

Abstract
ScFv‐h3D6 is a single chain variable fragment that precludes Aβ peptide‐induced cytotoxicity by withdrawing Aβ oligomers from the amyloid pathway to the worm‐like pathway. Production of scFv molecules is not a straightforward procedure because of the occurrence of disulfide scrambled conformations generated in the refolding process. Here, we separately removed the disulfide bond of each domain and solved the scrambling problem; and then, we intended to compensate the loss of thermodynamic stability by adding three C‐terminal elongation mutations, previously described to stabilize the native fold of scFv‐h3D6. Such stabilization occurred through stabilization of the intermediate state in the folding pathway and destabilization of a different, β‐rich, intermediate state driving to worm‐like fibrils. Elimination of the disulfide bridge of the less stable domain, V L, deeply compromised the yield and increased the aggregation tendency, but elimination of the disulfide bridge of the more stable domain, V H, solved the scrambling problem and doubled the production yield. Notably, it also changed the aggregation pathway from the protective worm‐like morphology to an amyloid one. This was so because a partially unfolded intermediate driving to amyloid aggregation was present, instead of the β‐rich intermediate driving to worm‐like fibrils. When combining with the elongation mutants, stabilization of the partially unfolded intermediate driving to amyloid fibrils was the only effect observed. Therefore, the same mutations drove to completely different scenarios depending on the presence of disulfide bridges and this illustrates the relevance of such linkages in the stability of different intermediate states for folding and misfolding.
Keywords: intermediate state, folding, misfolding, aggregation, amyloid fibrils, worm‐like fibrils, disulfide bonds
Abbreviations
- AD
Alzheimer's disease
- CD
circular dichroism
- DF
disulfide free
- FITR
Fourier‐transformed infrared spectroscopy
- scFv
single chain variable Fragment
- WL
worm‐like
Introduction
Alzheimer's disease (AD) is caused by the progressive accumulation of the Amyloid‐β (Aβ) peptide, which leads to neurofibrillary tangle formation, neuroinflammation, synaptic loss, neuron death and ultimately, dementia.1, 2, 3 Specially, Aβ oligomers and protofibrils are the molecular species that have been identified as responsible for initiating the pathological processes associated with AD.4, 5 Aβ‐immunotherapy has emerged as a promising strategy based on the reduction of these oligomeric forms of the Aβ peptide. For a long time, monoclonal antibodies have been studied to be used as therapies for different diseases (such as autoimmune disorders, cancer, transplant rejection or cardiovascular diseases) as they have been proven to be an excellent paradigm for the design of high‐affinity, protein‐based binding reagents.6, 7 In the case of AD, monoclonal antibodies have been shown to ameliorate cognitive symptoms, but some side‐effects (vasogenic edema, meningoencephalitis, microcerebral hemorrhages) were observed in clinical Phases (reviewed in Ref. 8). Single chain variable fragment (scFv) antibodies are considered to be safer than the whole antibody because they lack the Fc region responsible for activating the complement response.9 In addition, they are smaller in size and, thus, potentially easier to be delivered into the brain.10
ScFv‐h3D6 is a single chain variable fragment derived from the monoclonal antibody bapineuzumab, which targets the N‐terminal 1‐5 residues of the Aβ peptide11 known to be exposed to the solvent in the trimer conformation adopted by Aβ oligomers.12 ScFv‐h3D6 has been demonstrated to preclude Aβ peptide‐induced toxicity by withdrawing Aβ oligomers from the amyloid pathway towards the worm‐like (WL) pathway, a non‐toxic pathway featured by short and curved fibrils.13 The capability for forming WL fibrils is an intrinsic property of the scFv‐h3D6 itself, since they are assembled from a β‐rich intermediate state populating the thermal‐induced aggregation pathway, yet those made by the Aβ/scFv‐h3D6 complex are more stable than those made by the isolated scFv‐h3D6. We have found that administration of a single‐intraperitoneal dose of scFv‐h3D6 to 3xTg‐AD mice ameliorated first hallmarks of Alzheimer's disease at the behavioral, cellular and molecular levels.14, 15 Treatment improved cognition and reversed BPSD (behavioral and psychological symptoms of dementia)‐like symptoms, protected from cell‐death, decreased extracellular Aβ oligomers and restored apolipoproteins E and J concentrations. Hence, the therapeutic potential of this antibody fragment holds promise for the treatment of Alzheimer's disease.
We also previously showed that scFvh3D6 urea‐induced unfolding pathway is characterized by the presence of an intermediate state, different from that in the aggregation pathway, which is composed of the unfolded V L domain and the folded V H domain.16 Therefore, scFv‐h3D6 fits into Class I of the scFvs' classification by Wörn and Plückthun,17, 18 the intrinsic stability of one domain being significantly higher than the total stability (intrinsic plus interface contribution) of the other domain. Then, we concentrated our engineering efforts on the “weak part” of the molecule and redesigned the V L domain fold.16 This domain is located at the C‐terminus and graphical examination of its three‐dimensional model revealed that the side‐chain of the last residue, V L‐K107 (Kabat numbering19), was not properly performing the i, i‐2 electrostatic interaction required to stabilize the last β‐strand of the fold. This interaction was weakened by the attraction of the V L‐K107‐NZ to the OXT107‐O, so we decided to elongate the molecule by constructing elongation mutants V L‐el‐R108G (referred to as C1), V L‐el‐R108 (C2), and V L‐el‐R108T109 (C3). These mutants also showed a three‐state unfolding pathway, with a noticeably increased thermodynamic stability and lower aggregation tendency. Furthermore, the mutants maintained its capability of dragging Aβ‐oligomers away from the amyloid pathway.
