

Abstract
NagZ is an N‐acetyl‐β‐d‐glucosaminidase that participates in the peptidoglycan (PG) recycling pathway of Gram‐negative bacteria by removing N‐acetyl‐glucosamine (GlcNAc) from PG fragments that have been excised from the cell wall during growth. The 1,6‐anhydromuramoyl‐peptide products generated by NagZ activate β‐lactam resistance in many Gram‐negative bacteria by inducing the expression of AmpC β‐lactamase. Blocking NagZ activity can thereby suppress β‐lactam antibiotic resistance in these bacteria. The NagZ active site is dynamic and it accommodates distortion of the glycan substrate during catalysis using a mobile catalytic loop that carries a histidine residue which serves as the active site general acid/base catalyst. Here, we show that flexibility of this catalytic loop also accommodates structural differences in small molecule inhibitors of NagZ, which could be exploited to improve inhibitor specificity. X‐ray structures of NagZ bound to the potent yet non‐selective N‐acetyl‐β‐glucosaminidase inhibitor PUGNAc (O‐(2‐acetamido‐2‐deoxy‐d‐glucopyranosylidene) amino‐N‐phenylcarbamate), and two NagZ‐selective inhibitors – EtBuPUG, a PUGNAc derivative bearing a 2‐N‐ethylbutyryl group, and MM‐156, a 3‐N‐butyryl trihydroxyazepane, revealed that the phenylcarbamate moiety of PUGNAc and EtBuPUG completely displaces the catalytic loop from the NagZ active site to yield a catalytically incompetent form of the enzyme. In contrast, the catalytic loop was found positioned in the catalytically active conformation within the NagZ active site when bound to MM‐156, which lacks the phenylcarbamate extension. Displacement of the catalytic loop by PUGNAc and its N‐acyl derivative EtBuPUG alters the active site conformation of NagZ, which presents an additional strategy to improve the potency and specificity of NagZ inhibitors.
Keywords: NagZ, N‐acetyl‐β‐d‐glucosaminidase, GH3, family 3, AmpC, PUGNAc, EtBuPUG, trihydroxyazepane
Short abstract
Introduction
Induction of AmpC β‐lactamase expression is regulated by metabolites of peptidoglycan (PG) recycling in many Gram‐negative bacteria. Blocking PG recycling is therefore a potential strategy to suppress AmpC‐mediated β‐lactam resistance in these bacteria.1, 2 NagZ is an exo‐N‐acetyl‐β‐glucosaminidase found within the Gram‐negative PG recycling pathway that removes GlcNAc from GlcNAc‐1,6‐anhydromuramoyl‐peptides that have been excised from the PG layer for recycling [Fig. 1(A)].2, 3 When bacteria are exposed to β‐lactams, PG metabolism is disrupted and the PG recycling intermediate generated by NagZ, 1,6‐anhydromuramoyl‐peptide, accumulates to levels sufficient to convert the transcriptional regulator AmpR into an activator of ampC gene expression.4, 5, 6 Blocking NagZ function, either by genetic inactivation7, 8 or using small molecule inhibitors1, 7, 8, 9, 10, 11 has been found to suppress ampC expression and increase the susceptibility of inducible AmpC expressing bacteria to β‐lactams. Thus, blocking NagZ activity with small molecule inhibitors is a promising strategy to suppress AmpC mediated β‐lactam antibiotic resistance in Gram‐negative bacteria.
Figure 1.
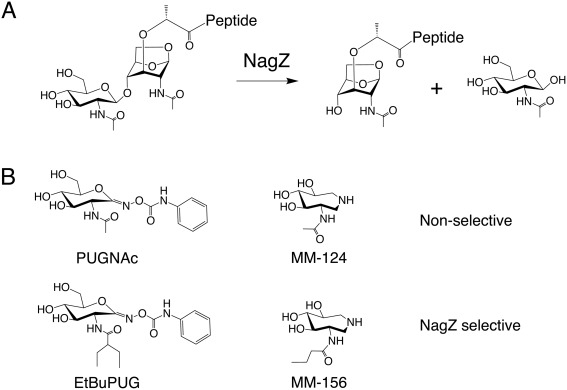
NagZ substrate and inhibitors. (A) NagZ substrate (GlcNAc‐1,6‐anhydromuramoyl‐peptide) and products (GlcNAc and 1,6‐anhydromuramoyl‐peptide), (B) non‐selective PUGNAc and trihydroxyazepane (MM‐124) NagZ inhibitors (top), and their NagZ‐selective N‐acetamido derivatives, EtBuPUG and MM‐156 (bottom).
NagZ enzymes belong to the glycoside hydrolase family 3 (GH3) of the CAZy classification system.12, 13 They possess a conserved active site aspartate that serves as a catalytic nucleophile14 and a unique consensus motif that contains a histidine general acid/base catalyst.15, 16 The two residues participate in removing GlcNAc from GlcNAc‐1,6‐anhydromuramoyl‐peptide using a two‐step double‐displacement mechanism.14, 17 In the first step, the aspartate acts as a nucleophile to attack the anomeric carbon of GlcNAc, while the histidine works as a general acid to assist the departure of the anhydromuramoyl‐peptide leaving group. This results in a covalent glycosyl‐enzyme intermediate that is broken down in the second step, where the catalytic histidine now acts as a general base to activate an incoming water that cleaves the glycosyl‐enzyme linkage and releases GlcNAc from the enzyme with retention of anomeric stereochemistry. Use of histidine as a general acid/base, as opposed to glutamate or aspartate,18 appears to enable GH3 N‐acetyl‐β‐glucosaminidases to accommodate a negatively charged phosphate in the active site, that can take the place of water to lead to the formation of GlcNAc‐1‐P as a product.19, 20 The presence of a histidine general acid/base, however, is not diagnostic of phosphorolytic activity in GH3 enzymes21 and it remains to be determined which products most GH3 NagZ enzymes produce.
