2014) and cardiovascular diseases (Streblow et al., 2001; Yi et al., 2008).
As an opportunistic agent, it is evident that the mechanisms that allow the virus to escape the humoral and cellular immune responses contribute to pathogenesis (Powers et al., 2008; La Rosa and Diamond, 2012; Noriega et al., 2012). Moreover, it is postulated that genetic variability may affect HCMV virulence. HCMV strains have polymorphisms in several regions (Pignatelli et al., 2004; Renzette et al., 2011, 2013; Sijmons et al., 2014) and in particular, the envelope glycoproteins are of interest as they are involved in events such as cell attachment and entry, and are important targets of the host immune system (Isaacson et al., 2008; Vanarsdall and Johnson, 2012).
The effect of genetic variability within the HCMV glycoproteins on many aspects of the viral life cycle has been investigated. One of the most explored polymorphic regions is the UL55 gene that encodes glycoprotein B (gB) (Pignatelli et al., 2004). gB is an essential component of the virus (Isaacson and Compton, 2009), made as a precursor of 160 kDA, that is cleaved by a cellular furin-like protease generating two subunits termed gp58 and gp116. These subunits are present in the viral envelope as a disulfide-linked complex, termed gCI (Britt and Auger, 1986; Spaete et al., 1988; Britt and Vugler, 1989; Vey et al., 1995). gB is the second most abundant protein in the viral particle (Varnum et al., 2004) and during natural infection it is a major target of neutralizing antibodies (Britt et al., 1988; Britt and Vugler, 1990; Marshall et al., 1992; Navarro et al., 1997; Macagno et al., 2010; Potzsch et al., 2011; Spindler et al., 2013). At the viral envelope, it is present as a trimer, similarly to homologs from HSV-1, an alphaherpesvirus, and EBV, a gammaherpesvirus (Sharma et al., 2012). Further, the HCMV gB trimeric domain possesses a high degree of structural homology to the HSV-1 and EBV gB homologs, despite low homology at the sequence level. However, HCMV gB has more glycosylation sites than its other herpesvirus homologs, suggesting that gB uses glycans as shields to protect neutralizing epitopes (Burke and Heldwein, 2015). gB is involved in viral attachment to the cell (Boyle and Compton, 1998), membrane fusion, entry, and cell to cell transfer of the virus (Navarro et al., 1993; Vanarsdall et al., 2008; Isaacson and Compton, 2009). It has also been demonstrated that interaction of gB with PDGFR (Soroceanu et al., 2008), integrins (Feire et al., 2004; Wang et al., 2005), and TLR2 (Boehme et al., 2006) results in diverse intracellular signaling pathways that coordinate cellular events in the host cell.
The gB protein has 906 amino acids, containing a cytoplasmic domain (Cy), a transmembrane region (TM), a membrane proximal region (MP), and a ectodomain (ED) (Sharma et al., 2013). It has three variable regions, which are present in the N-terminus, C-terminus and at the furin cleavage site (aa 460–461) (Chou and Dennison, 1991; Chou, 1992). The gB furin cleavage site is variable, and HCMV strains were first classified into genotypes gB1 through gB4 according to changes in the motif (Chou and Dennison, 1991). More recently, additional rare genotypes were described: gB5, gB6, and gB7 (Trincado et al., 2000).
The clinical relevance of these different genotypes has been evaluated by several studies, in different parts of the world, which investigated a correlation between gB genotypes and tropism, viral loads or clinical disease. While a number of studies found some possible association between gB genotype and infection course (Fries et al., 1994; Shepp et al., 1996; Rasmussen et al., 1997; Meyer-König et al., 1998; Cunha Ade et al., 2002; Wu et al., 2007; Roubalová et al., 2010; Madi et al., 2011; Xia et al., 2012; Vogel et al., 2013), other studies did not found any correlation (Lurain et al., 1999; Trincado et al., 2000; Sarcinella et al., 2002; Carraro and Granato, 2003; Tarragó et al., 2003; Tanaka et al., 2005; Yamamoto et al., 2007; Görzer et al., 2010; Wu et al., 2011; Paradowska et al., 2012). Some reports also indicated that infection with mixed genotypes could be related to disease progression (Sarcinella et al., 2002; Coaquette et al., 2004; Pang et al., 2008; Deckers et al., 2009). However, presently it is not clear if the gB genotype plays a role in disease prognosis. Regarding the requirement for gB cleavage, one study demonstrated that gB processing by furin is not essential for virus production in fibroblasts (Strive et al., 2002). However, the requirement for cleavage in vivo and in other cell types was not been determined.
Populations of organisms, including viruses, have genetic variability among their individuals, which can be selected by different environmental pressures. In the context of viral infection, the pressures imposed by the cellular environment positively select the variations that confer a higher fitness to the virus. The selective pressure can be estimated site by site using nucleotide multiple alignment of target regions among different populations and analyzing how it translates to amino acids in the protein primary structure. The identification of genomic regions under positive selection can be measured by estimating the non-synonymous (dN) and synonymous (dS) nucleotide substitutions and their ratio (ω = dN/dS). Rates of (ω) = 1 mean neutral evolution, (ω) < 1 purifying or negative selection, and (ω) > 1 diversifying or positive selection. Biologically, sites under positive selection could indicate regions that might be important for adaptation of the virus, augmenting viral fitness.
In this work, we performed an extensive analysis of the occurrence of positive selection in the variable region of the UL55 ORF that contains the furin cleavage site. We report the occurrence of sites under positive selection pressure. Analysis of the cleavage efficiency by furin, using FRET peptides, indicates that the all mutations on residue 462 (gB2) increase the quality of gB as a furin substrate in terms of processing velocity and afinity while the mutation on residue 461 (gB3) increased the processing velocity, but decreased the affinity. Moreover, sequences that characterize different gB genotypes are differentially cleaved.
Results
Determination of the gB Genotype in Clinical Samples and Sequence Data
In order to determine the gB genotypes present in clinical strains obtained from Brazil, and to establish their relationship to sequences from different geographical locations, DNA amplification of the gB variable region was performed in blood samples obtained from 22 renal recipients, 2 allogeneic transplant recipients, 1 premature newborn and tumor tissues tissue from 6 glioblastoma patients (Supplementary Table S1).
The gB2 genotype was the most prevalent (78%), followed by gB3 (18%) and gB1 (4%). Genotypes gB4, gB5, gB6, and gB7 were not found.
Multiple Alignment and Phylogenetic Analysis
In addition to the sequences obtained from Brazilian patients, 181 sequences from different groups of patients (HIV positive and transplant recipients) from distinct geographic regions were retrieved from GeneBank. The total 213 sequences were aligned and the 100% homologous sequences, at nucleotide level, were excluded (Supplementary Table S1 and Figure S4).
