

Abstract
Circadian clocks are ubiquitous timing systems that induce rhythms of biological activities in synchrony with night and day. In cyanobacteria, timing is generated by a posttranslational clock consisting of KaiA, KaiB, and KaiC proteins and a set of output signaling proteins, SasA and CikA, which transduce this rhythm to control gene expression. Here, we describe crystal and nuclear magnetic resonance structures of KaiB-KaiC, KaiA-KaiB-KaiC, and CikA-KaiB complexes. They reveal how the metamorphic properties of KaiB, a protein that adopts two distinct folds, and the post–adenosine triphosphate hydrolysis state of KaiC create a hub around which nighttime signaling events revolve, including inactivation of KaiA and reciprocal regulation of the mutually antagonistic signaling proteins, SasA and CikA.
Circadian clocks are an ancient evolutionary adaptation found in archaea (1, 2), bacteria (3), and eukaryotes (4, 5) that synchronize genetic, epigenetic, and metabolic activities to Earth’s rotation (6–8). In humans, chronic desynchronization of cellular clocks with the environmental light/dark cycle increases susceptibility to disease (9–11). In the cyanobacterium Synechococcus elongatus, the circadian clock orchestrates gene expression for the majority of its genes (12) to regulate daytime and nighttime metabolic processes that enhance fitness (13) and acutely affect survival of these obligate phototrophs at night (14, 15).
The cyanobacterial circadian clock is an ideal model to elucidate mechanisms of biological timekeeping because the oscillator consists of only three proteins—KaiA, KaiB, and KaiC (16)—that generate circadian rhythms of KaiC phosphorylation in the presence of adenosine triphosphate (ATP) (17). Two sensor histidine kinases SasA (18) and CikA (19) self-assemble into day- and night-specific complexes with Kai protein complexes, based on the phosphorylation state of KaiC (20) to regulate the activity of the response regulator and transcription factor RpaA (21) (Fig. 1). However, the structural basis by which the clock autonomously transitions from its daytime to nighttime signaling state has remained elusive.
Fig. 1. Model of circadian formation of cyanobacterial clock protein complexes.
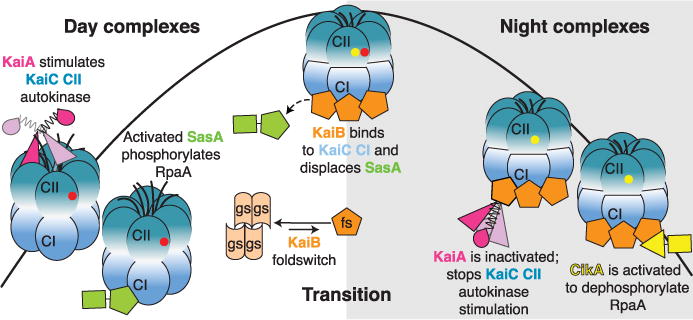
During the day, KaiA binds to the C-terminal extensions of KaiC to enhance the autokinase activity of the CII domain of KaiC, leading to sequential autophosphorylation. Yellow and red circles represent phosphorylated S431 (pS) and T432 (pT), respectively. The sensor histidine kinase, SasA, is activated by KaiC. During the day, the hexameric CII ring is loose and unstacked from the hexameric CI ring. At dusk, phosphorylation of S431 tightens the CII ring, leading to withdrawal of the KaiA binding sites on CII. The tightened CII ring interacts and stabilizes the CI ring in its post-ATP hydrolysis state, which now has its KaiB binding sites exposed. The rare fold-switched state of KaiB, KaiBfs, cooperatively binds CI, displacing SasA. KaiBfs sequesters the autoinhibited conformation of KaiA, allowing autodephosphorylation of KaiC. KaiBfs also binds and activates the phosphatase activity of CikA. Upon autodephosphorylation of S431, the CII ring loosens, causing unstacking of the rings. The CI ring returns to its pre-ATP hydrolysis state, letting go of KaiB, KaiA, and CikA to begin a new day.
KaiC uses multiple enzymatic activities to act as the central circadian pacemaker (22). It has two homologous domains (CI and CII) that belong to the AAA+ ATPase (adenosine triphosphatase) family (23), with each domain associating into a hexameric ring (Fig. 1). The CII domain possesses autokinase (24) and phosphotransferase activities (25, 26). Autophosphorylation of two residues on the CII domain, S431 and T432, tighten and loosen the CII ring (27), respectively, which in turn regulates accessibility of a KaiA binding site on CII (28) and a KaiB binding site on CI (29, 30). Positive and negative feedback by phosphoryl T432 and phosphoryl S431, respectively, governs an ordered phosphorylation cycle throughout the day: ST → S/pT → pS/pT → pS/T → S/T (S, serine; T, threonine; p, phosphorylated) (31, 32). KaiA is an obligate dimer with no known enzymatic activities (33, 34) that stimulates KaiC CII autophosphorylation during the day (35, 36) by maintaining the C-terminal A loops of CII in their exposed state (28, 37, 38) (Fig. 1). Over the course of the day, the sensor histidine kinase SasA binds to and is activated by the CI domain of KaiC to phosphorylate RpaA, which then activates transcription of class 1 genes that show peak expression at dusk (20, 28, 29, 39, 40) (Fig. 1).
