

Abstract
The human skin microbiota is quantitatively dominated by Gram-positive bacteria, detected by both culture and metagenomics. However, metagenomics revealed a huge variety of Gram-negative taxa generally considered from environmental origin. For species affiliation of bacteria in skin microbiota, clones of 16S rRNA gene and colonies growing on diverse culture media were analyzed. Species-level identification was achieved for 81% of both clones and colonies. Fifty species distributed in 26 genera were identified by culture, mostly belonging to Actinobacteria and Firmicutes, while 45 species-level operational taxonomic units distributed in 30 genera were detected by sequencing, with a high diversity of Proteobacteria. This mixed approach allowed the detection of 100% of the genera forming the known core skin Gram-negative microbiota and 43% of the known diversity of Gram-negative genera in human skin. The orphan genera represented 50% of the current skin pan-microbiota. Improved culture conditions allowed the isolation of Roseomonas mucosa, Aurantimonas altamirensis and Agrobacterium tumefaciens strains from healthy skin. For proteobacterial species previously described in the environment, we proposed the existence of skin-specific ecotypes, which might play a role in the fine-tuning of skin homeostasis and opportunistic infections but also act as a shuttle between environmental and human microbial communities. Therefore, skin-associated proteobacteria deserve to be considered in the One-Health concept connecting human health to the health of animals and the environment.
Keywords: Proteobacteria, Skin microbiota, Species-level identification, Culture, 16S rRNA gene
1. Introduction
Various human microbiota are now deciphered in depth thanks to metagenomics and new generation DNA sequencing (NGS) [1], [2]. These methods allowed comparative microbial ecology by examining the influence of environmental factors, body sites and pathology on the diversity of microbiome [3], [4], [5], [6], [7], [8], [9], [10], [11], [12]. Compared to culture-based approaches, NGS has generally extended the range of microbial diversity of complex communities and detected yet-to-be-described bacterial taxa. However, culture-based approaches detect living microorganisms and may be more sensitive for the detection of minority bacteria able to grow on artificial media in monoculture [13], [14]. Polyphasic studies associating molecular and culture-based analyses remained scarce [15], [16] despite the development of high-throughput methods named culturomics [13].
Human skin is colonized by a complex microbial community, considered for a long time as dominated by Gram-positive bacteria such as staphylococci, micrococci, corynebacteria, Propionibacterium spp., Brevibacterium spp., and members of the genus Acinetobacter being the most frequently encountered Gram-negative bacteria in human skin microbiota. These bacteria belong to the long-term resident microbiota, based on the frequency with which they have been detected [2], [4], [15], [17], [18], [19], [20], [21]. Beside these well-described bacteria, culture-independent approaches demonstrate that Gram-negative bacteria, particularly Proteobacteria, represent an important component of the skin microbiota [2], [12], [19], [20], [21], [22]. Despite their detection in numerous metagenomic studies and diverse physio-pathological conditions, cutaneous Proteobacteria remained poorly described, mainly because isolates were not available and sequences generated by NGS were generally too short to obtain an accurate species affiliation.
This study aimed to precise the phylogenetic relationships and taxonomy of Proteobacteria from healthy human skin microbiota by analysis of 16S rRNA gene sequences of more than 800 bp and by strain cultivation. Phylotypes and isolates will be described to the species and/or genotype level in order to compare skin-associated Proteobacteria with related environmental ecotypes.
2. Materials and Methods
2.1. Cutaneous samples, isolates and clones
The present study was an ancillary study proposed beside clinical study on atopy (Institut de Recherche Pierre Fabre, unpublished data). Briefly, in the main study, donors are tertiary workers (no healthcare workers) in urban areas without particular exposure to animals and soil. They took a shower using mild soap between 4 and 6 h before sampling. For each donor, one sample was taken from the inner forearm protected by clean personal garments until sampling. Sampling was performed according to the method described by Fleurette using a transfer fluid able to maintain the viability and to avoid proliferation of the microbiota [23]. Briefly, the open end of a sterile glass cylinder, with an area of 3.14 cm2 was manually placed on the skin. Two milliliters of RTF medium [23] sterilized by 0.22 μm filtration was used to collect skin microbiota. Four successive spots were realized each during 1 min with the same liquid or a total forearm skin area of 12.56 cm2. One tenth of the liquid was used for microbial culture, the remaining was stored at − 20 °C for molecular analysis.
A total of 311 isolates and 278 16S rRNA gene clones obtained from two healthy-donors included in the clinical study were analyzed herein. Isolates were obtained by culture on Columbia agar supplemented with 5% sheep blood (Biomérieux) incubated under aerobic and anaerobic (Anoxomat) conditions at 37 °C for 5 days. One colony of each morphotype observed was harvested, sub-cultured and stored at − 20 °C in cryopreservative medium (Eugon broth + 10% glycerol). A selective isolation of Gram-negative bacilli was performed at 30 °C for 5 days using culture media implemented by vancomycin (7.5 mg/L): R2A agar (Pronadisa), Schaedler agar (Difco) and Chocolat agar (Difco) for 8 samples from 2 other healthy donors.
The 16S rRNA clones library had been obtained after total DNA extraction directly from samples (MagNA Lyser Green beads, Roche Molecular Biochemicals), and purification (QIAamp DNA micro purification kit, Qiagen, Germany). The amplification of a 863 bp-sequence of 16SrRNA gene was performed using the universal primers, Universel1 (5′ AGCAGCCGCGGTRATWC 3′) and Universel2 (5′ ACGGGCGGTGTGTAC 3′) [24], [25]. The purified amplicons (QIAquick PCR Purification kit, Qiagen) were ligated into the plasmid vector pGEM®-T Easy, then transformed into JM109 High Efficiency competent cells using the pGEM®-T Easy Vector Systems kit (Promega). JM109 transformed cells were streaked onto Luria–Bertani agar plates containing 100 μg/mL ampicillin, 40 μg/mL X-gal, 0.5 mM IPTG for blue/white screening as previously described [15]. The insert of each selected white clone was amplified and sequenced (ABI PRISM 3130 Genetic Analyzer, Applied Biosystems, USA) using T7 and SP6 primers as previously described [26].