Figure 1 shows the alignment of 36 aa, corresponding to aa 456–492 of gB, in the resulting 35 unique sequences, representing the genotypes 1–4 (brackets) and 5, 6, and 7 (dots).
FIGURE 1.

Multiple sequence alignment of the 35 unique sequences viewed by amino acids. gB1 to gB4 sequences are grouped (brackets). gB 5, 6, and 7 (dots). gBext_35 corresponds to a Gorilla gorilla cytomegalovirus gB sequence, used as external group to root the phylogenetic tree. Arrow indicates the furin cleavage site. Colors represent amino acids properties as shown in the legend.
The cleavage by furin occurs between an arginine (R) and a serine (S) at residues 5 and 6 in the sequence, as indicated by an arrow. As observed the canonical sequence recognized by furin RXR(K)R/S is well conserved, however, mutations outside this region occur frequently and characterize the different genotypes.
To infer a phylogenetic relatedness, among different HCMV isolates worldwide, regarding the variable region of gB gene, a phylogenetic tree was constructed resulting in four distinct groups corresponding to genotypes gB1 to gB4 marked in red, green, dark, blue, and orange, respectively. The genotypes gB5, gB6, and gB7, marked in light blue, do not represent a defined group since there is only one sequence of each variant published. Of note, as previously reported and shown here (Figure 2), gB6 and gB7 are close related to gB1 and gB3 (Trincado et al., 2000).
FIGURE 2.
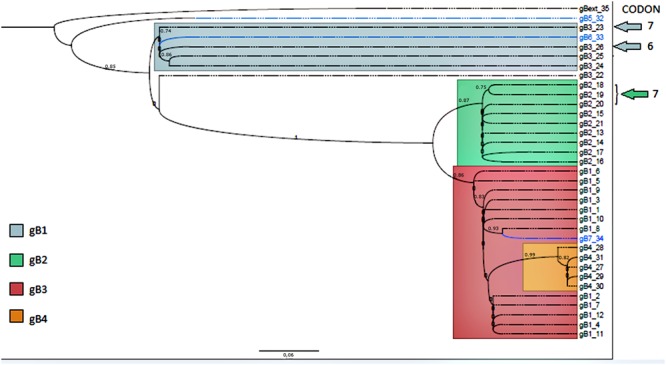
Phylogenetic tree containing the 35 unique sequences. Sequences corresponding genotypes gB1 to gB4 are in different colors as indicated. The blue group contains most of the gB3 and gB6; green group contains all the gB2; red group contains all the gB1 and the gB7. Orange group contains all the gB4. The sequences containing positive selective mutations are pointed by arrows and numbered with the codon (in the alignment) which presented the mutation. The bar indicates the phylogenetic distances in terms of genetic change (bar distance = 0.06). Numbers in each node represent genetic change among branches and are expressed as the number of changes divided by the length of the sequence.
Detection of Positive Selection
The rate of non-synonymous and synonymous substitutions (dN/dS or B+/α) was used to evaluate the strength and direction of selection. Codons with higher B+/α rates are under a stronger positive selective pressure than codons with lower B+/α rates (Murrell et al., 2012).
The output revealed the occurrence of positive selection on codons 6 and 7 on the alignment (Figure 1), which corresponds to codons 461 and 462 of the gB.
Notably, codon 461 is under a significantly stronger positive pressure than codon 462 as demonstrated by its higher B+/α rate (Table 1).
Table 1.
Mixed Effects Model Evolution (MEME) tool output.
| p-value | α/B+ | B+ | α | Codon |
|---|---|---|---|---|
| 0.052 | 24.142 | 16.782 | 0.695 | 6 |
| 0.098 | 6.523 | 13.913 | 2.132 | 7 |
Codons with the major selective pressure probability (6 and 7) and their α and B+ values.
These two codons are located near the furin cleavage site and correspond to the positions P1′ and P2′, respectively (Figure 3), according to the protease-substrate model from Schechter and Berger (1967). For codon 461 only one mutation was observed in genotype 3, where serine (S) is changed to aspartic acid (D), while for codon 462, while threonine (T) was mutated to methionine (M), arginine (R), and alanine (A) in genotype 2. (Figure 1). The other regions in the sequence did not exhibit significant positive selection.
FIGURE 3.
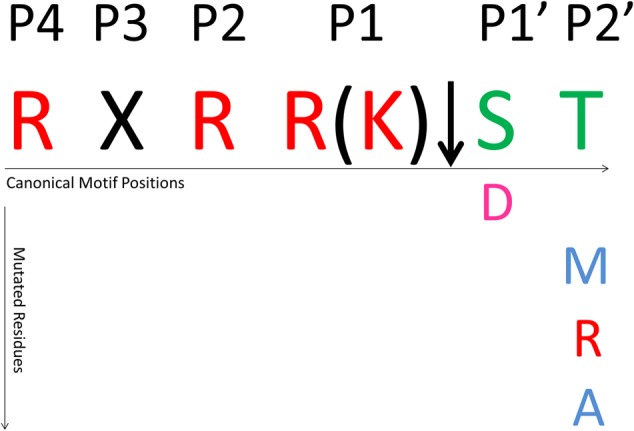
Furin cleavage site and sites under positive selective pressure. Red residues are positive polar, pink are negative polar, and green are neutral polar residues.
In order to strength our findings, a second analysis of selective pressure was performed codon by codon for each gB genotype by Bayesian Inference (BI), using the software MrBayes (Ronquist and Huelsenbeck, 2003). The BI confirmed the evidences of positive selection at codons 461 and 462, with a cut-off of 85% of positive selection probability (Figure 4).
FIGURE 4.
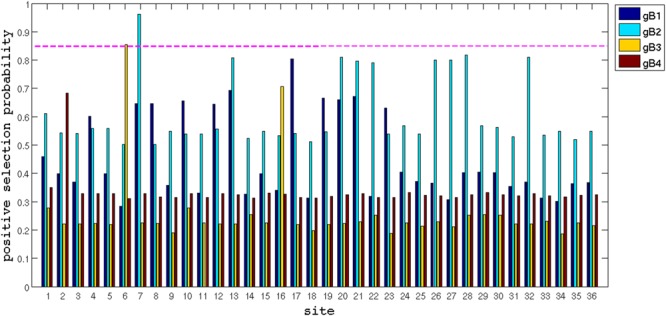
Bayesian Inference by MrBayes software. The probability of positive selective pressure (y-axis) plot for each site (x-axis). The cut-off of probability was 85%.