The most common problem when obtaining scFv molecules by recombinant protocols is the presence of two disulfide bridges, one in the core of each domain, which drives to scrambled conformations that must be fractionated from the native state.16 Then, the yield of production is dramatically reduced and the cost is increased. This problem is not exclusive of scFv molecules or antibodies, but also other recombinant therapeutic proteins such as receptors, hormones, or enzymes.20 Disulfide bridges are known to increase thermodynamic stability by lowering the conformational entropy of the unfolded state compared with the native state, which makes the de novo introduction of such linkages a strategy for protein stabilization.21 This strategy has already been proven as effective in scFv molecules as happens in the case of the cysteine‐restored variant of the A48 scFv, which is naturally missing the conserved disulfide bridge in VH.22 The fact that several disulfide‐free scFv molecules can reach the native state,23 allows using the removal of these linkages as the most straightforward strategy to preclude the appearance of scrambled conformations.
Here, we have separately get rid of both disulfide bridges and achieved our objective to solve the scrambling problem. Because removing a covalent bond resulted on an overall destabilization of the fold, we also combined these single‐disulfide variants with the stabilizing mutations derived from elongation of the V L domain. To understand the effect that such a combination produces, we have evaluated the stability and some structural features of the different constructs. Interestingly, the same mutations drove to completely different scenarios depending on the presence of disulfide bridges, illustrating the relevance of such linkages in the stability, not only of the native state, but also of the different intermediate states for folding and misfolding.
Results
Elimination of the V L disulfide bridge
First, we intended to eliminate the disulfide bridge of the V L domain by generating scFv‐h3D6 mutant V L‐C23V/C88A, for the sake of clarity henceforth referred to as V L‐DF (disulfide‐free). Surprisingly, the first consequence of removing the disulfide bridge of the V L domain was a dramatic decrease in the production yield. Then, it became evident that stability dramatically dropped upon mutation. In consonance, thermal denaturation showed that the conformational transition followed by fluorescence was advanced about 15°C [Fig. 1(A)]. Although the native state of V L‐DF was only different in the minimum at 230 nm featuring the V L domain, as seen by CD [Fig. 1(B)], thermal denaturation showed a wide and dispersed signal [Fig. 1(C)]. This may reflect a low stability of the fold, but the protein is well folded since, apart from showing a native CD spectrum, thermal unfolding followed by Trp‐fluorescence is cooperative. It is known that proteins subjected to a structural stress can be in a metastable, native‐like conformation. Thus, although changes in the overall spectrum cannot be seen, instability can drive to misfolding. This native conformation partially unfolds upon thermal denaturation driving to aggregation, but this is not likely to occur through a β‐rich intermediate as the spectra at 85ºC is not that of a pure β‐conformation [Fig. 1(B)].
Figure 1.
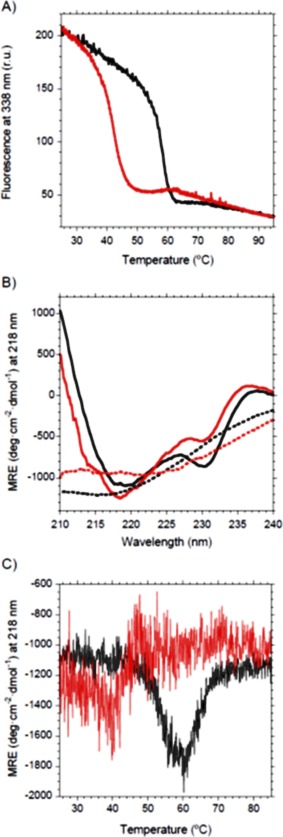
Thermal denaturation of VL‐DF followed by Trp‐fluorescence and Circular Dichroism (CD). Black, scFv‐h3D6, Red, V L‐DF. (A) Intensity of the fluorescence emission spectrum at 338 nm upon increasing temperature shows that the transition is advanced 15°C for the mutant. (B) Far‐UV CD spectra for the native states (25°C) of scFv‐h3D6 and V L‐DF mutant show differences in the V L signature (230 nm), whereas for the thermal‐denatured states (85°C) scFv‐h3D6 is in a β‐conformation and V L‐DF mutant displays a singular spectrum (dotted lines). (C) Ellipticity signal at 218 nm upon increasing temperature shows that the mutant partially unfolds. Data for scFv‐h3D6 are already published and are shown for comparative purposes.13, 16
Because we know from previous work that elongation mutations mainly increase the stability of the V L domain16 we tried to combine these mutations with the V L‐DF mutation, but no stabilization could be achieved (not shown). This confirms the necessity of the disulfide bridge of the V L domain to maintain a stable native fold and that the molecule does not endure the elimination of the disulfide bridge of the less stable domain. This may be also true for other class I scFv molecules.17, 18
Elimination of the V H disulfide bridge
Because the intermediate state in the unfolding pathway of scFv‐h3D6 is composed of the unfolded V L domain and the folded V H domain,16 we eliminated the disulfide bridge of the more stable domain, presuming that the loss of stability would not be as high as in the case of the less stable domain. For such a purpose, mutant V H‐C22V/C92A was generated. For the sake of clarity, this mutant will be henceforth referred to as V H‐DF (disulfide‐free). In fact, the first consequence of these mutations was doubling the production yield, while ensuring conformational homogeneity without the presence of scrambled conformations as assessed by cationic exchange and thermal denaturation (not shown). Interestingly, these mutations not only affected the stability of the molecule, but also its folding and misfolding and, consequently, its ability to form WL fibrils, as the following subsections describe.