2.2. Bacterial identification
Each colonial morphotype was submitted to identification by molecular methods based on 16S rRNA gene sequencing [15]. For members of the genus Staphylococcus, ITS 16S–23S and tuf gene sequencing were used for species affiliation [27], [28].
2.3. Phylogenetic analysis and taxon affiliation
Sequences used for further sequence analysis corresponded to high-quality sequences, i.e. presenting less than 0.5% undetermined positions. Nucleotide sequences were analyzed using the Blast program in NCBI and Greengenes database. Chimera was detected using Bellorophon software in the Greengenes website (greengenes.lbl.gov). For database comparison, we retained the stringent threshold value of 98.7% of similarity with a fully defined cultured strain (type or non-type) as recommended for bacterial species delineation [29], for the affiliation of a clone to a species- or a genus-level operational taxonomic unit (OTU). When a similarity level of more than 98.7% was obtained for an uncultured bacterial clone only, the sequence was classified as 16S rRNA gene clone and affiliated to a genus according Greengenes database. Beside sequences identified herein, the sequences used to reconstruct phylogenies were chosen by Blast analysis as follows. For each clone sequence, we included the most related deposited sequence and the most related sequences corresponding to (1) validated species and (2) human skin clone. The dataset of sequences was aligned using ClustalW software [30]. The most appropriate substitution model determined according to Akaike information criterion calculated with Modeltest (v.3.7) was GTR plus gamma distribution, plus invariant sites [31]. ML phylogenetic analysis was performed using PHYML v2.4.6, gamma shape parameter being estimated from the dataset [32]. ML bootstrap support was computed using PhyML after 100 reiterations.
The Shannon-Wiener (H′) and Simpson (D) diversity indexes were calculated for each phylum and according to the type of method (culture or uncultured approaches) [33], [34].
3. Results
3.1. Taxonomic diversity in the skin microbiota
Table 1 shows species-level identification of the bacterial isolates and clones. Fig. 1 summarizes the qualitative and quantitative diversity repartition of the skin microbiota according to phylum and type of cell wall structure. Our approach allowed species-level identification for 81% of both clones and colonies. The culture detected 50 species-level OTUs including 39 taxonomic species distributed in 26 genera among 311 colonies tested. The sequencing of 278 clones allowed the identification of 45 species-level OTUs in 30 genera, including 26 taxonomic species, 6 pairs of undifferentiated taxonomic species, 7 groups of related species and 5 unaffiliated OTUs (Table 1).
Table 1.
Bacterial species identified after culture by phenotypic and/or molecular methods, and genera or species-level OTUs obtained by 16S rRNA gene sequencing.
| Phyla | Firmicutes | Actinobacteria | Proteobacteria | Bacteroidetes | |
|---|---|---|---|---|---|
| Species or genus | Identification after culture | Bacillus sp.a | Actinomyces sp.a | Acinetobacter radioresistens | Chryseobacterium indologenes |
| Bacillus sphaericus | Brevibacterium sp. | Enterobacter sp.a | Prevotella buccae | ||
| Enterococcus faecalis | Cellulomonas sp.a | Enterobacter aerogenes | |||
| Eubacterium lentum | Cellulosimicrobium cellulans | Klebsiella oxytoca | |||
| Gemella morbillorum | Corynebacterium sp. | Moraxella osloensis | |||
| Staphylococcus sp.a | Corynebacterium amycolatum | Pasteurella sp.a | |||
| Staphylococcus aureus | Corynebacterium riegelii | Pseudomonas aeruginosa | |||
| Staphylococcus auricularis | Dermabacter hominis | Serratia ficaria | |||
| Staphylococcus capitis | Kocuria sp.a | Sphingomonas mucosissima | |||
| Staphylococcus caprae | Kocuria kristinae | ||||
| Staphylococcus cohnii | Micrococcus sp. | ||||
| Staphylococcus epidermidis | Micrococcus luteus | ||||
| Staphylococcus haemolyticus | Micrococcus muciloginosus | ||||
| Staphylococcus hominis | Propionibacterium sp.a | ||||
| Staphylococcus lugdunensis | Propionibacterium acnes | ||||
| Staphylococcus schleiferi | Propionibacterium avidum | ||||
| Staphylococcus sciuri | Propionibacterium granulosum | ||||
| Staphylococcus warnerii | Rothia dentocariosa | ||||
| Staphylococcus xylosus | |||||
| Streptococcus sp.a | |||||
| Streptococcus oralis | |||||
| No. of isolates | 175 | 106 | 14 | 16 | |
| No. of species | 21 | 18 | 9 | 2 | |
| No. of genera | 6 | 10 | 8 | 2 | |
| Identification by 16S rRNA gene sequencing | Bacillus sp. | Corynebacterium sp.c | Acidovorax delafieldii | Sphingobacterium siyangense | |
| Staphylococcus epidermidis groupa | Detzia papillomatosis/Detzia cinnameab | Acinetobacter lwoffii | |||
| Staphylococcus hominis | Micrococcus yunnanensis | Acinetobacter junii/Acinetobacter baumanniia | |||
| Streptococcus sp. | Propionibacterium acnes | Acinetobacter johnsonii | |||
| Streptococcus infantis | Propionibacterium granulosum | Aeromonas salmonicida and relateda | |||
| Veillonella rogosae | Pseudonocardia chlorethenivorans | Agrobacterium tumefaciens/Rhizobium pusensea | |||
| Alcanivorax dieselolei | |||||
| Alcanivorax venustensisb | |||||
| Aurantimonas coralicida | |||||
| Bradyrhizobium elkanii | |||||
| Escherichia coli | |||||
| Haemophilus parainfluenzae | |||||
| Halomonas neptunia/Halomonas alkantarticaa | |||||
| Halomonas aquamarina | |||||
| Idiomarina loihiensis | |||||
| Imtechium assamiensisb | |||||
| Marinobacter hydrocarbonoclasticus | |||||
| Neisseria perflava/Neisseria subflavaa | |||||
| Paracoccus haeundaensis | |||||
| Paracoccus seriniphilus | |||||
| Paracoccus yeeii | |||||
| Paracoccus sp. 1c | |||||
| Paracoccus sp. 2c | |||||
| Pelomonas puraquae | |||||
| Pseudomonas putida groupa | |||||
| Pseudomonas stutzeri groupa | |||||
| Pseudomonas fluorescens groupa | |||||
| Rasbo bacteriumc | |||||
| Sphingomonas asaccharolytica groupa | |||||
| Sphingomonas sp.c | |||||
| Sphingomonas aquatilis | |||||
| Stenotrophomonas maltophilia | |||||
| No. of clones | 81 | 33 | 127 | 1 | |
| No. of species level OTUs | 6 | 6 | 32 | 1 | |
| No. of genera | 4 | 5 | 20 | 1 | |
A group or a pair of described species that could not be discriminated by the markers used.