Cleavage by Furin
In order to see if the mutations have an influence in cleavage, peptides containing the original reference sequences and their derivative containing positive selected mutations, were synthesized and tested against recombinant human furin. All substrates were cleaved after the double basic residues RR or KR as analyzed by Liquid Chromatography mass spectrometry (LC-MS) (Supplementary Figures S1–S3). The kcat/km ratio relates to the catalytic efficiency, meaning how fast in M/s the enzyme reacts with the substrate once it encounters it. As a control, a peptide which contains a mutation previously shown to interfere with furin cleavage (Strive et al., 2002) was tested and as expected no cleavage was observed (Table 2 and Supplementary Figure S3).
Table 2.
gB2, gB3 peptides, and non-cleaved peptide (NCP) and their respective kcat, and km kinetics parameters.
| Peptide | Mutation | kcat (1/s) | SD | km (μmolar) | SD | kcat/km | Sequence name | Genotype |
|---|---|---|---|---|---|---|---|---|
| RTRR↓STSDNNT | – | 2.24 | 0.05 | 3.19 | 0.06 | 0.70 | gB2_13 | 2 |
| RTRR↓SRSDNNT | T462R | 5.06 | 0.08 | 0.23 | 0.01 | 22.00 | gB2_19 | |
| RTRR↓SMSDNNT | T462M | 3.01 | 0.29 | 1.65 | 0.14 | 1.82 | gB2_18 | |
| RTRR↓SASDNNT | T462A | 4.06 | 0.03 | 0.69 | 0.04 | 5.88 | gB2_20 | |
| RTKR↓STGNTTT | – | 0.25 | 0.02 | 0.02 | 0.01 | 12.5 | gB3_22 | 3 |
| RTKR↓DTGNTTT | S461D | 1.89 | 0.11 | 0.43 | 0.02 | 4.39 | gB3_26 | |
| ATRASTSDNNT | R457A/R460A | – | – | – | – | – | – | NCP |
Standard deviation (SD) were calculated and shown for each parameter.
As shown in Table 2 the substitution S461D in position P1′ of genotype 3, results in an increase of kcat, but a 20 times increase in the km, resulting in threefold decrease in kcat/km ratio. This mutation was found in three renal transplant recipients from São Paulo, Brazil (Supplementary Table S1). Mutations in position P2′, found in genotype 2, are shown in Table 2. All substitutions result in increase in kcat, compared to the reference sequence (gB2_13). The differences in km are notable, in particular in the S462R substitution, which increases 31-fold the kcat/km ratio. This mutation was found in two allogeneic transplant recipients and eight renal transplant recipients. Mutation S462M was found in one renal transplant recipient and S462A in eight real transplant recipients. All these patients are from São Paulo, Brazil (Supplementary Table S1). Interestingly, all peptides containing the mutations on sites under positive selective pressure are processed at higher velocities (kcat) in comparison to the reference sequences.
Discussion
The HCMV genome presents polymorphisms at both intra and inter host levels, and variability is found in many regions, in particular in genes that encode glycoproteins (Renzette et al., 2015). The mutations that arise can be advantageous to the virus resulting in increase in viral fitness and adaptation. In this regard, analysis of variability and selection pressure can identify sites under positive selection that might give benefits to the virus. In fact, HCMV positive selection appears to be a driver of genome evolution, associated with compartmentalization in the host (Renzette et al., 2013). Yet, studies of variability and selective pressure in HCMV sequences from individuals from different geographic regions are scarce.
Here we analyzed the occurrence of positive selection in the gB variable region that contains a furin cleavage site, in sequences from different populations worldwide.
Our analysis shows that the furin cleavage site is highly conserved in patient samples. Nevertheless, positive selection was evident in two codons at positions P1′ (aa 461) and P2′ (aa 462) (Figure 3). Notably, mutations that characterize positive selection were not found in regions outside the specific cleavage site.
In vitro analysis of the cleavage by furin using FRET peptides indicates that substrates containing the gB2 and gB3 genotypes reference sequences are cleaved with different efficiencies (Table 2), and the better-cleaved reference peptide among the genotypes is gB3. Furthermore, three positively selected mutations in genotype 2 significantly increase the efficiency of cleavage, while one mutation in genotype 3 decreases it (Table 2). This data reveal that variability in this regions affect cleavage and that mutations under positive selective pressure are indeed modifying the quality of the gB as a furin substrate, either increasing or decreasing the km/kcat ratio.
Positively selected mutations which alter the gB cleavage kinetics can indicate an augment of the viral fitness. Substrates with an increased kcat/km ratio are better cleaved by furin, suggesting that gB processing is important for viral replication in vivo. We also can speculate that in other context substrates for which the furin have high affinity and low reaction velocity, such as gB3_26, may trap the enzyme, preventing the cleavage of host substrates by furin, which may somehow favor to the virus. In this way, gB could act as a competitive inhibitor of furin. In fact, Bachis et al. (2012) reported that Human Immunodeficience Virus 1 (HIV-1) can reduce the cleavage of host proteins by furin, by reducing cellular levels of the enzyme. It is possible that HCMV acts by a different mechanism by reducing the proteolisis of host proteins.
The role of gB cleavage for HCMV life cycle in vivo is not recognized. A study by Strive et al. (2002), indicated that gB cleavage by furin is not essential for in vitro replication of the RVHB5 strain in fibroblasts. Analysis in other herpesviruses such as pseudorabies virus (PRV), Epstein-Barr virus (EBV), and BHV-1 gB also concluded that cleavage is not required for virus replication in vitro but is important for cell-to-cell fusion (Kopp et al., 1994; Okazaki, 2007; Sorem and Longnecker, 2009).
In murid herpesvirus, gB cleavage is not required for entry in fibroblasts and epithelial cells but is significantly important for entry in macrophages and bone marrow-derived dendritic cells (Glauser et al., 2013). Moreover, cleavage is required for lytic spread of the virus in the lungs (Glauser et al., 2013). Similarly, in varicella-zoster virus (VZV) the furin cleavage motif is not required for viral replication in vitro but contributes to pathogenesis in skin tissue in vivo (Oliver et al., 2009).
Interestingly, in many viruses that have fusion proteins which are cleaved by furin, such as the influenza hemagglutinin, Newcastle disease virus glycoprotein F0, and human immunodeficiency virus 1 glycoprotein gp160, the cleaved proteins tends to have more hydrophobic residues in P3′–P6′ in comparison to non-viral furin substrates. This fact seems to be related to the fusion efficiency, as the more hydrophobic the residues in P3′–P6′, the more efficient is the fusion to the host cell membrane. On the other hand, high hydrophobicity in this region can reduce the furin cleavage efficiency, since it may restrict access to solvent. Therefore, these viruses have to tune hydrophobicity and furin cleavage efficiency to succeed. One strategy to achieve this fine-tuning is to add small hydrophobic residues such as glycine, alanine, and proline in these positions (Tian, 2009).