The inhibition of KaiA at night permits autodephosphorylation of KaiC to reset the cycle. Little is known about how inhibition of KaiA is achieved by interaction with a KaiB-KaiC complex (35, 41, 42). KaiB belongs to a rare class of so-called metamorphic proteins, which reversibly switch between different folds under native conditions (43). KaiB transitions from a highly populated, inactive tetrameric ground-state fold (KaiBgs) to a rare, active-state monomeric fold (KaiBfs) that is captured by the CI ring of KaiC when CII is predominantly in the pS/pT or pS/T states in the evening (39) (Fig. 1). In addition, formation of the KaiBfs-KaiC complex requires ATPase activity in the CI domain (44). However, the structural basis for KaiBfs binding and the requirement of CI domain ATPase activity in regulating these interactions has been unclear.
The phosphorylation status of the hexameric CII ring is communicated to the CI ring via phosphoryl S431-dependent ring-stacking interactions (27, 29). When stacked with CII, the CI ring captures KaiBfs to displace SasA from KaiC via competition for overlapping binding sites on CI (28, 39), thereby turning off further stimulation of RpaA-mediated expression of class 1 genes (Fig. 1). While bound by CI, KaiBfs recruits and inhibits KaiA (39). The KaiBfs-KaiC complex also binds CikA to stimulate its phosphatase activity toward RpaA (21, 39), repressing class 1 genes and activating class 2 genes (peak expression at dawn) (Fig. 1). Therefore, the KaiBfs-KaiC complex forms a hub that regulates multiple interactions to disengage SasA, inhibit KaiA, and activate CikA.
Thermophilic clock proteins from Thermosynechococcus elongatus (fig. S1) are structurally and functionally similar to those from S. elongatus and more amenable for structural studies (45). Using mutations that stabilize the rare fold of KaiBfs from T. elongatus (39), here we present high-resolution structures of the KaiBfs-KaiC hub alone and in complex with domains of its interacting partners, KaiA and CikA. Together, these structures explain the requirement for CI ATPase activity and KaiB metamorphosis in the inactivation of KaiA and SasA and the recruitment of CikA.
Basis of SasA inactivation by KaiB
KaiB competes with SasA for an overlapping binding site on KaiC (46, 47) at the CI domain (28, 29) to regulate clock output signaling (39). Previously, we designed a monomeric form of the isolated CI domain, hereafter referred to as CImono (fig. S1), that forms a stable complex with KaiB (29). Here, we designed slightly truncated forms to favor crystallization, one on the N terminus of CImono, hereafter referred to as CIcryst (fig. S1), and another at the C terminus of a fold-switched KaiB mutant stabilized by G89A and D91R mutations (KaiBfs), hereafter referred to as KaiBfs-cryst (fig. S1). Y8A and Y94A substitutions were also incorporated to enhance the stability of KaiBfs-cryst. Using these optimized constructs, we obtained a 1.8-Å resolution crystal structure of the KaiBfs-cryst-CIcryst complex (Fig. 2A).
Fig. 2. Fold-switched KaiB binds to the posthydrolysis state of the CI domain of KaiC.