Taxonomic name not validly published.
Phylotypes corresponding to undescribed species.
Fig. 1.

Bacterial diversity of the skin microbiota according to phylum and type of cell wall structure. Number of colonies and clones assessed the quantitative representation of each phylum. Number of cultured species and number of uncultured OTUs assessed the species diversity in each phylum.
Gram-positive bacteria belonging to Firmicutes and Actinobacteria represented 90.3% of the colonial morphotypes studied. Genera belonging to Firmicutes (175 isolates) were Bacillus, Streptococcus, Enterococcus, Gemella, Eubacterium and Staphylococcus. The latter was the most diverse genus of the cultivable skin microbiota, with 15 different species identified. Actinobacteria appeared more diverse to the genus level since the 106 isolates affiliated to this phylum belonged to 10 different genera (Table 1; Fig. 1). Uncultured clones affiliated to Firmicutes and Actinobacteria were minority (Table 1; Fig. 1) compared to clones belonged to Gram-negative bacterial phyla (Table 1): Proteobacteria of the alpha (27.0%), beta (23.9%) and gamma (47.2%) subdivisions and Bacteroidetes (1.9%). Most of the Gram-negative genera identified by sequencing were not detected in culture, except for Acinetobacter, Pseudomonas and Sphingomonas. Indeed, only 30 of the total of 311 bacterial colonies tested (9.7%) corresponded to Gram-negative bacteria. Fourteen isolates belonged to Proteobacteria (Enterobacteriaceae and non-fermentative bacilli) (Table 1), and 16 isolates of Gram-negative anaerobes were represented by one unique species, Prevotella buccae in the phylum Bacteroidetes.
Fig. 1 revealed that the distribution of clones and colonies by phylum and type of cell wall structure differed markedly. Cultivating Gram-positive bacteria were quantitatively the most represented (175 colonies, Fig. 1) but by contrast their taxonomic diversity was low (21 species, H′ and D indexes of 0.3236 and 0.0057 respectively) (Fig. 1; Table 2). The ratio Gram-positive versus Gram-negative varied clearly according to the method used: the Gram-positive bacteria were mostly detected by culture (ratio of 9.37) but only partially by the molecular approach (ratio of 0.89) and vice versa for Gram-negative bacteria (Fig. 1). Moreover, most clones had no cultivable counterparts. These results showed major discrepancy between Gram-positive and Gram-negative diversities assessed by each approach. According to these results, the diversity was higher for Gram-negative bacteria retrieved by molecular approach than by culture, in particular for Proteobacteria (H′ = 0.3384 vs 0.1396 and D = 0.0088 vs 0.0769 for uncultured and culture approaches, respectively) (Table 2).
Table 2.
Phylum diversity indexes for culture and uncultured approaches. H′: Shannon-Wiener index, D: Simpson index.
| Culture approach |
Uncultured approach |
|||
|---|---|---|---|---|
| H′ | D | H′ | D | |
| Firmicutes | 0.323556959 | 0.005747126 | 0.366337084 | 0.0125 |
| Actinobacteria | 0.366860144 | 0.00952381 | 0.271695022 | 0.03125 |
| Gram positive | 0.091653203 | 0.003571429 | 0.354596189 | 0.008849558 |
| Proteobacteria | 0.139582952 | 0.076923077 | 0.338360873 | 0.007936508 |
| Bacteroidetes | 0.152653592 | 0.066666667 | 0.022681561 | 0 |
| Gram negative | 0.225587993 | 0.034482759 | 0.336876674 | 0.007874016 |
To address the lack of growing of cutaneous Proteobacteria on blood agar medium, we tested a posteriori their growth on Gram-negative selective media containing vancomycin. We tested the R2A medium, a medium developed to study bacteria that will not readily grow on rich and complex organic media [35] such as bacteria from water or other poor environments, as well as enriched media, chocolate agar and Scheadler broth. In this purpose, 8 additional skin samples were obtained from 2 additional healthy donors volunteers from the research team. Five samples were positive for Protebacteria: Roseomonas mucosa (n = 2), Agrobacterium tumefaciens (n = 2), Acinetobacter johnsonii (n = 1), Acinetobacter lwoffii (n = 1), Aurantimonas altamirensis (n = 2) and Pseudomonas psychrotolerans (n = 1). All strains were found onto chocolate plus vancomycin medium except Roseomonas mucosa that only grew in Schaedler medium. No growth was observed onto R2A agar plates.
3.2. Phylogenetic taxonomy of Proteobacteria in skin microbiota
Proteobacterial sequences were classified according to alpha, beta and gamma subdivisions that contained 11, 4 and 17 OTUs representing 43, 38 and 75 different clones, respectively. The phylogenetic trees showed the repartition of the clone sequences in the known proteobacterial diversity (Fig. 2A–C).
Fig. 2.


Phylogenetic tree showing the relationship of the 16S rRNA gene sequences.