Changes in the unfolding pathway of VH‐DF
Concerning the unfolding pathway, a two‐state model was enough to describe V H‐DF unfolding, instead of the more complex three‐state one showed by scFv‐h3D6 [Fig. 2(A)] and its elongation mutants.16 Thus, it seems that the intermediate state does not populate this pathway. This means that the lack of disulfide bridge in V H domain affects its stability so that differences between both domains are not apparent. Despite this destabilization, the maximum of the Trp‐fluorescence emission spectrum for the native state of V H‐DF was close to that of scFv‐h3D6, indicating that mutant V H‐DF is well folded. There was, however, a red‐shift of 1 nm in the native spectrum of the mutant, 339 nm versus 338 nm [Fig. 2(A)]. Such a small red‐shift is likely to be due to experimental error, but could also be indicative of faint differences in the packing of Trp residues within the mutant. On the other hand, since in both profiles the final plateau reached the emission maximum considered for Trp‐containing fully‐unfolded proteins, 355 nm,24 the urea‐denatured states were, in terms of tertiary structure, completely unfolded in both cases.
Figure 2.
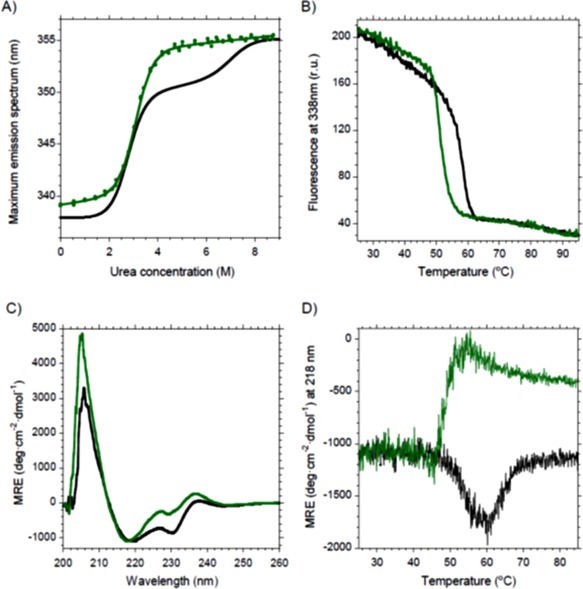
Changes in the unfolding pathway and the aggregation tendency of the V H‐DF mutant. Black, scFv‐h3D6; Green, V H‐DF. (A) Urea denaturation curve fits to a two‐state model for V H‐DF instead of the three‐state for scFv‐h3D6. (B) Thermal denaturation followed by Trp‐fluorescence shows a decrease in the mid‐point of about 6°C upon mutation. (C) Circular dichroism shows that the native state of V H‐DF was slightly different to that of scFv‐h3D6 in terms of secondary structure. (D) Thermal denaturation followed by CD indicates that V H‐DF unfolds before aggregation occurs, instead of directly reorganizing into a richer β‐sheet conformation that drives to aggregation.
Opposite to the three‐state behavior of scFv‐h3D6, the urea‐denaturation curve of V H‐DF does not show the two well‐defined transitions [Fig. 2(A)]. In fact, the fitting of the urea‐denaturation curve to the two‐state model rendered a very nice convergence with a Gibbs energy for unfolding of 21.8 ± 0.8 kJ mol−1 and a cooperativity of 7.1 ± 0.2 kJ mol−1 M−1 (Table 1). Because there are different theories on the physical meaning and contribution of these values, differences seen in Table 1 should be just qualitatively considered. Because the first transition has been attained to the V L domain and the second to V H,16 it appears that the latter has been dramatically destabilized in the V H‐DF mutant, as it was foreseen. In any case, and despite this appreciable destabilization, the domain seems to keep its folded arrangement according to the CD spectrum [Fig. 2(C); see next sub‐section for further details]. Thus, the native state spectrum is only different to the scFv‐h3D6 in the minimum at 230 nm, which is featuring the V L domain. In addition, the FTIR spectra show similar secondary structure distributions at 25°C (see also next sub‐section for further details). Perhaps, such a difference may indicate that elimination of the disulfide bridge of the V H domain not only influences its intrinsic stability but also that of the neighboring domain, probably through perturbation of the interfacial forces between domains. This inter‐domain cooperativity has been reported elsewhere studying other scFv molecules.22
Table 1.
Fitting of Urea Denaturation Curves to the Two‐State Model for Protein Folding of V H‐DF Mutant and its Elongation Mutants
| Parameter | ΔG F‐U | m F‐U | [D]F‐U |
|---|---|---|---|
| Variant | |||
| V H‐DF | −21.8 ± 0.8 | 7.1 ± 0.2 | 3.1 |
| V H‐DF/C1 | −21.5 ± 1.0 | 7.0 ± 0.3 | 3.1 |
| V H‐DF/C2 | −22.2 ± 0.8 | 7.1 ± 0.2 | 3.1 |
| V H‐DF/C3 | −23.0 ± 0.7 | 7.3 ± 0.2 | 3.2 |
C1 (V L‐el‐R108G), C2 (V L‐el‐R108), and C3 (V L‐el‐R108T109). ΔG in kJ mol−1; m in kJ mol−1 M−1. F = folded state; U = unfolded state; [D] between states = denaturant 50% in M.
Changes in the aggregation/misfolding pathway of VH‐DF
Thermal denaturation followed by Trp fluorescence showed that the conformational transition was advanced about 6°C, despite it displays a single cooperative transition as scFv‐h3D6 does [Fig. 2(B)]. To get insight into this transition circular dichroism (CD) was used. Figure 2(C) shows that the native state of V H‐DF was slightly different to that of scFv‐h3D6. It is known that the scFv‐h3D6 CD‐spectrum is featured, apart from the 218 nm minimum and 205 nm maximum characteristic for an all‐β fold, by a minimum at 230 nm and a positive shoulder at 237 nm.13 In a previous study, the minimum at 230 nm was attributed to the contribution of the conserved Trp in the V L domain, V L‐Trp 35, whereas the shoulder at 237 nm was attributed to the V H domain without any information for discriminating between the possible contribution of its conserved Trp, V H‐Trp 36, and that of the cystinyl side‐chain.25 Because this shoulder was even increased in the V H‐DF mutant, it became evident that V H‐Trp 36 generates such interference. In consonance with the interpretation of the urea denaturation curve, the minimum at 230 nm featuring the V L domain has been considerably decreased upon mutation, exhibiting that V L‐Trp 35 has also been reoriented upon eliminating the disulfide bridge within its neighboring domain.