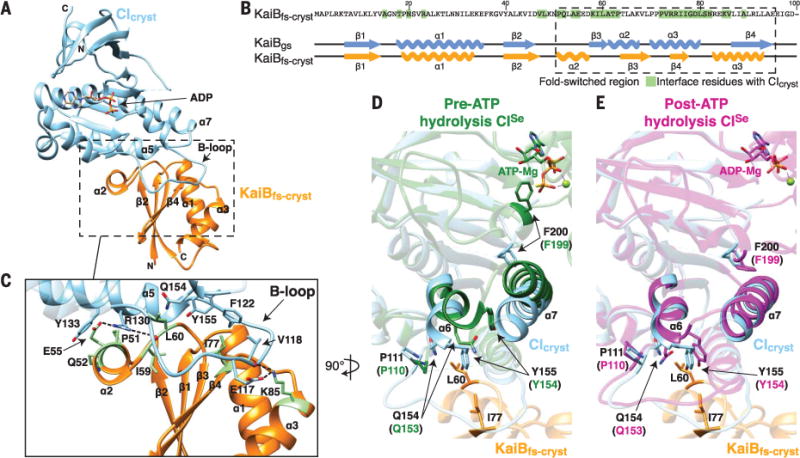
(A) Crystal structure of the KaiBfs-cryst-CIcryst complex at 1.8 Å. Orange, KaiBfs-cryst; sky-blue, CIcryst. (B) Secondary structures of KaiBgs (PDB 2QKE, subunit A) and KaiBfs-cryst bound to CIcryst. Residues of KaiBfs-cryst that interact with CIcryst are highlighted in green. (C) Zoomed-in view of the boxed region in (A). Representative interacting residues in (B) are shown in green. Dashed lines: electrostatic KaiBfs-cryst-CIcryst interactions. (D) Superposition of CI structures before ATP hydrolysis. Sky-blue, CIcryst; dark green, pre-ATP hydrolysis state of CISe (PDB 4TLC, subunit C), missing density for the B loop. Residue numbers in CISe are offset from those of T. elongatus CI by −1. (E) Superposition of CI structures after ATP hydrolysis. Sky blue, CIcryst; magenta, post-ATP hydrolysis state of CISe (PDB 4TLA, subunit E). Please see fig. S1 and table S1 for construct details.
The interface predominantly comprises residues from the fold-switched C-terminal half of KaiB (Fig. 2, B and C), explaining the requirement of fold-switching for KaiB-KaiC complex formation (39). The interface centers on the B loop of CIcryst, consistent with findings that deletion of the B loop and alanyl substitutions of B-loop residues E117, V118, and F122, as well as R130, abolished or weakened KaiB-KaiC binding (28, 39). Because KaiBfs has the same thioredoxin-like fold as the domain of SasA that binds KaiC (39), their modes of binding to the CI domain are likely to be similar. Indeed, SasA binding to CImono was shown to be weakened by the same set of mutations (28, 39). Thus, our KaiBfs-cryst-CIcryst structure provides insights into the competition between KaiB and SasA that explains the down-regulation of SasA activity at night.
The post-ATP hydrolysis state of KaiC CI domain captures KaiBfs
Mutations that tune the ATPase activity of the CI domain modulate the circadian period, but it is not yet known how this enzymatic activity exerts its intrinsic role in timekeeping (22, 44, 48). ATP hydrolysis by the CI domain is necessary for formation of the KaiB-KaiC complex (44, 48), suggesting that the hydrolysis activity is poised to control timing of the assembly of nighttime signaling complexes (49). Comparison of our adenosine diphosphate (ADP)–bound KaiBfs-cryst-CIcryst complex with structures of the CISe domain before and after ATP hydrolysis (superscript “Se” denotes proteins from S. elongatus) (22) revealed the structural basis for this obligate ATP hydrolysis. Residues at the CI ATP binding site display large conformational differences before and after hydrolysis (22) (Fig. 2, D and E). In attaining the posthydrolysis state, residue F200 near the ATP binding site rotates downward by ~130°, accompanied by the downward movement of helices α6 and α7. Our structure shows that α6 residues Q154 and Y155 drop down after ATP hydrolysis to form part of the KaiBfs-cryst-CIcryst interface (Fig. 2, D and E). Thus, ATP hydrolysis by the CI domain powers a conformational switch that is necessary to bind KaiBfs.
Previous reports have shown that KaiB forms a hexameric assembly on KaiC (50, 51). To study the KaiB-KaiC complex in its natural hexameric state, we used a KaiB fold-switch variant harboring an I88A substitution, hereafter referred to as KaiBfs-cryst* (fig. S1). Upon incubating KaiBfs-cryst* with a KaiCS431E mutant that mimics phosphoryl-S431 KaiC (fig. S1), we observed formation of a monodisperse hexameric assembly in solution (fig. S2), similar to that observed by native mass spectrometry with wild-type KaiB (50). To understand the structural basis for this hexameric assembly, we solved a 3.87-Å resolution crystal structure of the KaiBfs-cryst*-KaiCS431E complex (Fig. 3A). All previous crystal structures of full-length KaiC, including those with the S431D phosphomimetic mutation, depicted the prehydrolysis state of the CI and CII domains because the proteins were crystallized with ATP-γ-S (52). Our structure of the KaiBfs-cryst*-KaiCS431E hexamer was crystallized in the presence of ATP but had density for ADP between each CI subunit within the hexamer (Fig. 3B), as well as the concomitant conformational changes at CI helices α6 and α7 that accompany ATP hydrolysis (22) (fig. S3A). Thus, the fold-switched state of KaiB is captured by the post-ATP hydrolysis state of the CI ring within the KaiC hexamer.