ML phylogenetic tree showing the 16S rRNA gene sequence relationships of the clones obtained in this study (in blue) with cultured and uncultured members of Alphaproteobacteria (A), Betaproteobacteria (B) and Gammaproteobacteria (C). The sequences used to reconstruct this tree were obtained from the GenBank database (accession numbers are indicated in brackets). Sequences from uncultured bacteria obtained from skin human samples were in green. Sequences of strains of validate species and some not validate but published species were in black bold type. Neisseria perflava was used as outgroup for Alphaproteobacteria, Agrobacterium tumefaciens C58 for Betaproteobacteria and Pelomonas aquatica for Gammaproteobacteria. The scale bar indicates substitutions per nucleotide position. Numbers given at the nodes represent bootstrap percentages.
The Alphaproteobacteria subdivision was represented by the genus Paracoccus (Rhodobacterales) (34.9%), and the orders Sphingomonadales (44.2%) and Rhizobiales (20.9%). The phylogenetic analysis (Fig. 2A) allowed affiliation to the species Aurantimonas coralicida, A. tumefaciens/Rhizobium pusense, Bradyrhizobium elkanii, Paracoccus yeeii, Paracoccus haeundaensis, Paracoccus seriniphilus and Paracoccus marinus. Clones affiliated to the genus Sphingomonas were related but at less than 98% identity with several species not discriminated by 16S rRNA gene sequencing including Sphingomonas aquatilis (Fig. 2A). In the Paracoccus clade, two clones (PA + C_C06 and PA + C_C07) corresponded to a yet undescribed species. In Rhizobiales, one clone was related to “Rasbo bacterium”, an undescribed species detected in plasma samples during acute sepsis [36]. In all cases, a clone detected in metagenomic studies on skin microbiota was related to the clone detected herein (Fig. 2A).
The Betaproteobacteria subdivision was dominated by Pelomonas, which represented 70% of the clones of this sub-division with mainly the species Pelomonas puraquae. Other clones grouped with the non validate species Imtechium assamiensis. Published cutaneous clones grouped together with this non validate species but differed from Aquabacterium fontiphilum and other related species (Fig. 2B). Other clones grouped in Acidovorax spp. and Neisseria spp. but identification to the species level was not accurate for these genera. The most related but undistinguished species were indicated in the tree (Fig. 2B).
The gamma subdivision was mainly represented by the genera Acinetobacter (26.7%), Idiomarina (16%), Alcanivorax (12%) and Pseudomonas (9.4%) and the species Escherichia coli (20%). In the genus Acinetobacter, the clones were distributed among the species A. lwoffii, Acinetobacter baumanii, Acinetobacter junii and A. johnsonii. Seven clones were distributed among pseudomonads (Fig. 2C) mainly in Pseudomonas putida, Pseudomonas stutzeri and Pseudomonas fluorescens groups.
Finally, the phylogeny confirmed that four phylotypes (noted c in Table 1) corresponded to undescribed species of Alphaproteobacteria.
3.3. Core and pan Gram-negative skin microbiota
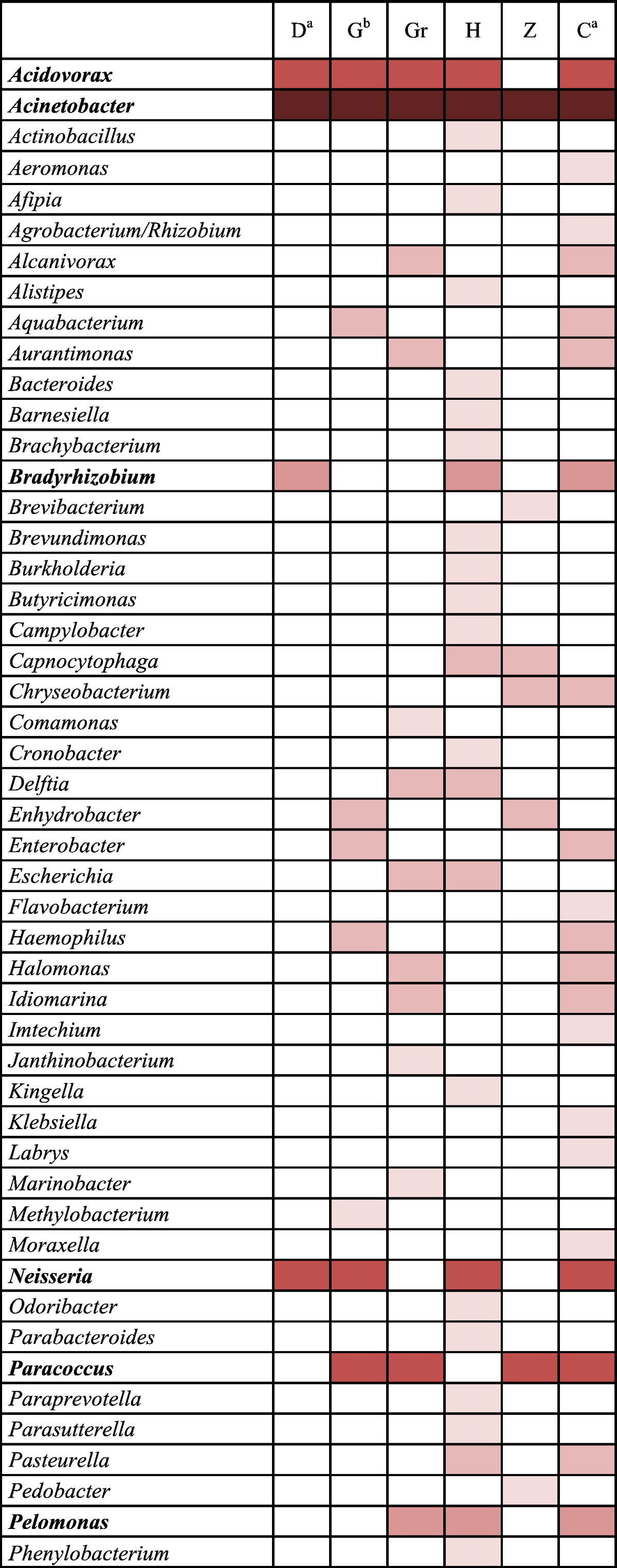
The recent studies of molecular ecology have enriched our knowledge about bacteria associated with the human skin. The Gram-negative diversity described in these studies and herein is compared in Table 3. The genera Acidovorax, Acinetobacter, Pseudomonas and Stenotrophomonas were isolated from independent donors and from different sampling sites (forearm, forehead, inner elbow and back) in all of the 6 studies or in all studies but one. Therefore, one can hypothesize that these bacteria may belong to the permanent core skin microbiota. If we consider the bacteria detected in at least 50% of the studies namely core50 skin microbiota, ten genera are highlighted (in bold in Table 3). Our approach coupling culture and molecular approach detected 100% of the genera forming the current core50 skin microbiota (Table 3). Moreover, we detected from only 2 subjects 43% of the known diversity of Gram-negative genera in human skin (Table 3).