The most salient feature comes from CD thermal unfolding curves. It was previously reported that thermal denaturation of scFv‐h3D6, followed at 218 nm, the minimum characteristic for a β‐fold, showed an increase in ellipticity starting at 45°C and finishing at 60°C; thus, a thermally‐induced β‐sheet rich intermediate state is maximally populated at 60°C. This intermediate was also shown to lead to scFv‐h3D6 aggregation as WL fibrils.13 The behavior of V H‐DF emerges as “opposite” [Fig. 2(D)], since the ellipticity decreases at mid‐temperatures, indicating that the β‐sheet rich intermediate does not populate. Surprisingly, the V H‐DF mutant aggregates at around 55°C [Fig. 2(B)].
To delve into these differences, additional thermal denaturation experiments were done using CD and FTIR as spectroscopic probes.
We recorded a collection of CD spectra upon thermal unfolding [Fig. 3(A)]. In the case of scFv‐h3D6, as the temperature increases the CD signals at 230 and 237 nm decrease, and the minimum at 218 nm corresponding to β‐sheet increases. Up above 60°C a transition to a prone‐to‐aggregation conformation is observed and aggregation could be appreciated from the signal dropping. Therefore, it is at 60°C when the intermediary state is maximally populated. Opposite, in the V H‐DF mutant the intensity of the β‐sheet minimum decreases from the beginning, so that a partial unfolding process occurs before the transition to a prone‐to‐aggregation conformation takes place. Therefore, we have clear evidence that the elimination of the disulfide of the V H domain has changed the conformation of the intermediate state populating the aggregation pathway.
Figure 3.
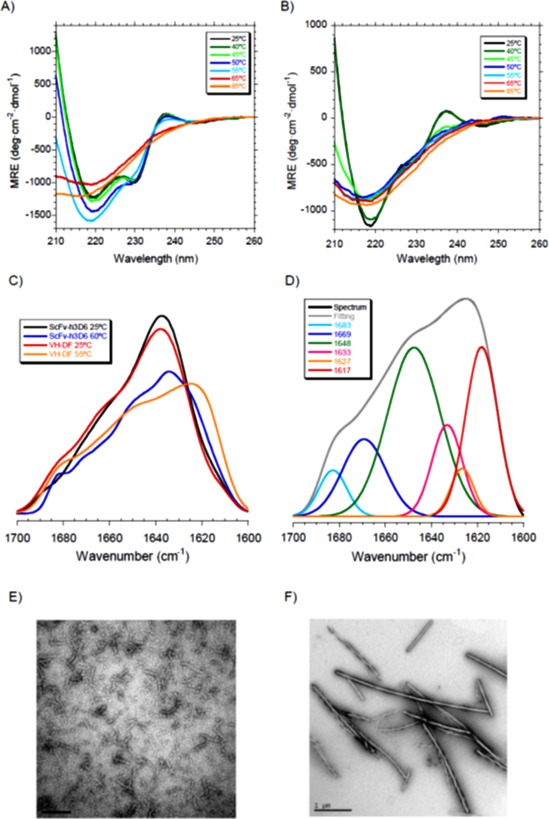
Changes in the aggregation pathway of V H‐DF. (A) CD at different temperatures of scFv‐h3D6 indicates that a conformational transition occurs between 55 and 65°C. (B) In the case of V H‐DF the conformational transition occurs from 45°C and the intensity of the β‐sheet minimum also decreases from the beginning, so that a partial unfolding process takes place before. (C) Amide I' FTIR spectra at 25°C shows no differences between scFv‐h3D6 and V H‐DF but the shift to lower wavenumbers of the maximum of the spectra upon heating at around 60°C is greater for V H‐DF. (D) Deconvolution of the V H‐DF spectrum reveals the presence of a prominent amyloid component at 1617 cm−1 at the temperature at which the intermediate state is maximally populated (55°C) (see Table II for quantification). (E) TEM micrographs of scFv‐h3D6 incubated at 60°C for 5 min shows the presence of WL fibrils. (F) TEM micrographs of V H‐DF incubated at 55°C for 5 min shows the presence of amyloid fibrils.
To gain structural insight of these intermediate states FTIR was used at the temperature at which they are maximally populated (60°C for the scFv‐h3D6, 55°C for the V H‐DF mutant). One of the advantages of FTIR is its ability to differentiate among different β‐sheet conformations because a shift to lower wavenumbers in the maximum of the amide I' spectrum can be interpreted as the result of increased hydrogen bonding within the β‐strands, of a more planar sheet or of a larger number of strands.26 Figure 3(C) shows that the spectra at 25°C were quite similar for scFv‐h3D6 and V H‐DF, with the maximum located at 1636 cm−1 for scFv‐h3D6 and 1638 cm−1 for V H‐DF (both attributable to a native β‐sheet main component), but the maximum in the spectra at 55–60°C were located at 1630 cm−1 for scFv‐h3D6 and 1624 cm−1 for V H‐DF. To delve in to the differences of these spectra deconvolution was performed (Table 2). At 25°C there are no differences but it becomes evident that at 55–60°C the WL component is smaller for V H‐DF (5% vs. 23%) and the amyloid component is rather bigger (24% vs. 9%), as well as the random coil component (39% vs. 14%). Therefore, it appears that the elimination of the disulfide bridge within the V H domain shifts the aggregation pathway from the WL to the amyloid one through the formation of a partially unfolded intermediate.
Table 2.