Fig. 3. KaiB assembles as a ring on the posthydrolysis state of KaiC.
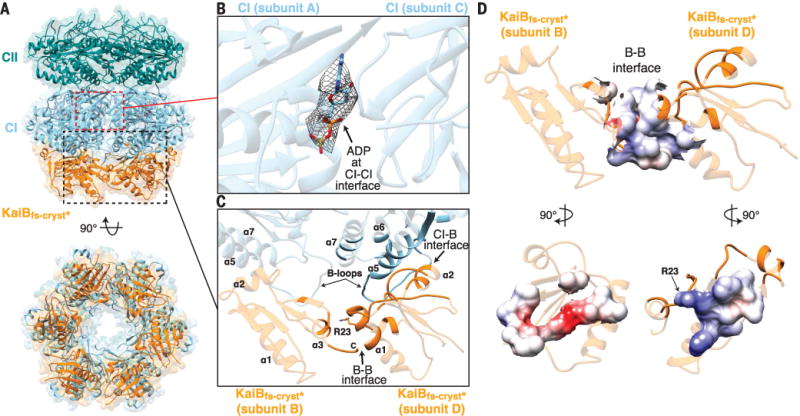
(A) The hexameric KaiBfs-cryst*-KaiCS431E complex at 3.87 Å. KaiCS431E includes CII hexamer (dark cyan) and CI hexamer (sky blue). Orange, KaiBfs-cryst*. (B) Zoomed-in view of the boxed region in (A) showing bound ADP highlighted by mesh representing the (Fobs − Fcalc) omit maps contoured at 2.5σ. Fobs is the observed structure-factor amplitude, and Fcalc is the calculated structure-factor amplitude. (C) Zoomed-in view of the boxed region in (A) showing KaiBfs-cryst* interfaces in the KaiBfs-cryst*-KaiCS431E hexamer. Interfacial residues are shown in nontransparent mode. (D) Columbic electrostatic surface map of the interfacial residues between two subunits of KaiBfs-cryst*. See fig. S1 and table S1 for construct details.
How does ATP hydrolysis by the CI domain functionally integrate with the phosphorylation cycle of the KaiC CII domain? When CII residue S431 is not phosphorylated, the CII ring is loose (27), the CII and CI rings are unstacked (29), CI ATPase activity is elevated (48), and KaiC is unable to bind KaiB (29). Upon S431 phosphorylation, the CII ring tightens (27), CII and CI rings stack (29), CI ATPase activity is suppressed (48), and KaiC becomes receptive to binding KaiB (29). Our crystal structures of the KaiBfs-KaiC complexes (Figs. 2 and 3) provide a plausible explanation for these observations: Based on the inherently low rate of ATP hydrolysis by the CI domain (~0.5 ATP h−1CI−1) (22), CI domains make infrequent and transient excursions from a nonreceptive prehydrolysis state to a KaiB-receptive posthydrolysis state, which is captured and stabilized by the CII ring tightened from S431 autophosphorylation (29). Therefore, the slow rate of ATP hydrolysis by the CI domain creates a time delay between CII ring tightening and CII-CI ring stacking, as well as encoding a conformational switch needed to bind KaiB. Indeed, mutations that decrease CI ATPase activity extend clock period to over 40 hours (22). However, because CI ATPase activity does not affect CII autophosphorylation and autodephosphorylation rates (44), increasing the hypothetical rate of ATP hydrolysis at the CI domain does not further shorten the period below ~22 hours (fig. S4). Hydrolyzing one ATP every ~2 hours, the CI ATPase tunes the period of the oscillator from a lower limit of ~22 hours to ~24 hours (~22 + ~2 hours). Therefore, ATP hydrolysis by the KaiC CI domain works in concert with the phosphorylation cycle at the KaiC CII domain to control KaiB binding and assembly of nighttime signaling complexes.
KaiB forms a hexameric ring on the CI domains of KaiC
The KaiBfs-cryst*-KaiCS431E CI interface observed in the full-length hexamer structure agrees well with that of the isolated KaiBfs-cryst-CIcryst heterodimer (fig. S3B). The hexameric complex is also consistent with a three-tiered envelope derived from a 16-Å-resolution cryogenic electron microscopy study (51) (fig. S3C). Monomers of KaiBfs-cryst* assemble in a ringlike structure on the bottom face of the KaiCS431E CI domain (Fig. 3), forming KaiB-KaiB interactions that are likely to promote the cooperative assembly observed in vitro (50). The fold-switched C-terminal α3 helix of one KaiBfs-cryst* protomer interacts with the N-terminal α1 helix of its clockwise neighbor, increasing the interfacial area by 206 Å2 over the average KaiBfs-cryst*-KaiCS431E CI interface area of 983 Å2, adding ~20% more binding surface (Fig. 3C). Our structure of the hexameric assembly shows that residue R23 is at the center of the interface between two adjacent KaiBfs-cryst* subunits, contributing to the electrostatic complementarity of the KaiB-KaiB interface (Fig. 3, C and D). Consistent with a role in cooperative assembly of the KaiB-KaiC hexamer (50, 53), an R23A mutation in KaiBAs (superscript “As” denotes proteins from Anabaena sp.) reduced its binding affinity for KaiCAs (54). Cooperative assembly of the KaiB-KaiC hexamer may facilitate a robust transition from subjective day to night in the clock by helping to efficiently capture the rare fold-switched form of KaiB.