Table 3.
Data comparison with the 5 main published studies that characterized healthy skin microbiota by molecular approaches.
This table compiles all the genus of Gram-negative bacteria that have been detected on the skin. Data are obtained from 5 published studies and this study. Studies were based on molecular methods in combination or not with culture analysis. D for Dekio et al. [15], G for Gao et al. [19], Gr for Grice et al. [22], H for Human Microbiome Project Consortium [2], Z for Zeeuwen et al. [12], and C for this study. The color intensity is function of the frequency of detection. The core50 microbiota corresponded to genera found in at least 50% of the published studies. Genera belonged to the core50 are in bold face. % of Gram negative skin pan-microbiota is the proportion of genera identified in each study among total number of known genera indentified on the skin in the 6 studies, namely skin pan-microbiota (62 genera). % Gram-negative orphan is the proportion of orphan genera identified in each study among the skin pan-microbiota. % of Gram negative genera of the core50 microbiota is the proportion of genera belonging to the core50 in each study among the current core50 microbiota.

4. Discussion
Current metagenomics based on NGS explores in depth the bacterial diversity and avoids the bias of cultivability. It has changed our vision of human microbiota but several limitations worth to be underlined. Particularly, NGS presents a lack of sensitivity for the detection of minority OTUs present at less than 106 bacteria per sample [37]. The size of sequenced DNA fragments varied greatly among techniques and studies rending comparison hard to perform and explaining some discrepancies [38]. In the case of the gut microbiota, the relative abundance of the phyla Bacteroidetes and Firmicutes depends on the 16S rRNA gene hypervariable region analyzed [39]. These discrepancies observed to the phylum level are probably magnified when genera or species are considered. Generally, in NGS studies, the quality of taxonomic affiliation is low and limited to the genus due to the size of generated sequences. Herein, we used classical methods such as Sanger sequencing and culture with the aim to affiliate to taxonomic species a collection of clones and bacterial colonies isolated from healthy skin. This combined approach was targeted on Proteobacteria that present high diversity in skin microbiota but remain scarcely described to the species-level in previous studies on skin metagenome.
Dekio et al. in 2005 described for the first time the presence of Pseudomonas, Stenotrophomonas, Acidovorax, Bradyrhizobium and Neisseria as proteobacteria inhabiting the skin microbial ecosystem [15]. Then, Gao et al. (2007) and Grice et al. (2008) reported two major molecular analyses confirming that Gram-negative bacteria are common residents and not contaminants from environment or other microbiota [19], [22]. They reported also the presence of bacteria usually found in the environment such as Methylobacterium, Sphingobium, Diaphorobacter, Enhydrobacter, Serratia, Pedomicrobium, Paracoccus, Halomonas and Delftia. Proteobacteria appears qualitatively the more diverse phylum [22] even if the quantitative predominance of the Gram-positive genera classically detected in culture is confirmed [20]. Recently, Probst et al. characterized another underestimated biodiversity, the Archaea of the human skin microbiota using the cloning of 16S rRNA gene PCR products [40].
Our study, limited to the forearm of two subjects, confirmed that the skin bacterial community is dominated in diversity by Gram-negative species, which participate to the skin pan-microbiota expanse. In this study, orphan genera represented 50% of the current pan-microbiota. Similarly, they represented 58% of the pan-microbiota in the study of Grice [22]. Moreover, about 70% (with 84.4% of Proteobacteria) of the genera detected by Gao et al. on the forearm were specific to an individual donor [19]. This high variability among subjects and studies could suggest that members of Proteobacteria are temporary environmental contaminants of the human skin. The question if Proteobacteria are transient or resident on human skin is not definitively settled in this pilot study performed on two subjects without longitudinal follow-up and without comparison according environmental conditions. However, the consistency of results between methodologically different studies suggests that very diverse Proteobacteria actually reside in skin microbiota and that beside the 10 genera found to belong to the core50 microbiota, Gram-negative diversity participates probably to the inter individual and inter body site variations [41].
By the approaches proposed herein, we detected all the members of the core50 Gram-negative skin microbiota and 43% of the Gram-negative skin pan-microbiota. These data validate our strategy, in spite of the lack of NGS data. It is particularly noteworthy that only a magnitude of 2 was observed between the 87 skin-associated bacterial genera detected in Human Metagenomic Project (4 cutaneous site and 242 subjects) [2] versus 48 genera detected by cloning and culture from one cutaneous site from 2 subjects in this study.
Since long ago, Gram-positive bacteria (Firmicutes and Actinobacteria) are recognized as the major component of the culturable skin microbiota [17]. When blood agar culture at 37 °C was used, we detected 92.0% of Gram-positive bacteria. Conversely, the same culture conditions yielded only few Gram-negative bacteria whereas most of those that had been detected by sequencing are considered to be culturable. The richness of the medium and the incubation temperature could be the causes of growth defect. The incubation at 30 °C of rich and poor media with vancomycin that inhibited Gram-positive bacteria led to the growth of R. mucosa, A. tumefaciens, A. altamirensis, A. johnsonii, A. lwoffii and P. psychrotolerans. Growth is observed only for rich media but not onto R2A whereas these strains belong to taxa that generally grow onto poor media such as R2A medium (Bergey's manual). This result suggests that the species or strains associated to skin microbiota may present particular requirements. Therefore, the existence of particular ecotypes of Proteobacteria specialized in human skin mutualism might be hypothesized.