In contrast to GH3 NagZ enzymes, functionally related human N‐acetyl‐β‐glucosaminidases from GH20 and GH84 use a substrate‐assisted catalytic mechanism that does not proceed through a glycosyl‐enzyme intermediate.17, 22, 23 Instead, the carbonyl oxygen of the 2‐acetamido group of the GlcNAc moiety acts as the nucleophile, attacking the anomeric carbon of the sugar to yield an oxazolinium ion intermediate. Departure of the leaving group is assisted by general acid catalysis from an active site carboxylic acid residue, which acts as a general base in a second step to activate an incoming water molecule that attacks the anomeric carbon of oxazolinium ion intermediate to release GlcNAc with retained anomeric stereochemistry.
The mechanistic differences between GH3 NagZ and N‐acetyl‐β‐glucosaminidases from GH20 and GH84 have been exploited to develop bacterial PG recycling inhibitors that selectively block NagZ over functionally related human enzymes from GH20 and GH84.1, 24 N‐acetyl‐β‐glucosaminidases from all three families are potently inhibited by the small molecule inhibitor PUGNAc25, 26 [Fig. 1(B)]; however, the active site architectures of GH20 and GH84 differ sufficiently from NagZ to allow bulky substituents to be installed on the 2‐acetamido group of PUGNAc to make the inhibitor selective for NagZ over GH20 and GH84 enzymes,24 such as the PUGNAc derivative EtBuPUG [Fig. 1(B)].1, 27 The active sites of GH20 and GH84 N‐acetyl‐β‐glucosaminidases must envelop the 2‐acetamido group to guide it toward the anomeric center of GlcNAc, which leaves minimal space around this moiety for functionalization.24 This is not the case for NagZ, where the 2‐acetamido group projects into an open pocket in the NagZ active site, where functionalization of this moiety can be accommodated.15, 24 Similarly, 3‐acetamido‐4,5,6‐trihydroxyazepane (MM‐124)28 [Fig. 1(B)] is a general N‐acetyl‐β‐glucosaminidase inhibitor that blocks the activity of NagZ, GH20 and GH84 enzymes.9 Analogous to PUGNAc, addition of bulky substituents to the acetamido moiety of this inhibitor also makes it selective for NagZ over GH20 and GH84 enzymes as seen in MM‐156 [Fig. 1(B)],9 but less so as compared with PUGNAc.
Unlike the typically rigid active site structure of glycosidases, GH3 NagZ enzymes position their histidine general acid/base residue on a highly mobile loop that moves in and out of the active site during catalysis in coordination with substrate distortions that promote glycosidic bond cleavage.16 Remarkably, the catalytic histidine can move as far as 20 Å away from the position it must adopt to participate in acid/base catalysis.16 In this work, structural analyses of NagZ from the Gram‐negative bacteria Burkholderia cenocepacia in complex with the inhibitors PUGNAc, EtBuPUG, and MM‐156 reveal that in addition to an active site that can accommodate N‐acyl substitutions that impart selectivity toward NagZ, the plasticity of the NagZ active site could also be exploited to improve the design of inhibitors that selectively bind NagZ over functionally related human N‐acetyl‐β‐glucosaminidases.
Results and Discussion
The phenylcarbamate extension on PUGNAc and EtBuPUG displaces the catalytic loop from the BcNagZ active site
To investigate the effects of inhibitor binding on the orientation of the catalytic loop in BcNagZ, crystals of the enzyme were soaked with 1 mM PUGNAc, EtBuPUG or MM‐156 for 24 h and structures of the complexes determined by molecular replacement using an unliganded model of BcNagZ (PDB 4G6C) from which solvent had been removed. BcNagZ is a single domain enzyme that adopts a characteristic TIM‐barrel (β/α)8 (Fig. 2). Interestingly, we found that MM‐156 allows the catalytic loop to enter the active site and correctly position the general acid/base residue His183 within ∼6 Å of the active site nucleophile (Asp 253) [Fig. 2(A)], which is consistent with the distance between the analogous residues of most retaining glycosidases.29 The position of the loop is also consistent with a previous structure of BcNagZ bound to the non‐selective N‐acetyl‐β‐glucosaminidase inhibitor 3‐acetamido‐4,5,6‐trihydroxyazepane MM‐124 (PDB ID: 4MSS)9 and the product GlcNAc (PDB ID: 4GNV), indicating that addition of a propyl extension off the 3‐acetamido group does not affect correct positioning of the catalytic loop and His183 within the enzyme active site [Fig. 3(A,B)].
Figure 2.