Band Decomposition of FTIR Amide I′ Band at Different Temperatures
| Secondary structure | scFv‐h3D6a | DF‐VH | ||||||
|---|---|---|---|---|---|---|---|---|
| 25°C | 60°C | 25°C | 55°C | |||||
| Center (cm−1) | % Area | Center(cm−1) | % Area | Center (cm−1) | % Area | Center (cm−1) | % Area | |
| ↑ Fr. antip. β‐sheet | 1681 | 9 | 1683 | 4 | 1684 | 5 | 1683 | 6 |
| ↑ Fr. loops/turns | – | – | 1671 | 11 | – | – | 1669 | 15 |
| ↓ Fr. loops/turns | 1660 | 30 | 1662 | 6 | 1663 | 33 | – | – |
| α‐helix | – | – | 1653 | 15 | – | – | – | – |
| Random coil | – | – | 1644 | 14 | – | – | 1648 | 39 |
| ↓ Fr. antip. β‐sheet | 1636 | 60 | 1636 | 16 | 1637 | 60 | 1633 | 12 |
| WL fibril | – | – | 1626 | 23 | – | – | 1627 | 5 |
| Amyloid fibril | 1612b | 1 | 1615 | 9 | 1611b | 2 | 1617 | 24 |
Data for the scFv‐h3D6 are already published and are shown only for comparative purposes 13. ↑ Fr, high frequency; ↓ Fr, low frequency; antip, antiparallel.
These correspond to side‐chains rather than to amyloid fibrils.
TEM analysis of scFv‐h3D6 and V H‐DF samples incubated at 60 or 55°C, respectively, confirmed the FTIR evidence, as seen in Figure 3(E,F), where the WL fibrils formed by scFv‐h3D6 [Fig. 3(E)] and the amyloid fibrils formed by the V H‐DF mutant [Fig. 3(F)] could be seen. Amyloid and WL fibrils populate different aggregation pathways: amyloid fibrils being straight and long, and forming through a nucleated‐dependent kinetics; whereas WL fibrils being curved and short, and forming through a non‐nucleation‐dependent kinetics.13, 27
Combination of VH‐DF with elongation mutants
Combination of elongation mutations with V H‐DF rendered a good yield but no differences were found. Albeit these elongation mutations were reported to increase the stability of scFv‐h3D6, this was not likely the case when combined with the V H‐DF mutant [Fig. 4(A)]. Table 1 shows that stability tend to slightly increase in the order C1, C2, C3, as described for the combination with scFv‐h3D6, but these differences cannot be taken as reliable since they are within the experimental error.
Figure 4.
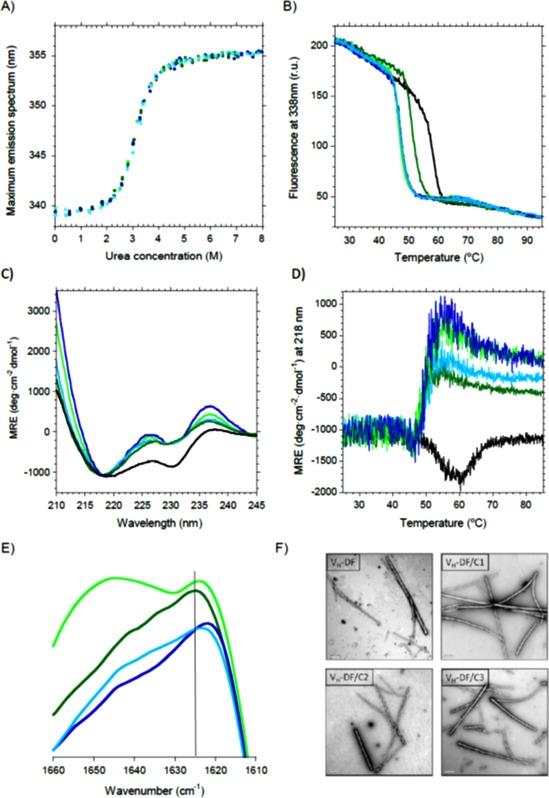
Effect of elongation mutations on V H‐DF. Black, scFv‐h3D6; Dark green, V H‐DF; Light green, V H‐DF/C1; Dark blue, V H‐DF/C2; Light blue, V H‐DF/C3. (A) Urea denaturation curves show no differences with V H‐DF. (B) Thermal denaturation curves followed by Trp‐fluorescence display a decrease in the mid‐point of about 10°C for all the combinations, instead of 6°C for V H‐DF. (C) Circular dichroism shows that both the V H and V L signatures were more evident for the combined mutants. (D) Thermal denaturation followed by CD indicates that V H‐DF unfolds before aggregation occurs, instead of directly reorganizing into a richer β‐sheet conformation that drives to aggregation as in the case of scFv‐h3D6. (E) Amide I' FTIR spectra at 55°C shows that all elongation mutants on V H‐DF display a shift to lower wavenumbers of the maximum of the spectra, indicating that the aggregation tendency in the form of amyloid fibrils has increased. (F) TEM micrographs of V H‐DF and its elongation mutants incubated at 55°C for 5 min show the presence of amyloid fibrils.
Thermal denaturation followed by Trp‐fluorescence was advanced 10°C for all the combinations, instead of the 6°C for the V H‐DF mutant [Fig. 4(B)]. This was also observed when following thermal denaturation by CD, where the unfolding before aggregation was even more evident [Fig. 4(D)] and the initial spectra were more affected, both at the V H and V L CD signatures [Fig. 4(C)]. FTIR analysis confirmed the increase in the aggregation tendency to amyloid fibrils, as the maximum of the spectra is shifted to lower wavenumbers in the mutants [Fig. 4(E)]. Finally, TEM micrographs showed the formation of amyloid fibrils by such variants [Fig. 4(F)].