Sequencing and phylogeny reconstruction detected many Proteobacteria considered to be environmental belonging to the orders Rhizobiales, Sphingomonadales, Burkholderiales, Oceanospirillales, Alteromonadales and Pseudomonadales. However, lifestyle of donors, that are tertiary workers in urban area, excluded regular or recent exposure to animals, plants, fresh or marine waters and soil. Most clones assumed to belong to environmental species have closest relatives sequences detected previously from the human skin (clones in green in Fig. 2) rather than from other ecosystems [4], [19], [22]. These clones being detected in independent analysis, one can hypothesize again that some of these sequences represent specific ecotypes in the human cutaneous microbiota. The metagenomic study of Mathieu et al. demonstrated functions that clearly illustrate the unique life style of the skin microbial communities and reinforce the hypothesis skin-specific ecotypes [42]. Microbiota description at the species-level completed by strain isolation is a way to study these ecotypes.
Some phenotypic and metabolic traits appear common to several species detected in skin microbiota and could help to define the proteobacterial skins ecotypes. Mainly, several clones correspond to halophilic genera and species of Gammaproteobacteria isolated in marine environments: Alcanivorax dieseoli, Alcalinivorax venustensis, Halomonas aquamarina, Halomonas neptunia or Halomonas alkantartica, Marinobacter hydrocarbonoclasticus and Idiomarina loihiensis. Halophily is consistent with the salt rich environment of the skin. Some of halophilic species in Gammaproteobacteria are also hydrocarbonoclastic: Alcanivorax dieseoli, Halomonas alkantartica, and Marinobacter hydrocarbonoclasticus. Such proprieties are also described for the genera Acinetobacter and Pseudomonas. Other clones grouped with the non validate species Imtechium assamiensis were described as forming biofilms on polychlorinated biphenyls surfaces [43]. Out of gammaproteobacteria Pseudonocardia chlorethenivorans and Sphingomonas spp. are other alkane-degrading bacteria detected herein [44], [45], [46].
The cutaneous microbiota is a major bacterial reservoir involved in opportunistic infections, particularly in health-care associated infections (HAI). Consequently, hand washing and skin antisepsis are now considered the most important interventions to prevent the spread of HAI agents [47]. Among Proteobacteria detected in this study, some species such as Stenotrophomonas, Aeromonas, Pseudomonas and Acinetobacter have been associated with opportunistic infections [48], [49], [50]. To our knowledge, the opportunistic pathogen Aeromonas has never been detected in the normal skin microbiota so far. Beside these well-known opportunistic pathogens, bacteria assumed to be environmental are more and more described in opportunistic infections. For instance, A. altamirensis was described as part of the microbial community that produces deleterious colonization of Paleolithic paintings in Altamira Cave [51] and since then, it has been mainly described in human infections [52]. For A. tumefaciens, a well-known phytopathogen agent, the population structure showed a genetic sub-population associated with human beings and involved in infections, clearly apart from environmental and plant-associated strains [53]. Finally, the genus Roseomonas gathers species mainly isolated from environment but R. mucosa and Roseomonas gilardii are frequently described in human infections [54]. By analogy with the main lifestyle observed for most members of Roseomonas spp., the source of human infections caused by R. mucosa is searched into environment and not among endogen microbial community [54], [55]. These cases are particularly emblematic of the need of species-level identification in human microbiota in order to assess infectious risk and prevent opportunistic infections.
The association of known Gram-positive species to disease or healthy states has been previously established, for example, acne and atopic dermatitis correlated with the prevalent colonization of Propionibacterium acnes and Detzia maris, respectively [10], [56]. Concerning Gram-negative bacteria, Proteobacteria seem more abundant in biopsies of psoriasis lesions and in chronic wounds than in healthy skin, without knowing whether this skin microbiota disequilibrium is a cause or a consequence of these dermatologic disorders [7], [9], [12]. Considering the variations in diversity among subjects and body sites of Proteobacteria in skin pan-microbiota, it is probable that the diversity by itself is not directly correlated to dermatologic disorders [41]. However, the Human Metagenome Project showed that metabolic variations among individuals might indicate pathways for facing personalized immune and environmental or behavioral exposures. The wide Gram-negative bacteria repertory and its interindividual variations in skin microbiome might participate to these metabolic variations.
5. Conclusion
Inter-ecosystem comparisons suggest that the human skin communities possess strong capacities for interacting with their environment [42]. Beyond their large diversity, skin-associated Proteobacteria slightly differed from their environmental counterpart. The genetic relatedness between skin and environmental ecotypes in the same proteobacterial species might act as shuttles linking environmental and human microbial communities. In this context, virulence or resistance genes exchange could be of great concern in the emergence of multiresistant pathogenic bacteria.
The One Health concept recognizes that the health of humans is connected to the health of animals and environment. Microbiota and particularly skin microbiota probably have a pivotal role in the continuity between human-associated bacteria and other microbial communities in animals, plants and environments. Therefore, microbiota relationships and overlaps with other communities need to be explored in the One Health approach beside other means of pathogenic exchanges such as vectors and direct transmission of pathogens.
Acknowledgments
This work was supported by the CERPER, Pierre Fabre Dermo-Cosmétique, the French Research Ministry and ADEREMPHA association. The authors are grateful to Pr. Devine from Leeds Dental Institute for critical reading of this manuscript.