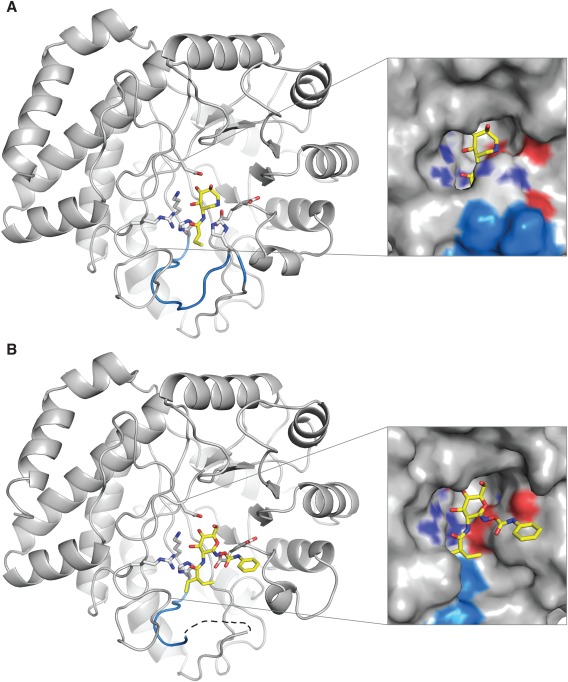
BcNagZ in complex with MM‐156 and EtBuPUG. (A) The general acid/base histidine‐bearing catalytic loop is highlighted in blue in the BcNagZ: MM‐156 complex, (B) and was disordered in both the EtBuPUG and PUGNAc‐bound complexes (PUGNAc not shown for clarity). The inset surface representations reveal the dramatic change in the NagZ active site due to the catalytic loop displacement caused by the phenylcarbamate extension of EtBuPUG (or PUGNAc). MM‐156 and EtBuPUG are shown as yellow sticks, and nitrogen and oxygen atoms are coloured dark blue and red, respectively.
Figure 3.
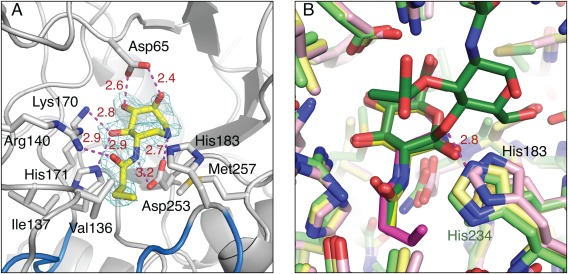
Active site interactions of BcNagZ with MM‐156. (A) Maximum‐likelihood weighted F obs–F calc map contoured at 3.5σ for MM‐156 (prior to ligand modeling and refinement; carbon atoms shown as yellow sticks) bound to BcNagZ. (B) Superposition of BcNagZ‐MM‐156 (pink), BcNagZ‐GlcNAc (yellow, PDB 4GNV), and BsNagZ‐GlcNAc‐MurNAc (green, PDB 4GYJ). MM‐124 is not shown for clarity, but binds very similarly to MM‐156. Hydrogen bonds are shown by dashed magenta lines. Nitrogen and oxygen atoms are coloured blue and red, respectively.
Moreover, the catalytic loop in these complexes adopts the same position as found in the GH3 NagZ from the Gram‐positive bacteria Bacillus subtilis (BsNagZ) bound to GlcNAc‐MurNAc substrate (PDB ID: 4GYJ), where the histidine (His234) of the catalytic loop of BsNagZ interacts directly with the glycosidic oxygen of the disaccharide through a 2.8 Å hydrogen bond [Fig. 3(B)].16 Unlike the single domain structure of NagZ enzymes of Gram‐negative bacteria, BsNagZ is a two‐domain enzyme, as are most NagZ enzymes from Gram‐positive bacteria. Nevertheless, the flexibility of the catalytic loop and its ability to place the catalytic histidine within hydrogen bonding distance of the oxygen of the scissile glycosidic bond appears conserved in GH3 NagZ enzymes from both Gram‐negative and ‐positive bacteria.15, 16
In contrast to the lack of influence of MM124 or MM156 on the positioning of the catalytic loop within the active site of BcNagZ, both PUGNAc and EtBuPUG completely displace the catalytic loop from the enzyme active site [Fig. 2(B)]. Superposition of BcNagZ structures bound to NagZ selective inhibitors MM‐156 and EtBuPUG reveal that the position of the phenylcarbamate extension off C1 of EtBuPUG (or PUGNAc) would result in a direct steric clash with His183 of the catalytic loop, which is likely responsible for displacing the loop from the active site (Fig. 4). Interestingly, the N‐acyl group of EtBuPUG, which occupies the same region in the active site as the propyl extension of the 3‐acetamido group of MM156, was found to have no appreciable effect on the conformation of nearby residues in the enzyme, indicating that even though it is larger than the N‐butyryl group of MM156, it does not participate in loop displacement. Additionally, MM‐156 and EtBuPUG both have low micromolar binding affinities against NagZ enzymes from Gram‐negative bacteria (KI = ∼7 μM and ∼3 μM, respectively)1, 9 and PUGNAc remains the most potent NagZ inhibitor known (KI = ∼0.05 μM);1 thus, displacement of the catalytic loop does not appear to incur a major binding penalty. The additional space in the active site that is opened up by the presence of the phenylcarbamate group on PUGNAc and EtBuPUG [Fig. 2(B)], and consequent displacement of the catalytic loop, could therefore be exploited to improve selectivity of these inhibitors for NagZ enzymes, such as the generation of a locked cyclic derivative where extensions off the 2‐acetamido group could be linked to a phenylcarbamate group.