In HCMV gB the residues in P3′–P6′ positions are mostly polar, such as aspartic acid (D), asparagine (N), and Threonine (T) (Figure 1). In gB1, gB3, gB4, gB6, and gB7, glycine (G), a small non-polar residue, is the only hydrophobic residue present in this region. These observation suggests that in HCMV gB this region is not directly related to host-cell membrane fusion, as its hydrophobicity is low when compared to viruses which have fusion proteins with hydrophobic P3′–P6′ regions. In fact, previous analysis indicate that the HCMV gB fusion loops comprise aa 174, 179, 259, 261, and 262 (Vanarsdall et al., 2016).
Regardless we show that the HCMV gB cleavage site is well conserved implying that cleavage is important for the biological function of gB in vivo, similar to other herpesviruses. Perhaps cleavage is required for infection of specific cells types. In fact, the main targets of HCMV in vivo are epithelial and endothelial cells and consequently studies need to be done to determine the significance of proteolytic cleavage in these cells.
For the first time we show that variability in the HCMV gB cleavage site is related to distinct degrees of cleavage by furin. All positive selected sites occurred in viral sequences from Brazilian transplanted patients and in more than one host, indicating that the mutations arose and became established in a population. This is the first study showing that selective pressures on the HCMV gB furin cleavage site are driving evolution toward a faster processing of the protein. Our findings open possibilities for further studies, ideally using recombinant viruses containing the mutations, aiming the elucidation of the biological implications of the mutations in vitro and in vivo.
Materials and Methods
Sequence Data Set
A total of 213 sequences corresponding to the gB variable region of HCMV were analyzed in this study. Thirty two sequences were obtained through DNA sequencing of blood from 22 renal transplant, 3 allogeneic transplant recipients, 1 premature newborn and tumor tissues of 6 glioblastoma patients from Brazil. The sequences KX859110 to KX859141, were deposited in NCBI’s GenBank1. One hundred eighty one sequences were retrieved from GenBank, which numbered 8 AIDS patients, 8 congenitally infected newborns, 76 renal transplant recipients, 34 hematopoietic cell transplant recipients, 35 unknown cases, and 19 HCMV reference sequences. One sequence from gorilla CMV was used as external group. These sequences represent different geographic regions of the world (France, Brazil, United States, South Korea, and Australia). The accession numbers are shown in Supplementary Table S1.
This study has approval of the research ethics committee (REC) no. 04171312.0.0000.5415 of the Medical School of São José do Rio Preto- FAMERP – São Paulo. The patients were anonymized and the written consent was waived by REC. This study also has approval of the REC 0654/10 of the Neurology Center of Clínicas Hospital – São Paulo. The patients were informed of the procedure and signed a consent form.
DNA Extraction
DNA from peripheral blood was extracted using the Wizard® Genomic DNA Purification Kit (PromegaTM) and DNA from glioblastoma tumor tissues was extracted using the Pure Link genomic DNA mini kit (Invitrogen), according manufacturer’s instructions.
Hemi-Nested PCR
To amplify the gB variable region a hemi-nested PCR (nPCR) was performed using primers previously described by Zheng et al. (2002). In the first round of amplification, the reactions were carried out in 50 μL volume containing 2 μL DNA, Taq Polymerase buffer 1x, 2 mM MgCl2, 2.5 U Platinum Taq Polymerase High Fidelity (Invitrogen), 200 μM dNTPs, and 0.15 μM of each primer.
The thermo cycling program was set as 94°C for 14 min, 35 cycles at 95°C for 30 s; 56.2°C for 30 s; 72°C for 45 s; 72°C for 10 min. All the reactions were performed at least three times for confirmation and to avoid false positive and negative results. No positive controls were used during the HCMV detection in nPCR to minimize the possibility of laboratory contamination with HCMV DNA. As a positive control for cellular DNA, primers for the Glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) gene were used (5′-ACC CAC TCC TCC ACC TTT GAC-3′ and 5′-CTG TTG CTG TAG CCA AAT TCG T-3′).
Amplified products were purified using the Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare) and sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit with an ABI-3130 (Applied BiosystemsTM) at the Serviço de Sequenciamento de DNA–SSDNA IQUSP. Confirmation of the HCMV sequences, and the determination of the gB genotypes were performed using the National Center for Biotechnology Information (NCBI-USA) Blast tool.
Sequence Alignment and Phylogenetic Analysis
The gB sequences were aligned using the method MUSCLE v3.8.312 (Edgar, 2004) and edited with the Bio edit software3 (Hall, 1999). Sequences with 100% similarity were excluded from the analyses.
The best phylogenetic tree substitution method was determined by performing tests on the jModelTest2 software4 (Darriba et al., 2012). A phylogenetic tree of the 35 resulting unique sequences was constructed using the GTR model of the software PhyML5 (Guindon et al., 2010).
The gB genotypes were assigned by comparison of obtained sequences with reference sequences previously published (Supplementary Table S1).
Positive Selection Analysis
The search for positive selection was performed using the Mixed Effects Model Evolution (MEME) (Murrell et al., 2012) via Datamonkey web Server (Pond and Frost, 2005)6, which detect episodic diversifying selection affecting individual codon sites in branch by branch. Selective pressure is estimated codon-by-codon based in the ratio of mutations that lead to non-synonymous (dN) or synonymous (dS) substitutions. The algorithm attributes different weights for each substitution, according to the probability of occurrence and their impact in the protein translation. In this manner scores are calculated for synonymous (α) and non-synonymous (β+) substitutions and the ratio is used to estimate the selective pressure site by site. A second analysis of the probability of selection was done for each gB genotype by BI, using the software MrBayes (Ronquist and Huelsenbeck, 2003) with a 85% probability cut-off (Figure 4).
Recombinant Furin Production
Recombinant human furin was expressed and purified as previously described (Kacprzak et al., 2004). The enzyme concentration was determined by active site titration using the inhibitor decanoyl-RVKR-CMK inhibitor in 10 mM MES buffer, 1 mM CaCl2, pH 7.0 and the substrate Abz-GIRRKRSVSHQ-EDDnp.