References
- 1.NIH HMP Working Group, Peterson J., Garges S., Giovanni M., McInnes P., Wang L. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Human Microbium Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207-4. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McBride M.E., Duncan W.C., Knox J.M. The environment and the microbial ecology of human skin. Appl. Environ. Microbiol. 1977;33:603–608. doi: 10.1128/aem.33.3.603-608.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fierer N., Hamady M., Lauber C.L., Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl. Acad. Sci. 2008;105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costello E.K., Lauber C.L., Hamady M., Fierer N., Gordon J.I., Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grice E.A., Kong H.H., Conlan S., Deming C.B., Davis J., Young A.C. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han A., Zenilman J.M., Melendez J.H., Shirtliff M.E., Agostinho A., James G. The importance of a multifaceted approach to characterizing the microbial flora of chronic wounds. Wound Repair Regen. 2011;19:532–541. doi: 10.1111/j.1524-475X.2011.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staudinger T., Pipal A., Redl B. Molecular analysis of the prevalent microbiota of human male and female forehead skin compared to forearm skin and the influence of make-up. J. Appl. Microbiol. 2011;110:1381–1389. doi: 10.1111/j.1365-2672.2011.04991.x. [DOI] [PubMed] [Google Scholar]
- 9.Fahlén A., Engstrand L., Baker B.S., Powles A., Fry L. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch. Dermatol. Res. 2012;304:15–22. doi: 10.1007/s00403-011-1189-x. [DOI] [PubMed] [Google Scholar]
- 10.Kong H.H., Oh J., Deming C., Conlan S., Grice E.A., Beatson M.A. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ursell L.K., Clemente J.C., Rideout J.R., Gevers D., Caporaso J.G., Knight R. The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J. Allergy Clin. Immunol. 2012;129:1204–1208. doi: 10.1016/j.jaci.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeeuwen P.L., Boekhorst J., van den Bogaard E.H., de Koning H.D., van de Kerkhof P.M., Saulnier D.M. Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biol. 2012;13:R101. doi: 10.1186/gb-2012-13-11-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greub G. Culturomics: a new approach to study the human microbiome. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2012;18:1157–1159. doi: 10.1111/1469-0691.12032. [DOI] [PubMed] [Google Scholar]
- 14.Alexeyev O.A. Bacterial landscape of human skin: seeing the forest for the trees. Exp. Dermatol. 2013;22:443–446. doi: 10.1111/exd.12160. [DOI] [PubMed] [Google Scholar]
- 15.Dekio I., Hayashi H., Sakamoto M., Kitahara M., Nishikawa T., Suematsu M. Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J. Med. Microbiol. 2005;54:1231–1238. doi: 10.1099/jmm.0.46075-0. [DOI] [PubMed] [Google Scholar]
- 16.Mathieu A., Vogel T.M., Simonet P. The future of skin metagenomics. Res. Microbiol. 2014;165:69–76. doi: 10.1016/j.resmic.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Leyden J.J., McGinley K.J., Nordstrom K.M., Webster G.F. Skin microflora. J. Invest. Dermatol. 1987;88(3):65s–72s. doi: 10.1111/1523-1747.ep12468965. [DOI] [PubMed] [Google Scholar]
- 18.Chiller K., Selkin B.A., Murakawa G.J. Skin microflora and bacterial infections of the skin. J. Investig. Dermatol. 2001;6:170–174. doi: 10.1046/j.0022-202x.2001.00043.x. [DOI] [PubMed] [Google Scholar]
- 19.Gao Z., Tseng C., Pei Z., Blaser M.J. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. 2007;104:2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Z., Perez-Perez G.I., Chen Y., Blaser M.J. Quantitation of major human cutaneous bacterial and fungal populations. J. Clin. Microbiol. 2010;48:3575–3581. doi: 10.1128/JCM.00597-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dekio I., Sakamoto M., Hayashi H., Amagai M., Suematsu M., Benno Y. Characterization of skin microbiota in patients with atopic dermatitis and in normal subjects using 16S rRNA gene-based comprehensive analysis. J. Med. Microbiol. 2007;56:1675–1683. doi: 10.1099/jmm.0.47268-0. [DOI] [PubMed] [Google Scholar]
- 22.Grice E.A., Kong H.H., Renaud G., Young A.C., Bouffard G.G., Blakesley R.W. A diversity profile of the human skin microbiota. Genome Res. 2008;18:1043–1050. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleurette J., Transy M.J. Estimation of the activity of an antiseptic agent on skin microbial flora under various conditions. Ann. Biol. Clin. (Paris) 1976;34:203–210. [PubMed] [Google Scholar]
- 24.Khammar N., Malhautier L., Degrange V., Lensi R., Godon J.-J., Fanlo J.-L. Link between spatial structure of microbial communities and degradation of a complex mixture of volatile organic compounds in peat biofilters. J. Appl. Microbiol. 2005;98:476–490. doi: 10.1111/j.1365-2672.2004.02474.x. [DOI] [PubMed] [Google Scholar]
- 25.Trafny E.A., Kozłowska K., Szpakowska M. A novel multiplex PCR assay for the detection of Salmonella enterica serovar Enteritidis in human faeces. Lett. Appl. Microbiol. 2006;43:673–679. doi: 10.1111/j.1472-765X.2006.02007.x. [DOI] [PubMed] [Google Scholar]
- 26.Jeung J.U., Cho S.K., Shim K.S., Ok S.H., Lim D.S., Shin J.S. Construction of two pGEM 7Zf(+) phagemid T-tail vectors using AhdI-restriction endonuclease sites for direct cloning of PCR products. Plasmid. 2002;48:160–163. doi: 10.1016/s0147-619x(02)00122-1. [DOI] [PubMed] [Google Scholar]
- 27.Martineau F., Picard F.J., Roy P.H., Ouellette M., Bergeron M.G. Species-specific and ubiquitous-DNA-based assays for rapid identification of Staphylococcus aureus. J. Clin. Microbiol. 1998;36:618–623. doi: 10.1128/jcm.36.3.618-623.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martineau F., Picard F.J., Ke D., Paradis S., Roy P.H., Ouellette M. Development of a PCR assay for identification of staphylococci at genus and species levels. J. Clin. Microbiol. 2001;39:2541–2547. doi: 10.1128/JCM.39.7.2541-2547.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stackebrandt E., Brambilla E., Cousin S., Dirks W., Pukall R. Culture-independent analysis of bacterial species from an anaerobic mat from Lake Fryxell, Antarctica: prokaryotic diversity revisited. Cell. Mol. Biol. 2004;50:517–524. [PubMed] [Google Scholar]