Figure 4.
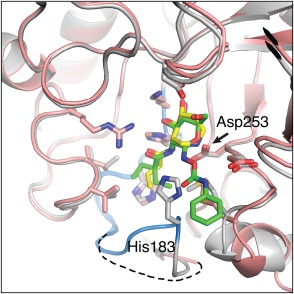
Superposition of BcNagZ structures bound to MM‐156 and EtBuPUG. The catalytic loop is highlighted in blue in the BcNagZ: MM‐156 complex (grey), and depicted as a dashed line in the EtBuPUG‐bound complex (salmon; PUGNAc not shown for clarity). The phenylcarbamate arm of EtBuPUG is oriented such that it would sterically clash with His183 from the MM‐156‐bound complex. MM‐156 and EtBuPUG are shown as yellow and green sticks, respectively; nitrogen and oxygen atoms are coloured dark blue and red.
Molecular interactions not involving the catalytic loop of BcNagZ underlie the binding potency of the NagZ‐selective inhibitors MM156 and EtBuPUG
The azepane of MM‐156 and GlcNAc component of PUGNAc and EtBuPUG all bind similarly within the active site of BcNagZ active site (Fig. 4). The azepane hydroxyl groups of MM‐156 form several hydrogen‐bonding interactions with the enzyme [Fig. 3(A)] that mimic the interaction of the enzyme with hydroxyl groups of the terminal GlcNAc residue of PG recycling intermediates and the GlcNAc moieties of PUGNAc and EtBuPUG. However, an additional 2.8 Å hydrogen bond occurs between the endocyclic nitrogen of MM156 and the catalytic nucleophile of BcNagZ (Asp253) [Figs. 3(A) and 5(A,B)], that likely contributes significantly to the binding potency of azepane‐based inhibitors to NagZ enzymes.9 The propyl extension to the 3‐acetamido group of MM‐156 provides excellent selectivity for GH3 NagZ over GH20 N‐acetyl‐β‐glucosaminidases (>1000‐fold) and modest selectivity over GH84 N‐acetyl‐β‐glucosaminidases (∼7‐fold) due to the steric constraints of the active sites of GH20 and GH84 enzymes.9 This extension also forms hydrophobic interactions within the BcNagZ binding pocket with residues Val136, Ile137 and Met257 [Fig. 3(A)]. Moreover, the pocket is able to accommodate even larger substituents as described below for EtBuPUG.
Figure 5.
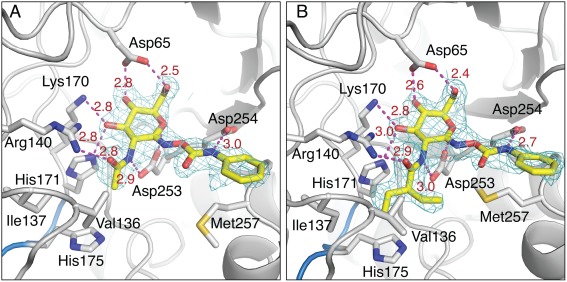
Active site interactions of BcNagZ with PUGNAc and EtBuPUG. (A) Maximum‐likelihood weighted F obs–F calc map contoured at 3.5σ for PUGNAc and (B) EtBuPUG bound to BcNagZ (prior to ligand modeling and refinement), with dashed lines indicating hydrogen‐bonding interactions with active site residues. The ethylbutyryl group appears to be easily accommodated within the active site. No electron density was observed for the catalytic His183 residue‐bearing loop. Carbon, nitrogen and oxygen atoms are coloured grey/yellow, blue and red, respectively.
EtBuPUG has excellent selectivity for NagZ over both GH20 (∼1000‐fold) and GH84 (∼100‐fold) N‐acetyl‐β‐glucosaminidases,1 which arises from its large, branched N‐ethylbutyryl group [Fig. 5(B)]. Similar to the propyl extension on MM‐156, the ethylbutyryl group of EtBuPUG extends into a pocket where it forms hydrophobic interactions with Val136, Ile137, and Met257 [Fig. 5(B)]. Notably, even though the ethylbutyryl extension is slightly larger than the propyl group on MM156, it does not significantly alter the conformation of the residues comprising the pocket into which it binds when compared with unliganded BcNagZ. The major conformational difference in BcNagZ that does arise from binding EtBuPUG or PUGNAc is the displacement of the catalytic loop by the phenylcarbamate extension, which forms a 2.7 Å hydrogen bonding interaction with Asp254 of the enzyme [Fig. 5(B)]. The presence of this interaction with Asp254 appears to restrict the phenylcarbamate extension from adopting an alternate conformation that might otherwise allow the catalytic loop to enter the active site, as observed for BsNagZ bound to PUGNAc15 (see below). Additional interactions of PUGNAc and EtBuPUG with BcNagZ include a hydrogen‐bonding interaction between the 2‐acylamido group nitrogen and the active site nucleophile Asp253 as well as hydrogen bonding interactions between the enzyme and hydroxyl groups of the carbohydrate moiety of the inhibitor. Finally, the geometric resemblance of PUGNAc and EtBuPUG to the proposed oxocarbenium ion‐like transition states that arise along the reaction coordinate is also believed to contribute significantly to binding potency of these inhibitors to NagZ enzymes.1, 19
The phenylcarbamate extension of PUGNAc adopts different conformations depending on the active site architecture of NagZ from Gram‐negative versus Gram‐positive bacteria