FRET-Peptide Synthesis and Enzymatic Kinetics Assays
Seven synthetic peptides were obtained by solid-phase strategy, as previously described (Izidoro et al., 2010) using the Fmoc-procedure in an automated multiple solid-phase peptide synthesizer (PSSM 8 system, Shimadzu, Japan) (Hirata et al., 1995; Korkmaz et al., 2008). Peptides were prepared in dimethylformamide and stock concentrations measured spectrophotometrically at 365 nm (e = 17,300 M-1.cm-1). The quantum yields of the FRET peptides and the products of their hydrolysis were unchanged in all tested conditions.
The FRET peptides were assayed in 10 mM MES, 1 mM CaCl2, pH 7.0, using a F-2500 spectrofluorimeter (Hitachi, Tokyo, Japan), at 37°C (Izidoro et al., 2010), as previously described. Fluorescence changes were monitored continuously at λex = 320 nm and λem = 420 nm. The enzyme concentrations for initial rate determinations were chosen at a level intended to hydrolyze less than 5% of the amount of added substrate over the time course of data collection.
Three kinetic parameters from the Michaelis-Menten model were analyzed, representing enzyme-substrate (km), the catalysis constant (kcat) and the kcat/km ratio. The kinetic parameters km and kcat were calculated by non-linear regression using Grafit software (Erithacus Software, Horley, Surrey, UK). km, kcat, and standard errors (less than 10%) were calculated using Grafit software (v.5.0.13, Erithacus Software, Horley, Surrey, UK) with a non-linear least squares Michaelis-Menten fit associated with a matrix invertion fitting routine to refine kcat, km and allowing estimation of the errors. Standard deviations greater than 10% are due to low values obtained.
Author Contributions
All the authors contributed with: substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo-FAPESP-Number 2013/14215-9.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00934/full#supplementary-material
HPLC of furin-processed FRET substrates. Each substrate was incubated with/without furin for 12 h and further analyzed by HLPC. Substrate and products were followed by fluorescence (Abz, ex320 nm; em420 nm) quenching (Eddnp, uv = 365 nm) indicating single cleavage site for all tested sequences. (A) gB3_22. (B) gB_20. (C) gB3_26 (D) gB2_18. (E) gB2_13. (F) gB2_19.
Mass spectra of furin-processed substrates. Furin (50 nM) were incubated with each substrate for 12 h and further analyzed by Maldi-TOF (Bruker Daltonics, Germany). (A) gB3_22 substrate and products (1549.3, 678.7, and 888.7 Da). (B) gB2_13 (1634.4, 706.7, and 945.7 Da). (C) gB_20 (1604.3, 706.7, and 915.6 Da). (D) gB2_18 (1664.4, 706.7, and 975.8 Da) (E) gB3_26 (1577.3, 678.7, and 916.7 Da). (F) gB2_19 (1689.4, 706.7, and 1000.8 Da).
Maldi-TOF spectrum of the synthetic FRET peptide ATRASTSDNNT (Strive et al., 2002), not processed by furin. FRET ATRASTSDNNT (5 μM, 1,592.2 Da) was tested as a furin substrate in spectrofluorimeter and Maldi-TOF after 2 h of incubation with furin.
Multiple sequence alignment of the 35 unique sequences viewed by nucleotides.
References
- Bachis A., Avdoshina V., Zecca L., Parsadanian M., Mocchetti I. (2012). Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J. Neurosci. 32 9477–9484. 10.1523/JNEUROSCI.0865-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme K. W., Guerrero M., Compton T. (2006). Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 177 7094–7102. 10.4049/jimmunol.177.10.7094 [DOI] [PubMed] [Google Scholar]
- Boyle K. A., Compton T. (1998). Receptor-binding properties of a soluble form of human cytomegalovirus glycoprotein B. J. Virol. 72 1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt W. J., Auger D. (1986). Synthesis and processing of the envelope gp55-116 complex of human cytomegalovirus. J. Virol. 58 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt W. J., Vugler L., Stephens E. B. (1988). Induction of complement-dependent and -independent neutralizing antibodies by recombinant-derived human cytomegalovirus gp55-116 (gB). J. Virol. 62 3309–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt W. J., Vugler L. G. (1989). Processing of the gp55-116 envelope glycoprotein complex (gB) of human cytomegalovirus. J. Virol. 63 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt W. J., Vugler L. G. (1990). Antiviral antibody responses in mothers and their newborn infants with clinical and subclinical congenital cytomegalovirus infections. J. Infect. Dis. 161 214–219. 10.1093/infdis/161.2.214 [DOI] [PubMed] [Google Scholar]
- Burke H. G., Heldwein E. E. (2015). Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog. 11:e1005227 10.1371/journal.ppat.1005227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M. J. (2009). Congenital cytomegalovirus (CMV) epidemiology and awareness. J. Clin. Virol. 46 S6–S10. 10.1016/j.jcv.2009.09.002 [DOI] [PubMed] [Google Scholar]
- Carraro E., Granato C. F. (2003). Single human cytomegalovirus gB genotype shed in multiple sites at the time of diagnosis in renal transplant recipients. J. Med. Virol. 70 240–243. 10.1002/jmv.10383 [DOI] [PubMed] [Google Scholar]
- Chou S. (1992). Molecular epidemiology of envelope glycoprotein H of human cytomegalovirus. J. Infect. Dis. 166 604–607. 10.1093/infdis/166.3.604 [DOI] [PubMed] [Google Scholar]
- Chou S. W., Dennison K. M. (1991). Analysis of interstrain variation in cytomegalovirus glycoprotein B sequences encoding neutralization-related epitopes. J. Infect. Dis. 163 1229–1234. 10.1093/infdis/163.6.1229 [DOI] [PubMed] [Google Scholar]
- Coaquette A., Bourgeois A., Dirand C., Varin A., Chen W., Herbein G. (2004). Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin. Infect. Dis. 39 155–161. 10.1086/421496 [DOI] [PubMed] [Google Scholar]
- Cobbs C., Khan S., Matlaf L., McAllister S., Zider A., Yount G., et al. (2014). HCMV glycoprotein B is expressed in primary glioblastomas and enhances growth and invasiveness via PDGFR-alpha activation. Oncotarget 5 1091–1100. 10.18632/oncotarget.1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha Ade A., Marin L. J., Aquino V. H., Figueiredo L. T. M. (2002). Diagnosis of cytomegalovirus infections by qualitative and quantitative PCR in HIV infected patients. Rev. Inst. Med. Trop. Saƥo Paulo 44 127–132. 10.1590/S0036-46652002000300003 [DOI] [PubMed] [Google Scholar]