Structural basis of cooperative assembly of the KaiA-KaiB-KaiC CI complex
The CII domain of KaiC autophosphorylates under stimulation by KaiA during the day (35, 36), whereas it autodephosphorylates at night when KaiA is inhibited by KaiB (41), presumably sequestered in a KaiA-KaiB-KaiC complex (39, 55) (Fig. 1). Both full-length KaiA or KaiAΔN (fig. S1), a construct missing the N-terminal domain yet retaining the ability to stimulate KaiC autophosphorylation and be inhibited by KaiB (28), are capable of cooperatively forming a KaiA-KaiB-KaiC ternary complex (28) (fig. S5, A to C) and promoting disassembly of the SasA-KaiC output signaling complex (28).
To begin to examine molecular determinants of the KaiAΔN-KaiBfs-KaiC CI ternary complex in solution, we collected the methyl-TROSY (transverse relaxation-optimized spectroscopy) nuclear magnetic resonance (NMR) spectrum of U-[15N, 2H]-Ile-δ-[13C, 1H]–labeled KaiAΔN in complex with KaiBfs-cryst and CImono. We found that it was virtually identical to the spectrum bound to wild-type KaiB and CImono (fig. S5B), demonstrating that the conformationally locked KaiB variant and natively captured KaiB both interact with KaiAΔN in a similar manner. We also used one additional mutation (C272S) in KaiAΔN, hereafter referred to as KaiAcryst (fig. S1), to prevent intramolecular disulfide formation within the KaiA dimer during the crystallization process (56). We then solved a 2.6-Å resolution crystal structure of the ternary complex KaiAcryst-KaiBfs-cryst-CIcryst (Fig. 4A), consistent with the stoichiometry of 2 KaiA:1 KaiB:1 CI that we previously observed in solution (28).
Fig. 4. The mechanism of KaiA autoinhibition is revealed by the KaiAcryst-KaiBfs-cryst-CIcryst complex.
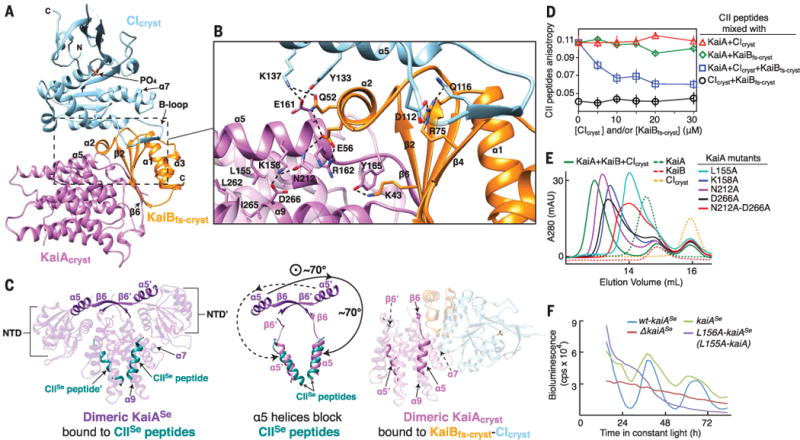
(A) The ternary KaiAcryst-KaiBfs-cryst-CIcryst complex at 2.6 Å. Orange, KaiBfs-cryst; sky blue, CIcryst; orchid, KaiAcryst. (B) Zoomed-in view of the boxed region in (A). Dashed lines: electrostatic interactions. (C) Conformational changes of dimeric KaiA. NTD, N-terminal domain. Prime symbols denote the other protomer within the dimer. (Left) Crystal structure (PDB 5C5E) of KaiASe (purple) bound to KaiC CIISe peptides (dark cyan). (Right) The KaiAcryst-KaiBfs-cryst-CIcryst complex, with same coloring scheme as in (A). (Middle) Superposition of KaiASe (left) and KaiAcryst in complex (right); only α5 helices and b6 strands are shown. (D) Fluorescence anisotropy of 6IAF-labeled CII peptides (0.05 μM). Open circles, 0 μM KaiA, titration with CIcryst and KaiBfs-cryst; triangles, 10 μM KaiA, titration with CIcryst; diamonds, 10 μM KaiA, titration with KaiBfs-cryst; squares, 10 μM KaiA, titration with equal molar of KaiBfs-cryst and CIcryst. Error bars, SD from triplicates. (E) Size-exclusion chromatography of ternary complex formation. Wild-type KaiA (green, dashed); wild-type KaiB (red, dashed); CIcryst (orange, dashed); KaiA +KaiB+CIcryst (green); L155A-KaiA+KaiB+CIcryst (cyan); K158A-KaiA+KaiB+CIcryst (blue); D212A-KaiA+ KaiB+CIcryst (purple); D266A-KaiA+KaiB+CIcryst (black); and N212A-D266A-KaiA+ KaiB+CIcryst (red). (F) Bioluminescence rhythms from strains of S. elongatus: wt-kaiASe (blue), complemented with kaiASe (green), L156A-kaiASe (purple), and kaiASe knockout (red). L156A of kaiASe is analogous to L155A in kaiA. See fig. S1 and table S1 for construct details.