Crystal structures of NagZ enzymes from both Gram‐negative and ‐positive bacteria have now been determined in complex with PUGNAc.1, 15 While NagZ enzymes from Gram‐negative bacteria are typically single‐domain enzymes that adopt a TIM‐barrel (β/α)8, NagZ enzymes from Gram‐positive bacteria have a two‐domain architecture; however, the N‐terminal domain adopts a TIM‐barrel fold that is conserved with NagZ enzymes from Gram‐negative bacteria.15 Although Gram‐positive bacteria lack the ampR‐ampC system that is blocked by NagZ inhibitors, they do appear to have a PG recycling pathway in which NagZ participates.30 Interestingly, superposing PUGNAc‐bound complexes of BsNagZ and BcNagZ reveals that in the latter complex, the conformation of the phenylcarbamate extension of PUGNAc differs significantly from the conformation it adopts when bound to BsNagZ [Fig. 6(A)]. This difference likely arises due to the presence of Ala319 in BsNagZ, which replaces the aspartate in this position (Asp254) in BcNagZ. The alanine residue creates sufficient space for the phenylcarbamate of PUGNAc to adopt a conformation that permits the accommodation of the catalytic loop of BsNagZ within the active site and correctly places His234 for catalysis [Fig. 6(A)]. Conversely, in BcNagZ, Asp254 prevents the phenylcarbamate of PUGNAc from adopting such a conformation and instead forms a hydrogen bonding interaction with the nitrogen atom of the extension, which holds it in a conformation that does not allow the catalytic loop to enter the active site [Fig. 6(A)]. Interestingly, the phenylcarbamate extension within the BcNagZ active site adopts a nearly identical conformation when PUGNAc is bound to another single domain GH3 NagZ from the Gram‐negative bacteria Vibrio cholerae (VcNagZ) and is within 3.6 Å of Asp243, the analogous residue of Asp254 in BcNagZ [Fig. 6(B)].
Figure 6.
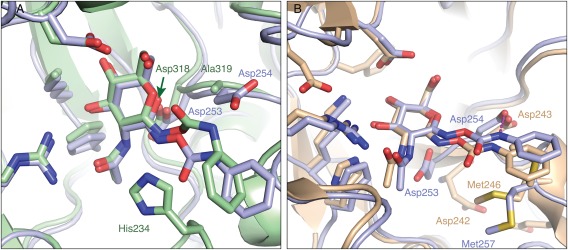
Active site superposition of PUGNAc‐bound NagZ enzymes. (A) Superposition of the NagZ: PUGNAc complex from B. cenocepacia (light blue) and B. subtilis (green, PDB 3NVD) reveals that the catalytic aspartate nucleophiles for each structure are present in a physiologically relevant conformation. The PUGNAc phenylcarbamate group distortion when bound to BsNagZ appears to be accommodated due to the presence of Ala319. Instead, in BcNagZ, there is Asp254 at this site which is within hydrogen‐bonding distance of the nitrogen bonded to the phenyl group. (B) Superposition of PUGNAc‐bound structures of B. cenocepacia (light blue) and V. cholera (light brown, PDB 2OXN). Unlike the BcNagZ complex, the catalytic nucleophile Asp242 is flipped away from the active site in VcNagZ, while the PUGNAc phenyl group competes for occupancy with Met246. Key active site residues are labelled in corresponding colours. Hydrogen‐bonding interactions between BcNagZ and PUGNAc are represented by dashed magenta lines. Nitrogen and oxygen atoms are indicated in dark blue and red, respectively.
Multiple sequence alignments of GH3 NagZ enzymes from Gram‐negative and ‐positive bacteria reveal Asp254 of BcNagZ (Asp243 VcNagZ) is highly conserved in Gram‐negative bacteria. However, this position in NagZ enzymes from Gram‐positive bacteria, while predominantly alanine, exhibits a higher degree of variability, including the presence of aspartate or cysteine (Fig. 7). It is noteworthy that the phenylcarbamate extension of PUGNAc can adopt different conformations depending on differences in the active site architectures of NagZ enzymes from Gram‐negative and ‐positive bacteria. Given that Asp254 of BcNagZ is highly conserved among NagZ enzymes from other Gram‐negative bacteria, we predict the catalytic loop will be displaced by PUGNAc in these enzymes, whereas, in NagZ from Gram‐positive bacteria that have an alanine in this position, we expect the phenylcarbamate extension of PUGNAc will adopt a conformation that could allow the catalytic loop to enter the active site as observed for BsNagZ [Fig. 6(A)]. These findings reveal that amino acid heterogeneity exists within active site of NagZ enzymes from Gram‐negative versus Gram‐positive bacteria. However, since the inducible AmpC system appears exclusive to Gram‐negative bacteria, inhibitors that block this resistance mechanism should be optimized toward NagZ from Gram‐negative bacteria.
Figure 7.

Multiple sequence alignments of NagZ enzyme active site residues. Multiple sequence alignments are shown for a conserved active site region (boxed) within GH3 family NagZ proteins from Gram‐negative species (A) and Gram‐positive bacteria (B). Active site residues corresponding to Asp254 in BcNagZ are highly conserved in Gram‐negative bacteria, while residues homologous to Ala319 in BsNagZ exhibit greater variability (positions shown by arrow).