Therefore, an increase in the aggregation tendency has occurred upon elongation of the V H‐DF mutant. This is indicative of the stabilization of the intermediate state in the amyloid aggregation pathway of V H‐DF rather than the stabilization of the native state, as was the case for the elongated scFv‐h3D6.16
Discussion
ScFv‐h3D6 is an antibody fragment that precludes Aβ peptide‐induced toxicity by withdrawing Aβ oligomers from the amyloid pathway towards the WL pathway.13 Because its therapeutic potential has been successfully assessed in the 3xTg‐AD mouse model of Alzheimer's disease,14, 15 it is worthy to improve the main drawback of its recombinant production. Here, we separately removed the disulfide bond of each domain and solved the scrambling problem, and then, we intended to compensate the loss of thermodynamic stability by adding three C‐terminal elongation mutations previously described as stabilizing the native fold of scFv‐h3D6.
ScFv‐h3D6 does not endure the elimination of the disulfide bridge of the less stable domain, V L. Apart from the compromised low‐yield obtained, and although the CD spectrum was native‐like, thermal denaturation showed an evident decrease in stability. It is generally accepted that proteins subjected to a structural stress, here removing the V L disulfide bridge, can adopt a metastable conformation that do not change the overall structure but affects the stability and go to misfolding when subjected to environmental changes. Another consequence of removing this disulfide bridge was that aggregation did not occur through a β‐rich intermediate state. This behavior indicates that the disulfide containing V L domain is a structure involved in aggregation in the form of WL fibrils previously described for the scFv‐h3D6. The involvement of structured domains in scFv aggregates was postulated by Plünckthun from the observation that the presence of a stable V L domain in combination with a disulfide‐free V H domain was likely to promote aggregation.28 On the other hand, introduction of the already known as elongation mutations, V L‐el‐R108G (referred to as C1), V L‐el‐R108 (C2), and V L‐el‐R108T109 (C3), did not help to stabilize V L‐DF. It is calculated that the stabilizing contribution of the disulfide bridge in variable domains is around −19 kJ mol−1 29 and we know that the most stable of the three introduced mutations, V L‐el‐R108T109 (C3), stabilized the scFv‐h3D6 fold in −5.1 kJ mol−1,16 so that the combination of the elimination of the V L disulfide bridge with the stabilized mutants was already expected to be challenging.
When removing the disulfide bond of the more stable V H domain the fluorescence, CD and FTIR spectra were native‐like and the yield was doubled because scrambling conformers were not present, so we achieved our main goal. Two main effects of removing V H disulfide bridge were observed. First, the folding behavior apparently changed from a three‐state to a two‐state [Fig. 5(A)], so that the equilibrium intermediate was not detected. In addition, stability dropped much more than the calculated contribution of one disulfide bond in a variable domain, −19 kJ mol−1.29 The energy previously published for the folding of the scFv‐h3D6 V L domain plus interface contribution is −19 kJ mol−1 and that for the folding of the V H domain is −37 kJ mol−1. Because a value of −22 kJ mol−1 is reported here as the energy required to fold the complete V H‐DF mutant, it can be concluded that the lack of a properly folded V H domain also marginally affects V L domain because the native interface is somehow affected. This concurs with the observation by CD of the hydrophobic environment of Trp‐35 of V L domain being different in the V H‐DF mutant. Mutual stabilization of V L and V H in scFvs is already known,22 and it reflects the relevance of the interface in the folding of these molecules. Therefore, we can conclude that destabilization of the V H domain induced by the removal of its disulfide bridge has changed the folding behavior from a Class I scFv, with one domain much more stable than the sum of the other plus interface contribution, to a Class II, where the intrinsic stability of one domain is in the same range as the total stability of the other domain.17
Figure 5.
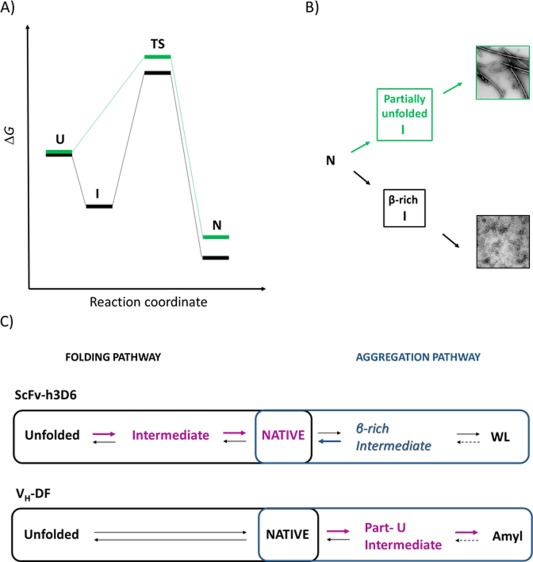
Diagrams for the folding and the aggregation pathways of scFv‐h3D6 and V H‐DF. ScFv‐h3D6, black, V H‐DF, grey, (A) Energy diagram for the folding reaction. ScFv‐h3D6 follows a three‐state folding reaction, whereas V H‐DF does not show any intermediate state. (B) Scheme of the aggregation pathway. ScFv‐h3D6 aggregates as WL fibrils through the formation of a β‐rich intermediate, whereas V H‐DF aggregates as amyloid fibrils through the formation of a partially unfolded intermediate. (C) Folding and aggregation pathways compete, such as at 25°C the folding pathway is the predominant one whereas at 60°C (scFv‐h3D6) or 54°C (V H‐DF) it is the aggregation pathway the one that predominates. The effect of the already described stabilizing mutations on scFv‐h3D6 is indicated with bold arrows, the stabilized states in magenta, and the destabilized states in blue. These mutations stabilize the intermediate state in the folding pathway of scFv‐h3D6 while destabilizing the β‐rich intermediate driving to the formation of WL fibrils. In the case of V H‐DF stabilization of the partially unfolded intermediate driving to amyloid fibrils is the only effect observed.