The structure of this isolated ternary complex demonstrates the molecular basis for cooperative assembly of KaiA-KaiBfs-KaiC complexes. First, conserved residue K137 of CIcryst forms a hydrogen bond with Q52 of KaiBfs-cryst and a salt bridge to E161 of KaiAcryst (Fig. 4B). Second, a triple junction of interactions was also observed between K137 and Y133 of CIcryst; E161, R162, and N212 of KaiAcryst; and Q52 and E56 of KaiBfs-cryst. Finally, the presence of KaiAcryst increased the number of KaiBfs-cryst-CIcryst hydrogen bonds from 13 to 19 (Figs. 2 and 4), and the KaiBfs-cryst-CIcryst interfacial surface area from 1000 to 1075 Å2 (Figs. 2 and 4), supporting previous observations that KaiA induces an increase in apparent affinity of KaiB for the CI domain (28). This cooperative assembly also promotes SasA displacement from KaiC (28), likely eliciting a switchlike behavior as the clock transitions from daytime to nighttime mode.
KaiA adopts an autoinhibited conformation in the KaiA-KaiB-KaiC complex
During the day, the KaiA homodimer uses two symmetrically related sites located on its α9 helices to bind C-terminal peptides emanating from the CII domain (28, 37, 38, 57) (Fig. 4C). At night, KaiA is somehow prevented from doing so upon sequestration into a KaiA-KaiBfs complex on the KaiC CI domain (28, 39) (Fig. 1). Because asymmetric sequestration of one KaiC binding site on KaiA could in principle still allow KaiA to bind and stimulate a second KaiC particle, both KaiC binding sites on KaiA should somehow be inactivated to allow KaiC to fully autodephosphorylate. However, it was unclear how this potent inactivation of KaiA could be achieved by binding to only one KaiBfs monomer docked onto the KaiC CI domain (28).
Our structure of the ternary complex reveals that the KaiAcryst dimer undergoes large-scale conformational changes when it binds to the KaiBfs-cryst-CIcryst complex, prohibiting itself from interacting with the CII domain (Fig. 4C). The b6 strands from KaiAcryst monomers rotate over 70° relative to their previous positions in KaiASe (fig. S6); a β6 strand from one of the KaiAcryst monomers then docks onto the β2 strand of KaiBfs-cryst to form an antiparallel β sheet (Fig. 4). This asymmetric binding interface with KaiBfs-cryst is also accompanied by a downward rotation of both a5 helices of KaiAcryst to pack onto the KaiC binding sites on a7 and a9 of the KaiAcryst dimer interface, thereby mimicking CII peptide binding (57) (Fig. 4C). Thus, both KaiC binding sites on KaiAcryst are effectively blocked by their own α5 helices in this autoinhibited conformation through a long-range allosteric mechanism. This structure, along with previous solution NMR studies (33, 35, 37, 38), supports a model in which free KaiA, like KaiB, is in a dynamic equilibrium between active and inactive conformations; in the case of KaiA, these dynamics tune the stimulatory effect of KaiA on KaiC autophosphorylation (35, 37). Phosphoryl S431-dependent hexameric CII ring tightening allows the CII ring to withdraw its A loops from KaiA (28), and the inactive conformation then becomes selectively bound by the KaiBfs-KaiC complex at night.