- Darriba D., Taboada G. L., Doallo R., Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers M., Hofmann J., Kreuzer K.-A., Reinhard H., Edubio A., Hengel H., et al. (2009). High genotypic diversity and a novel variant of human cytomegalovirus revealed by combined UL33/UL55 genotyping with broad-range PCR. Virol. J. 6 210 10.1186/1743-422X-6-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziurzynski K., Chang S. M., Heimberger A. B., Kalejta R. F., Dallas S. R. M., Smit M., et al. (2012). Consensus on the role of human cytomegalovirus in glioblastoma. Neuro. Oncol. 14 246–255. 10.1093/neuonc/nor227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feire A. L., Koss H., Compton T. (2004). Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. U.S.A. 101 15470–15475. 10.1073/pnas.0406821101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries B. C., Chou S., Boeckh M., Torok-Storb B. (1994). Frequency distribution of cytomegalovirus envelope glycoprotein genotypes in bone marrow transplant recipients. J. Infect. Dis. 169 769–774. 10.1093/infdis/169.4.769 [DOI] [PubMed] [Google Scholar]
- Gaytant M. A., Steegers E. A. P., Semmekrot B. A., Merkus H. M. M. W., Galama J. M. D. (2002). Congenital cytomegalovirus infection: review of the epidemiology and outcome. Obstet. Gynecol. Surv. 57 245–256. 10.1007/s11940-002-0039-8 [DOI] [PubMed] [Google Scholar]
- Glauser D. L., Milho R., Frederico B., May J. S., Kratz A.-S., Gillet L., et al. (2013). Glycoprotein B cleavage is important for murid herpesvirus 4 to infect myeloid cells. J. Virol. 87 10828–10842. 10.1128/JVI.00709-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görzer I., Kerschner H., Redlberger-Fritz M., Puchhammer-Stöckl E. (2010). Human cytomegalovirus (HCMV) genotype populations in immunocompetent individuals during primary HCMV infection. J. Clin. Virol. 48 100–103. 10.1016/j.jcv.2010.03.005 [DOI] [PubMed] [Google Scholar]
- Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hall T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41 95–98. [Google Scholar]
- Hirata I. Y., Sedenho Cezari M. H., Nakaie C. R., Boschcov P., Ito A. S., Juliano M. A., et al. (1995). Internally quenched fluorogenic protease substrates: solid-phase synthesis and fluorescence spectroscopy of peptides containing ortho-aminobenzoyl/dinitrophenyl groups as donor-acceptor pairs. Lett. Pept. Sci. 1 299–308. 10.1007/BF00119771 [DOI] [Google Scholar]
- Isaacson M. K., Compton T. (2009). Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J. Virol. 83 3891–3903. 10.1128/JVI.01251-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson M. K., Juckem L. K., Compton T. (2008). Virus entry and innate immune activation. Curr. Top. Microbiol. Immunol. 325 85–100. 10.1007/978-3-540-77349-8_5 [DOI] [PubMed] [Google Scholar]
- Izidoro M. A., Assis D. M., Oliveira V., Santos J. A. N., Juliano M. A., Lindberg I., et al. (2010). Effects of magnesium ions on recombinant human furin: selective activation of hydrolytic activity upon substrates derived from virus envelope glycoprotein. Biol. Chem. 391 1105–1112. 10.1515/BC.2010.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacprzak M. M., Peinado J. E., Than M. E., Appel J., Henrich S., Lipkind G., et al. (2004). Inhibition of furin by polyarginine-containing peptides: nanomolar inhibition by nona-D-arginine. J. Biol. Chem. 279 36788–36794. 10.1074/jbc.M400484200 [DOI] [PubMed] [Google Scholar]
- Kopp A., Blewett E., Misra V., Mettenleiter T. C. (1994). Proteolytic cleavage of bovine herpesvirus 1 (BHV-1) glycoprotein gB is not necessary for its function in BHV-1 or pseudorabies virus. J. Virol. 68 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz B., Attucci S., Juliano M. A., Kalupov T., Jourdan M.-L., Juliano L., et al. (2008). Measuring elastase, proteinase 3 and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat. Protoc. 3 991–1000. 10.1038/nprot.2008.63 [DOI] [PubMed] [Google Scholar]
- La Rosa C., Diamond D. J. (2012). The immune response to human CMV. Future Virol. 7 279–293. 10.2217/fvl.12.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurain N. S., Kapell K. S., Huang D. D., Short J. A., Paintsil J., Winkfield E., et al. (1999). Human cytomegalovirus UL144 open reading frame: sequence hypervariability in low-passage clinical isolates. J. Virol. 73 10040–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macagno A., Bernasconi N. L., Vanzetta F., Dander E., Sarasini A., Revello M. G., et al. (2010). Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 84 1005–1013. 10.1128/JVI.01809-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madi N., Al-Nakib W., Pacsa A., Saeed T. (2011). Cytomegalovirus genotypes gB1 and gH1 are the most predominant genotypes among renal transplant recipients in Kuwait. Transplant. Proc. 43 1634–1637. 10.1016/j.transproceed.2011.02.053 [DOI] [PubMed] [Google Scholar]
- Marshall G. S., Rabalais G. P., Stout G. G., Waldeyer S. L. (1992). Antibodies to recombinant-derived glycoprotein B after natural human cytomegalovirus infection correlate with neutralizing activity. J. Infect. Dis. 165 381–384. 10.1093/infdis/165.2.381 [DOI] [PubMed] [Google Scholar]
- Meyer-König U., Vogelberg C., Bongarts A., Kampa D., Delbrück R., Wolff-Vorbeck G., et al. (1998). Glycoprotein B genotype correlates with cell tropism in vivo of human cytomegalovirus infection. J. Med. Virol. 55 75–81. [DOI] [PubMed] [Google Scholar]
- Murrell B., Wertheim J. O., Moola S., Weighill T., Scheffler K., Kosakovsky Pond S. L., et al. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764 10.1371/journal.pgen.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro D., Lennette E., Tugizov S., Pereira L. (1997). Humoral immune response to functional regions of human cytomegalovirus glycoprotein B. J. Med. Virol. 52 451–459. [DOI] [PubMed] [Google Scholar]
- Navarro D., Paz P., Tugizov S., Topp K., La Vail J., Pereira L. (1993). Glycoprotein B of human cytomegalovirus promotes virion penetration into cells, transmission of infection from cell to cell, and fusion of infected cells. Virology 197 143–158. 10.1006/viro.1993.1575 [DOI] [PubMed] [Google Scholar]