The second main effect of removing the V H disulfide bridge was that the aggregation pathway changed from a WL morphology, through the formation of a β‐rich intermediate, to an amyloid morphology, through the formation of a partially unfolded intermediate [Fig. 5(B)], as demonstrated by CD, FTIR, and TEM. Interestingly, the disulfide bridge in the VL domain is also required for WL aggregation to occur. Therefore, it is likely that both disulfide bridges might stabilize the β‐rich intermediate driving to aggregation in the form of WL fibrils. Folding and misfolding are competing pathways because kinetic partitioning occurs,30 similarly WL and amyloid pathways compete.
When adding the elongation mutations on V H‐DF the stabilization of the native fold failed, in contrast to what was already reported for scFv‐h3D616 [Fig. 5(C)]. These mutations stabilize the native fold by stabilizing the intermediate state in the folding pathway of scFv‐h3D6, whereas on V H‐DF such an intermediate is not detected and no stabilization of the native state can be achieved. Stabilization of the folding intermediate favors the folding pathway at the expense of the aggregation one, as the β‐rich intermediate driving to WL fibrils is destabilized. However, in the absence of the V H disulfide bridge the partially unfolded intermediate driving to the formation of amyloid fibrils is the one that is stabilized.
As a general conclusion, the goals of getting rid of scrambling conformations and increasing the production yield have been achieved. The removal of the V L disulfide bridge was not endured by the molecule, but the removal of the V H disulfide bridge rendered a native‐like conformation. Both the unfolding and the aggregation pathways have changed, and elongation of the C‐terminus had no effect on the stability of the native state. Therefore, next step to further improve the molecule will be to explore evolutionary approaches on the V H disulfide‐free mutant.18
Material and Methods
Mutagenesis, protein expression, and purification
Mutagenesis was performed by QuickChange Lightning Site‐Directed Mutagenesis Kit (Agilent Technologies). Protein expression was carried out in Escherichia coli strain Origami 2 (DE3), as previously described for scFv‐h3D6.13 In brief, pETtrx‐1a was used for the reference scFv‐h3D6, V H‐DF mutants, whereas for the V L‐DF mutants' expression in pCri5a was used. These vectors generated scFv variants fussed to Trx and NusA carriers, respectively. Carriers were removed by TEV proteolysis and purification was performed as previously described.16
Secondary structure determination by CD
Protein secondary structure was monitored at different temperatures by far‐UV CD spectroscopy from 260 to 190 nm in a Jasco J‐715 spectrophotopolarimeter. Protein concentration was 20 μM, and 20 scans were recorded at 50 nm min−1 (response 2s) in a 0.2 cm pathlength cuvette.
Secondary structure determination by FTIR
FTIR analysis was performed as described previously.13 Briefly, protein at 100 μM was dialyzed at 4°C against deuterated‐PBS. Spectra were acquired at different temperatures ranging from 25 to 60°C in a Variant Resolutions Pro spectrometer using excavated cells with a 50 μm path (Reflex Analytical) and the series software licensed under OMNIC (Thermo Scientific).
Thermal denaturation
Thermal denaturation was followed up by far‐UV CD spectroscopy at 218 nm and tryptophan fluorescence emission at 338 nm, both at 20 μM protein concentration and 1°C min−1 heating rate.
Chemical denaturation
Chemical denaturation was performed by creating series of progressive urea concentrations with 2 μM final concentration of protein. Measurement of Trp‐fluorescence was performed at 25°C at an excitation wavelength of 290 nm and an emission spectrum recorded from 320 to 380 nm in a Varian Cary Eclipse fluorimeter. Excitation slit was set to 5 nm and the emission one at 10 nm, and 5 scans were averaged. The maximum of each emission spectrum was obtained by fitting to a three‐parametric polynomial, as described before.16
TEM
To visualize the aggregation extent and morphology of the variants incubation of 100 μM samples were carried out at 60°C for 10 min. Then, samples were diluted 1:10 in PBS and quickly adsorbed onto glow‐discharge carbon‐coated grids. Transmission electron microscopy was performed in a Jeol 120‐kV JEM‐1400 microscope, using 1% uranyl acetate for negative staining.
Statement: We previously designed scFv‐h3D6, a single chain variable fragment that ameliorates first hallmarks of Alzheimer's Disease. Because the presence of two disulfide bridges in the core of each domain hinders its recombinant production, we separately removed each bond and improved the yield. The addition of previously reported stabilizing mutations lead to completely different scenarios in the conformational landscape depending on the presence of disulfide bridges.