In support of KaiBfs-induced autoinhibition of KaiA, hydrogen-deuterium exchange mass spectroscopy (HDX-MS) revealed strong protection of the α5 helix of KaiAΔN and the β2 strand of KaiBfs-cryst upon formation of the KaiAΔN-KaiBfs-cryst-CIcryst complex in solution, matching the interfaces observed in our crystal structure (Fig. 4 and fig. S7A). To rule out that the autoinhibitory conformation is not influenced by our truncation of the N-terminal domain in KaiA, we used a fluorescence polarization–based displacement assay to probe for direct competition with the CII peptide that binds KaiA. In this assay, 6-iodoacetamidofluorescein (6IAF)–labeled CII peptides remained bound to full-length KaiA in the presence of either KaiBfs-cryst or the KaiC CIcryst domain but was efficiently competed away with the KaiBfs-cryst-CIcryst complex (Fig. 4D). Therefore, binding to the KaiBfs-KaiC CI complex is sufficient to stabilize the autoinhibitory conformation in native KaiA and block interaction with the KaiC CII domain.
We conducted structure-guided mutagenesis to test our model that the α7 and α9 helices of KaiAcryst appear to be crucial for stabilizing the α5 helices in the autoinhibited conformation that binds to the KaiBfs-KaiC CI complex (Fig. 4C). We identified four substitutions on the α5 helix of KaiA (L155A, K158A, L159A, and L163A) that weakened ternary complex formation as determined by size-exclusion chromatography (Fig. 4E and fig. S7B). Likewise, mutagenesis of α5-facing residues N212 and D266 on α7 and α9 of KaiA also weakened formation of the ternary complex (Fig. 4E), further demonstrating the importance of the KaiA conformational change in binding to the KaiBfs-KaiC CI complex.
Mutagenesis of A41D and K43E on the β2 strand of KaiBfs eliminated KaiAΔN binding to the KaiBfs-CImono complex by disrupting formation of the antiparallel β sheet (fig. S7C). The structural integrity of KaiBfs mutants remained intact because they were still able to form complexes with CImono (fig. S7C). Likewise, an 15N, 1H-HSQC (heteronuclear single-quantum coherence) spectrum of L155A-KaiAΔN showed similarly dispersed peaks as compared with the spectrum of KaiAΔN, indicating that the L155A mutation did not appreciably affect the structure of KaiAΔN (fig. S8A). We found that analogous mutations of the S. elongatus proteins, L156A in kaiASe, and A40D and K42E in kaiBSe, individually disrupted cellular circadian rhythms in vivo (Fig. 4F and fig. S8D). Moreover, the A40D and K42E substitutions in kaiBSe also produced abnormally long cells, consistent with the kaiB knockout phenotype (58) (fig. S8, E and F). Finally, superimposing the KaiAcryst-KaiBfs-cryst-CIcryst ternary complex onto our hexameric KaiBfs-cryst*-KaiCS431E assembly shows that KaiAcryst likely protrudes out from the side of the KaiCS431E hexamer (fig. S9), consistent with negative-stain electron microscopy images of a presumed complex of native KaiA-KaiB-KaiC (59).
CikA-KaiB interactions critical for CikA activation
At night, the phosphatase activity of CikA is stimulated upon binding the KaiBfs-KaiC complex to dephosphorylate and thereby repress RpaA-dependent regulation of class 1 genes and activate transcription of class 2 genes (21, 39) (Fig. 1). Although the molecular basis of this interaction has been unclear, the pseudoreceiver domain of CikA (CikAPsR) (fig. S1) is known to directly interact with the KaiBfs-KaiC complex (39, 60). Overexpression of this domain alone shortened cellular circadian rhythms by as much as 4 hours (60), and addition of the isolated CikAPsR domain to an in vitro cycling reaction with purified Kai proteins shortened the circadian period in a dose-dependent manner (39). These data suggest that the CikAPsR domain can interfere somehow with formation of Kai protein interactions that contribute to normal circadian timekeeping.
To probe how the PsR domain of CikA can influence circadian rhythms, we first investigated how CikAPsR interacts with KaiBfs-CImono complexes by NMR (fig. S5, D to I). Methyl-TROSY spectra of U-[15N, 2H]-Ile-δ-[13C, 1H]–labeled CikAPsR showed that it interacts with a complex of CImono bound to the fold-switched conformation induced from native KaiB but not to either protein separately (fig. S5, D and E). In support of this finding, NMR spectra of CikAPsR bound to complexes with the conformationally locked KaiB mutant, KaiBfs-CImono, and those originating from wild-type KaiB-CImono were similar (fig. S5E). We noted that CikAPsR adopts two conformations in the ternary complex with wild-type KaiB and CImono, but only one of these is shared with KaiBfs and CImono (fig. S5E). In the absence of CImono, KaiBfs interacted more weakly with CikAPsR (fig. S5F), suggesting that cooperativity is also important in formation of a CikAPsR-KaiB-KaiC CI ternary complex, as was observed with KaiA-KaiB-KaiC binding (28) (fig. S5, A to C).