- Noriega V., Redmann V., Gardner T., Tortorella D. (2012). Diverse immune evasion strategies by human cytomegalovirus. Immunol. Res. 54 140–151. 10.1007/s12026-012-8304-8 [DOI] [PubMed] [Google Scholar]
- Okazaki K. (2007). Proteolytic cleavage of glycoprotein B is dispensable for in vitro replication, but required for syncytium formation of pseudorabies virus. J. Gen. Virol. 88 1859–1865. 10.1099/vir.0.82610-0 [DOI] [PubMed] [Google Scholar]
- Oliver S. L., Sommer M., Zerboni L., Rajamani J., Grose C., Arvin A. M. (2009). Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J. Virol. 83 7495–7506. 10.1128/JVI.00400-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X., Humar A., Preiksaitis J. K. (2008). Concurrent genotyping and quantitation of cytomegalovirus gB genotypes in solid-organ-transplant recipients by use of a real-time PCR assay. J. Clin. Microbiol. 46 4004–4010. 10.1128/JCM.01341-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradowska E., Studziñska M., Nowakowska D., Wilczyñski J., Rycel M., Suski P., et al. (2012). Distribution of UL144, US28 and UL55 genotypes in Polish newborns with congenital cytomegalovirus infections. Eur. J. Clin. Microbiol. Infect. Dis. 31 1335–1345. 10.1007/s10096-011-1447-z [DOI] [PubMed] [Google Scholar]
- Pignatelli S., Dal Monte P., Rossini G., Landini M. P. (2004). Genetic polymorphisms among human cytomegalovirus (HCMV) wild-type strains. Rev. Med. Virol. 14 383–410. 10.1002/rmv.438 [DOI] [PubMed] [Google Scholar]
- Pond S. L. K., Frost S. D. W. (2005). Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21 2531–2533. 10.1093/bioinformatics/bti320 [DOI] [PubMed] [Google Scholar]
- Potzsch S., Spindler N., Wiegers A. K., Fisch T., Rucker P., Sticht H., et al. (2011). B cell repertoire analysis identifies new antigenic domains on glycoprotein b of human cytomegalovirus which are target of neutralizing antibodies. PLoS Pathog. 7:e1002172 10.1371/journal.ppat.1002172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers C., DeFilippis V., Malouli D., Früh K. (2008). Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 325 333–359. 10.1007/978-3-540-77349-8_19 [DOI] [PubMed] [Google Scholar]
- Rasmussen L., Hong C., Zipeto D., Morris S., Sherman D., Chou S., et al. (1997). Cytomegalovirus gB genotype distribution differs in human immunodeficiency virus-infected patients and immunocompromised allograft recipients. J. Infect. Dis. 175 179–184. 10.1093/infdis/175.1.179 [DOI] [PubMed] [Google Scholar]
- Renzette N., Bhattacharjee B., Jensen J. D., Gibson L., Kowalik T. F. (2011). Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 7:e1001344 10.1371/journal.ppat.1001344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N., Gibson L., Bhattacharjee B., Fisher D., Schleiss M. R., Jensen J. D., et al. (2013). Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection. PLoS Genet. 9:e1003735 10.1371/journal.pgen.1003735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N., Pokalyuk C., Gibson L., Bhattacharjee B., Schleiss M. R., Hamprecht K., et al. (2015). Limits and patterns of cytomegalovirus genomic diversity in humans. Proc. Natl. Acad. Sci. U.S.A. 112 E4120–E4128. 10.1073/pnas.1501880112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19 1572–1574. 10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- Roubalová K., Strunecký O., Zufanová S., Procházka B., Vitek A. (2010). Genotyping of viral glycoprotein B (gB) in hematopoietic stem cell transplant recipients with active cytomegalovirus infection: analysis of the impact of gB genotypes on the patients’ outcome. Epidemiol. Mikrobiol. Imunol. 59 92–99. [PubMed] [Google Scholar]
- Sarcinella L., Mazzulli T., Willey B., Humar A. (2002). Cytomegalovirus glycoprotein B genotype does not correlate with outcomes in liver transplant patients. J. Clin. Virol. 24 99–105. 10.1016/S1386-6532(01)00238-4 [DOI] [PubMed] [Google Scholar]
- Schechter I., Berger A. (1967). On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27 157–162. 10.1016/S0006-291X(67)80055-X [DOI] [PubMed] [Google Scholar]
- Sharma S., Wisner T. W., Johnson D. C., Heldwein E. E. (2012). HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435 239–249. 10.1016/j.virol.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Wisner T. W., Johnson D. C., Heldwein E. E. (2013). HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435 239–249. 10.1016/j.virol.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepp D. H., Match M. E., Ashraf A. B., Lipson S. M., Millan C., Pergolizzi R. (1996). Cytomegalovirus glycoprotein B groups associated with retinitis in AIDS. J. Infect. Dis. 174 184–187. 10.1093/infdis/174.1.184 [DOI] [PubMed] [Google Scholar]
- Sijmons S., Van Ranst M., Maes P. (2014). Genomic and functional characteristics of human cytomegalovirus revealed by next-generation sequencing. Viruses 6 1049–1072. 10.3390/v6031049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg-Naucler C. (2006). Human cytomegalovirus persists in its host and attacks and avoids elimination by the immune system. Crit. Rev. Immunol. 26 231–264. 10.1615/CritRevImmunol.v26.i3.30 [DOI] [PubMed] [Google Scholar]
- Sorem J., Longnecker R. (2009). Cleavage of Epstein-Barr virus glycoprotein B is required for full function in cell-cell fusion with both epithelial and B cells. J. Gen. Virol. 90 591–595. 10.1099/vir.0.007237-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroceanu L., Akhavan A., Cobbs C. S. (2008). Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nature 455 391–395. 10.1038/nature07209 [DOI] [PubMed] [Google Scholar]
- Soroceanu L., Cobbs C. S. (2011). Is HCMV a tumor promoter? Virus Res. 157 193–203. 10.1016/j.virusres.2010.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaete R. R., Thayer R. M., Probert W. S., Masiarz F. R., Chamberlain S. H., Rasmussen L., et al. (1988). Human cytomegalovirus strain Towne glycoprotein B is processed by proteolytic cleavage. Virology 167 207–225. 10.1016/0042-6822(88)90071-2 [DOI] [PubMed] [Google Scholar]