References
- 1. Selkoe DJ (2000) Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta‐protein. Ann NY Acad Sci 924:17–25. [DOI] [PubMed] [Google Scholar]
- 2. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 3. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. [DOI] [PubMed] [Google Scholar]
- 4. De Kimpe L, Scheper W (2010) From alpha to omega with Abeta: targeting the multiple molecular appearances of the pathogenic peptide in Alzheimer's disease. Curr Med Chem 17:198–212. [DOI] [PubMed] [Google Scholar]
- 5. Robert R, Dolezal O, Waddington L, Hattarki MK, Cappai R, Masters CL, Hudson PJ, Wark KL (2009) Engineered antibody intervention strategies for Alzheimer's disease and related dementias by targeting amyloid and toxic oligomers. Protein Eng Des Sel 22:199–208. [DOI] [PubMed] [Google Scholar]
- 6. Holliger P, Hudson PJ (2005) Engineered antibody fragments and the rise of single domains. Nat Biotechnol 23:1126–1136. [DOI] [PubMed] [Google Scholar]
- 7. Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9:269–277. [DOI] [PubMed] [Google Scholar]
- 8. Montoliu‐Gaya L, Villegas S (2016) Aβ‐Immunotherapeutic strategies: a wide range of approaches for Alzheimer's disease treatment. Expert Rev Mol Med 18:e13. [DOI] [PubMed] [Google Scholar]
- 9. Huang L, Su X, Federoff H (2013) Single‐chain fragment variable passive immunotherapies for neurodegenerative diseases. Int J Mol Sci 14:19109–19127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y, Chen X, Liu J, Zhang Y (2015) The protective effects and underlying mechanism of an anti‐oligomeric Aβ42 single‐chain variable fragment antibody. Neuropharmacology 99:387–395. [DOI] [PubMed] [Google Scholar]
- 11. Panza F, Frisardi V, Imbimbo BP, Seripa D, Paris F, Santamato A, D'Onofrio G, Logroscino G, Pilotto A, Solfrizzi V (2011) Anti‐β‐amyloid immunotherapy for Alzheimer's disease: focus on bapineuzumab. Curr Alzheimer Res 8:808–817. [DOI] [PubMed] [Google Scholar]
- 12. Lu J‐X, Qiang W, Yau W‐M, Schwieters CD, Meredith SC, Tycko R (2013) Molecular structure of β‐amyloid fibrils in Alzheimer's disease brain tissue. Cell 154:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marín‐Argany M, Rivera‐Hernández G, Martí J, Villegas S (2011) An anti‐Aβ (amyloid β) single‐chain variable fragment prevents amyloid fibril formation and cytotoxicity by withdrawing Aβ oligomers from the amyloid pathway. Biochem J 437:25–34. [DOI] [PubMed] [Google Scholar]
- 14. Giménez‐Llort L, Rivera‐Hernández G, Marin‐Argany M, Sánchez‐Quesada JL, Villegas S (2013) Early intervention in the 3xTg‐AD mice with an amyloid β‐antibody fragment ameliorates first hallmarks of Alzheimer disease. MAbs 5:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esquerda‐Canals G, Marti J, Rivera‐Hernández G, Giménez‐Llort L, Villegas S (2013) Loss of deep cerebellar nuclei neurons in the 3xTg‐AD mice and protection by an anti‐amyloid β antibody fragment. MAbs 5:660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rivera‐Hernández G, Marín‐Argany M, Blasco‐Moreno B, Bonet J, Oliva B, Villegas S (2013) Elongation of the C‐terminal domain of an anti‐amyloid β single‐chain variable fragment increases its thermodynamic stability and decreases its aggregation tendency. MAbs 5:678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wörn A, Plückthun A (1999) Different equilibrium stability behavior of ScFv fragments: identification, classification, and improvement by protein engineering. Biochemistry 38:8739–8750. [DOI] [PubMed] [Google Scholar]
- 18. Wörn A, Plückthun A (2001) Stability engineering of antibody single‐chain Fv fragments. J Mol Biol 305:989–1010. [DOI] [PubMed] [Google Scholar]
- 19. Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C (1991) Variable region heavy chain sequences In Sequences of Proteins of Immunological Interest. Natl. Tech. Inf. Serv. (NTIS). NIH Publ. No. 91‐3242. [Google Scholar]
- 20. Zhang L, Chou CP, Moo‐Young M (2011) Disulfide bond formation and its impact on the biological activity and stability of recombinant therapeutic proteins produced by Escherichia coli expression system. Biotechnol Adv 29:923–929. [DOI] [PubMed] [Google Scholar]
- 21. Bulaj G (2005) Formation of disulfide bonds in proteins and peptides. Biotechnol Adv 23:87–92. [DOI] [PubMed] [Google Scholar]
- 22. Wörn A, Plückthun A (1998) Mutual stabilization of VL and VH in single‐chain antibody fragments, investigated with mutants engineered for stability. Biochemistry 37:13120–13127. [DOI] [PubMed] [Google Scholar]
- 23. Wörn A, Plückthun A (1998) An intrinsically stable antibody scFv fragment can tolerate the loss of both disulfide bonds and fold correctly. FEBS Lett 427:357–361. [DOI] [PubMed] [Google Scholar]
- 24. Schmid F (1997) Optical spectroscopy to characterize protein conformation and conformational changes: Protein structure: a practical approach, 2nd ed IRL Press Oxford, pp 261–267. [Google Scholar]
- 25. Sreerama N, Manning MC, Powers ME, Zhang JX, Goldenberg DP, Woody RW (1999) Tyrosine, phenylalanine, and disulfide contributions to the circular dichroism of proteins: circular dichroism spectra of wild‐type and mutant bovine pancreatic trypsin inhibitor. Biochemistry 38:10814–10822. [DOI] [PubMed] [Google Scholar]
- 26. Zandomeneghi G, Krebs MRH, McCammon MG, Fändrich M (2009) FTIR reveals structural differences between native β‐sheet proteins and amyloid fibrils. Protein Sci 13:3314–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gosal WS, Morten IJ, Hewitt EW, Smith DA, Thomson NH, Radford SE (2005) Competing pathways determine fibril morphology in the self‐assembly of beta2‐microglobulin into amyloid. J Mol Biol 351:850–864. [DOI] [PubMed] [Google Scholar]
- 28. Ramm K, Gehrig P, Plückthun A (1999) Removal of the conserved disulfide bridges from the scfv fragment of an antibody: effects on folding kinetics and aggregation. J Mol Biol 290:535–546. [DOI] [PubMed] [Google Scholar]
- 29. Frisch C, Kolmar H, Schmidt A, Kleemann G, Reinhardt A, Pohl E, Usón I, Schneider TR, Fritz HJ (1996) Contribution of the intramolecular disulfide bridge to the folding stability of REIv, the variable domain of a human immunoglobulin kappa light chain. Fold Des 1:431–440. [DOI] [PubMed] [Google Scholar]
- 30. Chiti F, Taddei N, Baroni F, Capanni C, Stefani M, Ramponi G, Dobson CM (2002) Kinetic partitioning of protein folding and aggregation. Nat Struct Biol 9:137–143. [DOI] [PubMed] [Google Scholar]