Conclusions
Inhibiting NagZ activity with small molecule inhibitors has been shown to suppress AmpC‐mediated β‐lactam antibiotic resistance in Gram‐negative bacteria by blocking PG recycling and the formation of PG recycling intermediates that induce ampC gene expression. Here, we demonstrate that the remarkable plasticity of NagZ enzymes enables them to adopt different conformations in response to various inhibitor designs. In the case of PUGNAc and its analogue EtBuPUG, the displacement of the catalytic loop by this inhibitor class opens up the entrance to the active site considerably (Fig. 2). This could be exploited to develop new inhibitors with substituents that occupy the open space to significantly enhance selectivity and potency for bacterial NagZ enzymes over functional related human N‐acetyl‐β‐glucosaminidases.
Materials and Methods
BcNagZ expression and purification
Escherichia coli BL21 (DE3) GOLD cells containing the expression plasmid pBCNagZ9 were grown to OD600 of 0.5 at 37°C in 500‐ml volumes of LB media supplemented with 35 μg/ml kanamycin, then induced with 1 mM isopropyl β‐d‐1‐thiogalactopyranoside for 3 h at 28°C. Cells were pelleted by centrifugation and stored at −80°C. Thawed pellets were resuspended in 20 ml lysis buffer (0.5 M NaCl, 5% glycerol, 25 mM HEPES pH 7, 1 μM PMSF), and the cells lysed using a French pressure cell press (Aminco). Following centrifugation, the soluble protein fraction of the lysate containing His‐tagged BcNagZ was incubated with nickel‐nitrilotriacetic acid (Ni‐NTA) resin (Qiagen, Canada) at 4°C for 1 h, prior to loading on a gravity column. Resin‐bound protein was subjected to washes with binding buffer (25 mM HEPES pH 7, 0.5 M NaCl and 5% glycerol) supplemented with 0, 10 and 20 mM imidazole, and eluted using wash buffer containing 250 mM imidazole. The eluted protein was dialyzed overnight against 2 L binding buffer, and concentrated to 13–20 mg/ml.
Crystallization and structure determination of BcNagZ with inhibitors
BcNagZ crystals were grown at 20°C using the hanging drop vapour‐diffusion method by mixing equal volumes of reservoir buffer (30–32% PEG8000, 0.1M MES pH 6.2–6.8) with protein solution (13–20 mg/ml). The inhibitors MM‐156, PUGNAc and EtBuPUG were prepared as described.9, 27 A single droplet containing several BcNagZ crystals in reservoir buffer was soaked for 24 h in MM‐156, PUGNAc or EtBuPUG at final concentrations of 1 mM to obtain the protein‐inhibitor complex. Prior to screening, crystals were cryo‐protected in 30% PEG3500, 15% PEG8000 and 0.1M MES pH 6.6, and flash‐cooled in liquid nitrogen. X‐ray data for the EtBuPUG and PUGNAc‐bound complexes were collected using a Rigaku R‐AXIS IV++ detector and 007HF MicroMax X‐ray generator at the University of Manitoba. Data for the BcNagZ‐MM‐156 complex were collected using beamline 08B1‐1 at the Canadian Light Source (Saskatoon, Canada). The X‐ray data were indexed using iMosflm,31 then scaled and averaged using SCALA (CCP4 package).32 The BcNagZ:inhibitor complex structures were determined by molecular replacement using PHASER (from within the PHENIX package)33 and a structure of BcNagZ (PDB ID: 4G6C) from which solvent had been removed prior to use as the search model. The MR model was subsequently rebuilt using PHENIX.AUTOBUILD.33 The ligand restraint file for MM‐156 and EtBuPUG was generated using PHENIX eLBOW33 and a model of the inhibitor was manually fitted into its electron density. Subsequent refinement of the complex and addition of solvent was carried out using PHENIX.REFINE33 and COOT.34 Crystallographic and refinement statistics are presented in Table 1.
Table 1.
Crystallographic Statistics for Structures of BcNagZ Bound to MM‐156, PUGNAc, and EtBuPUG
| Data collection | BcNagZ:MM‐156 | BcNagZ:PUGNAc | BcNagZ:EtBuPUG |
|---|---|---|---|
| X‐ray source | CLS 08B1‐1 | Rigaku MicroMax 007HF | Rigaku MicroMax 007HF |
| Space group | P 1 21 1 | P 1 21 1 | P 1 21 1 |
| Unit cell dimensions |
a = 48.66 Å, b = 87.82 Å, c = 66.97 Å; α = γ = 90°, β = 91.92° |
a = 48.67 Å, b = 87.67 Å, c = 66.70 Å; α = γ = 90° β = 91.75° |
a = 51.21 Å, b = 85.89 Å, c = 65.61 Å; α = γ = 90° β = 102.79° |
| Wavelength (Å) | 1.03 | 1.54 | 1.54 |
| Resolution (Å) | 66.93–2.15 (2.21–2.15) | 43.83–2.20 (2.27–2.20) | 49.94–2.20 (2.27–2.20) |
| R merge | 0.091 (0.264) | 0.124 (0.458) | 0.137 (0.249) |
| CC(1/2) | 0.993 (0.889) | 0.966 (0.647) | 0.950 (0.908) |
| Ι/σΙ | 9.7 (4.5) | 6.4 (2.0) | 5.1 (3.1) |
| Completeness (%) | 100 (100) | 99.4 (99.2) | 98.2 (98.0) |
| Redundancy | 3.4 (3.3) | 2.2 (2.2) | 2.9 (2.8) |
| Refinement | |||
| R work/R free | 0.14/0.19 | 0.16/0.21 | 0.17/0.23 |
| B‐factor (Å2) | 17.80 | 22.58 | 17.03 |
| Protein | 17.13 | 22.34 | 16.90 |
| Ligands | 16.57 | 30.71 | 18.60 |
| Solvent | 23.15 | 24.65 | 18.51 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.002 | 0.003 | 0.002 |
| Bond angles (° | 0.62 | 0.64 | 0.63 |
| Ramachandran favoured/allowed (%) | 96/4.0 | 95/5.3 | 96/4.2 |
Values in parentheses refer to the high‐resolution shell.