- Spindler N., Rucker P., Potzsch S., Diestel U., Sticht H., Martin-Parras L., et al. (2013). Characterization of a discontinuous neutralizing epitope on glycoprotein B of human cytomegalovirus. J. Virol. 87 8927–8939. 10.1128/JVI.00434-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger C. (2007). Clinical relevance of cytomegalovirus infection in patients with disorders of the immune system. Clin. Microbiol. Infect. 13 953–963. 10.1111/j.1469-0691.2007.01781.x [DOI] [PubMed] [Google Scholar]
- Streblow D. N., Orloff S. L., Nelson J. A. (2001). Do pathogens accelerate atherosclerosis? J. Nutr. 131 2798S–2804S. [DOI] [PubMed] [Google Scholar]
- Strive T., Borst E., Messerle M., Radsak K., Strive T., Borst E., et al. (2002). Proteolytic processing of human cytomegalovirus glycoprotein B is dispensable for viral growth in culture. J Virol. 76 1252–1264. 10.1128/JVI.76.3.1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Numazaki K., Tsutsumi H. (2005). Human cytomegalovirus genetic variability in strains isolated from Japanese children during 1983-2003. J. Med. Virol. 76 356–360. 10.1002/jmv.20366 [DOI] [PubMed] [Google Scholar]
- Tarragó D., Quereda C., Tenorio A. (2003). Different cytomegalovirus glycoprotein B genotype distribution in serum and cerebrospinal fluid specimens determined by a novel multiplex nested PCR. J. Clin. Microbiol. 41 2872–2877. 10.1128/JCM.41.7.2872-2877.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S. (2009). A 20 residues motif delineates the furin cleavage site and its physical properties may influence viral fusion. Biochem. Insights 2 9–20. 10.4137/BCI.S2049 [DOI] [Google Scholar]
- Trincado D. E., Scott G. M., White P. A., Hunt C., Rasmussen L., Rawlinson W. D. (2000). Human cytomegalovirus strains associated with congenital and perinatal infections. J. Med. Virol. 61 481–487. [DOI] [PubMed] [Google Scholar]
- Vanarsdall A. L., Howard P. W., Wisner T. W., Johnson D. C. (2016). Human cytomegalovirus gH/gL forms a stable complex with the fusion protein gB in virions. PLoS Pathog. 12:e1005564 10.1371/journal.ppat.1005564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanarsdall A. L., Johnson D. C. (2012). Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2 37–42. 10.1016/j.coviro.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanarsdall A. L., Ryckman B. J., Chase M. C., Johnson D. C. (2008). Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82 11837–11850. 10.1128/JVI.01623-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum S. M., Streblow D. N., Monroe M. E., Smith P., Auberry K. J., Paša-Tolic L., et al. (2004). Identification of proteins in human cytomegalovirus (HCMV) Particles: the HCMV Proteome. J. Virol. 78 10960–10966. 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey M., Schäfer W., Reis B., Ohuchi R., Britt W., Garten W., et al. (1995). Proteolytic processing of human cytomegalovirus glycoprotein B (gpUL55) is mediated by the human endoprotease furin. Virology 206 746–749. 10.1016/S0042-6822(95)80002-6 [DOI] [PubMed] [Google Scholar]
- Vogel J.-U., Otte J., Koch F., Gümbel H., Doerr H. W., Cinatl J. (2013). Role of human cytomegalovirus genotype polymorphisms in AIDS patients with cytomegalovirus retinitis. Med. Microbiol. Immunol. 202 37–47. 10.1007/s00430-012-0244-3 [DOI] [PubMed] [Google Scholar]
- Wang X., Huang D. Y., Huong S.-M., Huang E.-S. (2005). Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med. 11 515–521. 10.1038/nm1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.-G., Hung M.-C., Chang Y.-T., Chen C.-J., Yang S.-P., Liu C.-Y., et al. (2011). Occurrence of human cytomegalovirus glycoprotein B genotypes in immunocompetent and immunosuppressed Taiwanese patients. Intervirology 54 196–201. 10.1159/000322382 [DOI] [PubMed] [Google Scholar]
- Wu Y.-M., Yan J., Ojcius D. M., Chen L.-L., Gu Z.-Y., Pan J.-P. (2007). Correlation between Infections with different genotypes of human cytomegalovirus and Epstein-Barr Virus in subgingival samples and periodontal status of patients. J. Clin. Microbiol. 45 3665–3670. 10.1128/JCM.00374-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C., Zhao X., Sun Y., Zhang Z. (2012). Human cytomegalovirus glycoprotein B genotypes in Chinese hematopoietic stem cell transplant recipients. Intervirology 55 342–348. 10.1159/000330303 [DOI] [PubMed] [Google Scholar]
- Yamamoto A. Y., Mussi-Pinhata M. M., de Deus Wagatsuma V. M., Marin L. J., Duarte G., Figueiredo L. T. M. (2007). Human cytomegalovirus glycoprotein B genotypes in Brazilian mothers and their congenitally infected infants. J. Med. Virol. 79 1164–1168. 10.1002/jmv [DOI] [PubMed] [Google Scholar]
- Yi L., Wang D.-X., Feng Z.-J. (2008). Detection of human cytomegalovirus in atherosclerotic carotid arteries in humans. J. Formos. Med. Assoc. 107 774–781. 10.1016/S0929-6646(08)60190-4 [DOI] [PubMed] [Google Scholar]
- Zheng S.-S., Zhou L., Qian J., Cai T., Fan J., Ma W.-H. (2002). Cytomegalovirus glycoprotein B sequence variation in Chinese liver transplant recipients. Hepatobiliary Pancreat. Dis. Int 1 26–29. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HPLC of furin-processed FRET substrates. Each substrate was incubated with/without furin for 12 h and further analyzed by HLPC. Substrate and products were followed by fluorescence (Abz, ex320 nm; em420 nm) quenching (Eddnp, uv = 365 nm) indicating single cleavage site for all tested sequences. (A) gB3_22. (B) gB_20. (C) gB3_26 (D) gB2_18. (E) gB2_13. (F) gB2_19.
Mass spectra of furin-processed substrates. Furin (50 nM) were incubated with each substrate for 12 h and further analyzed by Maldi-TOF (Bruker Daltonics, Germany). (A) gB3_22 substrate and products (1549.3, 678.7, and 888.7 Da). (B) gB2_13 (1634.4, 706.7, and 945.7 Da). (C) gB_20 (1604.3, 706.7, and 915.6 Da). (D) gB2_18 (1664.4, 706.7, and 975.8 Da) (E) gB3_26 (1577.3, 678.7, and 916.7 Da). (F) gB2_19 (1689.4, 706.7, and 1000.8 Da).
Maldi-TOF spectrum of the synthetic FRET peptide ATRASTSDNNT (Strive et al., 2002), not processed by furin. FRET ATRASTSDNNT (5 μM, 1,592.2 Da) was tested as a furin substrate in spectrofluorimeter and Maldi-TOF after 2 h of incubation with furin.
Multiple sequence alignment of the 35 unique sequences viewed by nucleotides.