- 30.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Posada D., Crandall K.A. MODELTEST: testing the model of DNA substitution. Bioinforma. Oxf. Engl. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 32.Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 33.Hill T.C.J., Walsh K.A., Harris J.A., Moffett B.F. Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 2003;43:1–11. doi: 10.1111/j.1574-6941.2003.tb01040.x. [DOI] [PubMed] [Google Scholar]
- 34.Gafan G.P., Lucas V.S., Roberts G.J., Petrie A., Wilson M., Spratt D.A. Statistical analyses of complex denaturing gradient gel electrophoresis profiles. J. Clin. Microbiol. 2005;43:3971–3978. doi: 10.1128/JCM.43.8.3971-3978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reasoner D.J., Blannon J.C., Geldreich E.E. Rapid seven-hour fecal coliform test. Appl. Environ. Microbiol. 1979;38:229–236. doi: 10.1128/aem.38.2.229-236.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blomqvist G., Wesslén L., Påhlson C., Hjelm E., Pettersson B., Nikkilä T. Phylogenetic placement and characterization of a new alpha-2 proteobacterium isolated from a patient with sepsis. J. Clin. Microbiol. 1997;35:1988–1995. doi: 10.1128/jcm.35.8.1988-1995.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagier J.-C., Armougom F., Million M., Hugon P., Pagnier I., Robert C. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin. Miicrobiol. Infect. 2012;18:1185–1193. doi: 10.1111/1469-0691.12023. [DOI] [PubMed] [Google Scholar]
- 38.Liu L., Li Y., Li S., Hu N., He Y., Pong R. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Claesson M.J., Wang Q., O'Sullivan O., Greene-Diniz R., Cole J.R., Ross R.P. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Probst A.J., Auerbach A.K., Moissl-Eichinger C. Archaea on human skin. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grice E.A., Segre J.A. The skin microbiome. Nat. Rev. Microbiol. 2011;9:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathieu A., Delmont TO, Vogel T.M., Robe P., Nalin R., Simonet P. Life on human surfaces: skin metagenomics. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macedo A.J., Timmis K.N., Abraham W.-R. Widespread capacity to metabolize polychlorinated biphenyls by diverse microbial communities in soils with no significant exposure to PCB contamination. Environ. Microbiol. 2007;9:1890–1897. doi: 10.1111/j.1462-2920.2007.01305.x. [DOI] [PubMed] [Google Scholar]
- 44.Kämpfer P., Kohlweyer U., Thiemer B., Andreesen J.R. Pseudonocardia tetrahydrofuranoxydans sp. nov. Int. J. Syst. Evol. Microbiol. 2006;56:1535–1538. doi: 10.1099/ijs.0.64199-0. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H., Lee Y.K., Zhang W., Lee H.K. Culturable actinobacteria from the marine sponge Hymeniacidon perleve: isolation and phylogenetic diversity by 16S rRNA gene-RFLP analysis. Antonie Van Leeuwenhoek. 2006;90:159–169. doi: 10.1007/s10482-006-9070-1. [DOI] [PubMed] [Google Scholar]
- 46.Little A.E.F., Currie C.R. Symbiotic complexity: discovery of a fifth symbiont in the attine ant–microbe symbiosis. Biol. Lett. 2007;3:501–504. doi: 10.1098/rsbl.2007.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milstone A.M., Passaretti C.L., Perl T.M. Chlorhexidine: expanding the armamentarium for infection control and prevention. Clin. Infect. Dis. 2008;46:274–281. doi: 10.1086/524736. [DOI] [PubMed] [Google Scholar]
- 48.Teo W.Y., Chan M.Y., Lam C.M., Chong C.Y. Skin manifestation of Stenotrophomonas maltophilia infection—a case report and review article. Ann. Acad. Med. 2006;35:897–900. [PubMed] [Google Scholar]
- 49.Pardesi K.R., Yavankar S.P., Chopade B.A. Plasmid distribution & antimicrobial susceptibility patterns of Acinetobacter genospecies from healthy skin of a tribal population in western India. Indian J. Med. Res. 2007;125:79–88. [PubMed] [Google Scholar]
- 50.Yavankar S.P., Pardesi K.R., Chopade B.A. Species distribution and physiological characterization of Acinetobacter genospecies from healthy human skin of tribal population in India. Indian J. Med. Microbiol. 2007;25:336–345. doi: 10.4103/0255-0857.37335. [DOI] [PubMed] [Google Scholar]
- 51.Jurado V., Gonzalez J.M., Laiz L., Saiz-Jimenez C. Aurantimonas altamirensis sp. nov., a member of the order Rhizobiales isolated from Altamira Cave. Int. J. Syst. Evol. Microbiol. 2006;56:2583–2585. doi: 10.1099/ijs.0.64397-0. [DOI] [PubMed] [Google Scholar]
- 52.Luong M.-L., Békal S., Vinh D.C., Lauzon D., Leung V., Al-Rawahi G.N. First report of isolation and characterization of Aurantimonas altamirensis from clinical samples. J. Clin. Microbiol. 2008;46:2435–2437. doi: 10.1128/JCM.00337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aujoulat F., Jumas-Bilak E., Masnou A., Sallé F., Faure D., Segonds C. Multilocus sequence-based analysis delineates a clonal population of Agrobacterium (Rhizobium) radiobacter (Agrobacterium tumefaciens) of human origin. J. Bacteriol. 2011;193:2608–2618. doi: 10.1128/JB.00107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bard J.D., Deville J.G., Summanen P.H., Lewinski M.A. Roseomonas mucosa isolated from bloodstream of pediatric patient. J. Clin. Microbiol. 2010;48:3027–3029. doi: 10.1128/JCM.02349-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsai S.-F., Chen C.-H., Shu K.-H., Wu M.-J. Peritonitis caused by Roseomonas in a patient undergoing automated peritoneal dialysis: case report and literature review. Intern. Med. 2012;51:1721–1724. doi: 10.2169/internalmedicine.51.6737. [DOI] [PubMed] [Google Scholar]
- 56.Dessinioti C., Katsambas A.D. The role of Propionibacterium acnes in acne pathogenesis: facts and controversies. Clin. Dermatol. 2010;28:2–7. doi: 10.1016/j.clindermatol.2009.03.012. [DOI] [PubMed] [Google Scholar]