Multiple sequence alignments
The amino acid sequences of all GH3 family NagZ enzymes from Gram‐positive bacteria, and selected Gram‐negative species, were obtained from the CAZy database (http://www.cazy.org/). Multiple sequence alignments were performed comparing Gram‐negative species, and Gram‐positive species, respectively, using the Clustal Omega program from EMBL‐EBI bioinformatics services (http://www.ebi.ac.uk/Tools/msa/clustalo/).
PDB accession codes
The atomic coordinates and structure factors for BcNagZ in complex with PUGNAc, EtBuPUG, and MM‐156 have been deposited in the Protein Data Bank (http://wwpdb.org/) under the codes 5UTQ, 5UTP, and 5UTR, respectively.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Veronica Larmour for technical assistance and Shaun Labiuk and the staff of the Canadian Macromolecular Crystallography Facility (Canadian Light Source) for assistance with X‐ray data collection. MM‐156 was provided by Y.B. PUGNAc and EtBUPUG were provided by K.A.S. and D.J.V. G.V. conducted all protein crystallography experiments. G.V., K.A.S., Y.B., D.J.V., and B.L.M. conceived the experiments and contributed to manuscript writing.
Statement: NagZ is an enzyme within the peptidoglycan recycling pathway required for the activation of AmpC β‐lactamase production in several pathogenic Gram‐negative bacteria. AmpC confers resistance to numerous clinically important β‐lactam antibiotics. Thus, blocking NagZ activity with small‐molecule inhibitors has garnered interest as a therapeutic strategy to suppress AmpC‐mediated β‐lactam resistance. Here, we provide a clear structural framework for improving the potency and selectivity of NagZ inhibitors for use in enhancing the efficacy of β‐lactam antibiotics.
References
- 1. Stubbs KA, Balcewich M, Mark BL, Vocadlo DJ (2007) Small molecule inhibitors of a glycoside hydrolase attenuate inducible AmpC‐mediated beta‐lactam resistance. J Biol Chem 282:21382–21391. [DOI] [PubMed] [Google Scholar]
- 2. Vötsch W, Templin MF (2000) Characterization of a β‐N‐acetylglucosaminidase of Escherichia coli and elucidation of its role in muropeptide recycling and β‐lactamase induction. J Biol Chem 275:39032–39038. [DOI] [PubMed] [Google Scholar]
- 3. Cheng Q, Li H, Merdek K, Park JT (2000) Molecular characterization of the β‐N‐acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J Bacteriol 182:4836–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT (1994) Bacterial cell wall recycling provides cytosolic muropeptides as effectors for β‐lactamase induction. EMBO J 13:4684–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacobs C, Frère JM, Normark S (1997) Cytosolic intermediates for cell wall biosynthesis and degradation control inducible β‐lactam resistance in Gram‐negative bacteria. Cell 88:823–832. [DOI] [PubMed] [Google Scholar]
- 6. Dietz H, Pfeifle D (1997) The signal molecule for β‐lactamase induction in Enterobacter cloacae is the anhydromuramyl‐pentapeptide. Antimicrob Agents Chemother 41:2113–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asgarali A, Stubbs KA, Oliver A, Vocadlo DJ, Mark BL (2009) Inactivation of the glycoside hydrolase NagZ attenuates antipseudomonal beta‐lactam resistance in Pseudomonas aeruginosa . Antimicrob Agents Chemother 53:2274–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zamorano L, Reeve TM, Deng L, Juan C, Moyá B, Cabot G, Vocadlo DJ, Mark BL, Oliver A (2010) NagZ inactivation prevents and reverts beta‐lactam resistance, driven by AmpD and PBP 4 mutations, in Pseudomonas aeruginosa . Antimicrob Agents Chemother 54:3557–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mondon M, Hur S, Vadlamani G, Rodrigues P, Tsybina P, Oliver A, Mark BL, Vocadlo DJ, Blériot Y (2013) Selective trihydroxyazepane NagZ inhibitors increase sensitivity of Pseudomonas aeruginosa to β‐lactams. Chem Commun 49:10983–10985. [DOI] [PubMed] [Google Scholar]
- 10. Stubbs KA, Bacik J‐P, Perley‐Robertson GE, Whitworth GE, Gloster TM, Vocadlo DJ, Mark BL (2013) The development of selective inhibitors of NagZ: increased susceptibility of Gram‐negative bacteria to β‐lactams. ChemBioChem 14:1973–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamaguchi T, Blázquez B, Hesek D, Lee M, Llarrull LI, Boggess B, Oliver AG, Fisher JF, Mobashery S (2012) Inhibitors for bacterial cell‐wall recycling. ACS Med Chem Lett 3:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Henrissat B, Bairoch A (1996) Updating the sequence‐based classification of glycosyl hydrolases. Biochem J 316(Pt 2):695–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate‐active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vocadlo DJ, Mayer C, He S, Withers SG (2000) Mechanism of action and identification of Asp242 as the catalytic nucleophile of Vibrio furnisii N‐acetyl‐beta‐d‐glucosaminidase using 2‐acetamido‐2‐deoxy‐5‐fluoro‐alpha‐l‐idopyranosyl fluoride. Biochemistry 39:117–126. [DOI] [PubMed] [Google Scholar]
- 15. Litzinger S, Fischer S, Polzer P, Diederichs K, Welte W, Mayer C (2010) Structural and kinetic analysis of Bacillus subtilis N‐acetylglucosaminidase reveals a unique Asp‐His dyad mechanism. J Biol Chem 285:35675–35684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bacik J‐P, Whitworth GE, Stubbs KA, Vocadlo DJ, Mark BL (2012) Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem Biol 19:1471–1482. [DOI] [PubMed] [Google Scholar]
- 17. Vocadlo DJ, Withers SG (2005) Detailed comparative analysis of the catalytic mechanisms of beta‐N‐acetylglucosaminidases from families 3 and 20 of glycoside hydrolases. Biochemistry 44:12809–12818. [DOI] [PubMed] [Google Scholar]
- 18. Vocadlo DJ, Davies GJ (2008) Mechanistic insights into glycosidase chemistry. Curr Opin Chem Biol 12:539–555. [DOI] [PubMed] [Google Scholar]
- 19. Macdonald SS, Blaukopf M, Withers SG (2015) N‐acetylglucosaminidases from CAZy family GH3 are really glycoside phosphorylases, thereby explaining their use of histidine as an acid/base catalyst in place of glutamic acid. J Biol Chem 290:4887–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su H, Sheng X, Liu Y (2016) Insights into the catalytic mechanism of N‐acetylglucosaminidase glycoside hydrolase from Bacillus subtilis: a QM/MM study. Org Biomol Chem 290:3432–3442. [DOI] [PubMed] [Google Scholar]
- 21. Ducatti DRB, Carroll MA, Jakeman DL (2016) On the phosphorylase activity of GH3 enzymes: a β‐N‐acetylglucosaminidase from Herbaspirillum seropedicae SmR1 and a glucosidase from Saccharopolyspora erythraea . Carbohydr Res 435:106–112. [DOI] [PubMed] [Google Scholar]
- 22. Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE (1996) Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay‐Sachs disease. Nat Struct Biol 3:638–648. [DOI] [PubMed] [Google Scholar]
- 23. Mark BL, Vocadlo DJ, Knapp S, Triggs‐Raine BL, Withers SG, James MN (2001) Crystallographic evidence for substrate‐assisted catalysis in a bacterial beta‐hexosaminidase. J Biol Chem 276:10330–10337. [DOI] [PubMed] [Google Scholar]
- 24. Balcewich MD, Stubbs KA, He Y, James TW, Davies GJ, Vocadlo DJ, Mark BL (2009) Insight into a strategy for attenuating AmpC‐mediated beta‐lactam resistance: structural basis for selective inhibition of the glycoside hydrolase NagZ. Protein Sci 18:1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horsch M, Hoesch L, Vasella A, Rast DM (1991) N‐acetylglucosaminono‐1,5‐lactone oxime and the corresponding (phenylcarbamoyl)oxime. Novel and potent inhibitors of beta‐N‐acetylglucosaminidase. Eur J Biochem 197:815–818. [DOI] [PubMed] [Google Scholar]
- 26. Beer D, Maloisel J‐L, Rast DM, Vasella A (1990) Synthesis of 2‐acetamido‐2‐deoxy‐d‐gluconhydroximolactone‐ and chitobionhydroximolactone‐derived N‐phenylcarbamates, potential inhibitors of beta‐N‐acetylglucosaminidase. Helv Chim Acta 73:1918–1922. [Google Scholar]
- 27. Stubbs KA, Zhang N, Vocadlo DJ (2006) A divergent synthesis of 2‐acyl derivatives of PUGNAc yields selective inhibitors of O‐GlcNAcase. Org Biomol Chem 4:839–845. [DOI] [PubMed] [Google Scholar]
- 28. Li H, Marcelo F, Bello C, Vogel P, Butters TD, Rauter AP, Zhang Y, Sollogoub M, Blériot Y (2009) Design and synthesis of acetamido tri‐ and tetra‐hydroxyazepanes: potent and selective beta‐N‐acetylhexosaminidase inhibitors. Bioorg Med Chem 17:5598–5604. [DOI] [PubMed] [Google Scholar]
- 29. Zechel DL, Withers SG (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc Chem Res 33:11–18. [DOI] [PubMed] [Google Scholar]
- 30. Reith J, Mayer C (2011) Peptidoglycan turnover and recycling in Gram‐positive bacteria. Appl Microbiol Biotechnol 92:1–11. [DOI] [PubMed] [Google Scholar]
- 31. Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW (2011) iMOSFLM: a new graphical interface for diffraction‐image processing with MOSFLM. Acta Cryst D67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Evans P (2006) Scaling and assessment of data quality. Acta Cryst D62:72–82. [DOI] [PubMed] [Google Scholar]
- 33. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]