During our investigation, we identified an N29A substitution on KaiBfs, hereafter referred to as KaiBfs-nmr (fig. S1), that promoted virtually complete binding to CikAPsR even without CImono (fig. S5G). We solved a solution structure of a CikAPsR-KaiBfs-nmr complex (Fig. 5) as a model to understand how CikA might bind to the KaiBfs-KaiC complex. This complex has a binding interface of 1230 Å2 that includes β2 of CikAPsR and β2 of KaiBfs-nmr, which interact in parallel to build a nine-stranded β sheet that spans both proteins (Fig. 5A). Residue A29 on KaiBfs-nmr was found to interact with hydrophobic residues I641 and L654 on CikAPsR (Fig. 5A), suggesting that the observed enhancement in binding arising from the N29A substitution was likely due to an increased hydrophobic effect. Residue I641 of CikAPsR sits at the center of the β2-β2 heterodimeric interface (Fig. 5A), consistent with our observations that it experienced the largest chemical shift perturbation (fig. S5I) and was protected against hydrogen-deuterium exchange upon binding KaiBfs-nmr in solution (fig. S10A).
Fig. 5. Structure of the CikAPsR-KaiBfs-nmr complex reveals intermolecular contacts essential for output signaling.
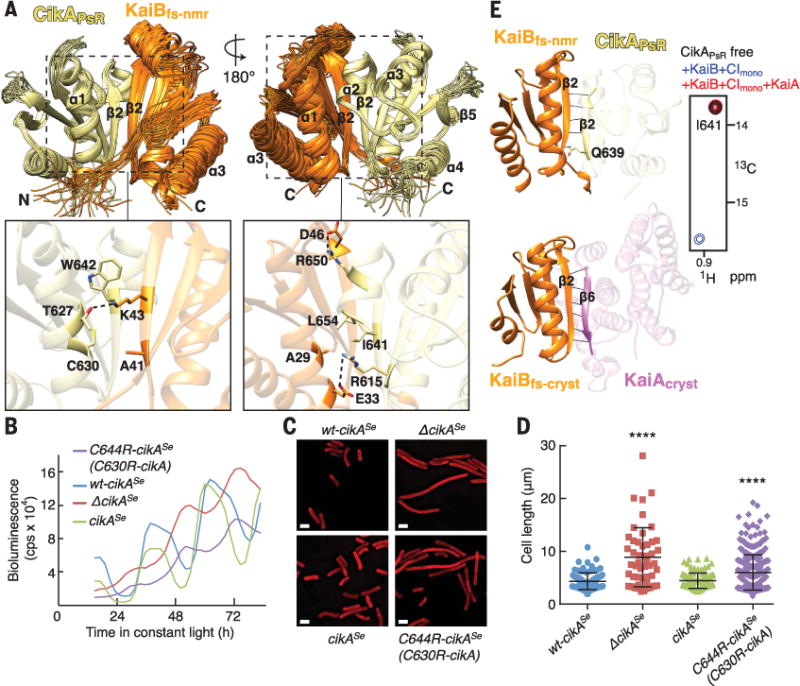
(A) Top, ensemble of the 20 lowest-energy NMR structures of the CikAPsR-KaiBfs-nmr complex. Orange, KaiBfs-nmr; khaki, CikAPsR. Bottom, zoomed-in view of boxed regions above using average minimized structure from the ensemble. Dashed lines, intermolecular electrostatic interactions. (B) Bioluminescence rhythms from S. elongatus. wt-cikASe (blue), complemented with cikASe (green), C644R-cikASe (purple), or cikASe knockout (red). C644R of cikASe is analogous to C630R of cikA. (C) Representative cell micrographs. wt-cikASe, complemented with cikASe, C644R-cikASe, or lacking cikASe. Red, membrane autofluorescence. Scale bars, 2.5 μm. (D) Scatter plots of cell lengths. wt-cikASe (blue) (n = 87), complemented with cikASe (green) (n = 110), C644R-cikASe (purple) (n = 353), or lacking cikASe (red) (n = 48). One-way analysis of variance of log10-transformed cell length data produced P < 0.0001. ****, Bonferroni-corrected P values (< 0.0001) for pairwise comparisons to wt-cikASe (α = 0.05). (E) CikAPsR and KaiAcryst compete for the β2 strand of KaiBfs. (Left) Orange, KaiBfs-nmr (top) and KaiBfs-cryst (bottom); khaki, CikAPsR; orchid, KaiAcryst; black lines, backbone-backbone or sidechain-backbone hydrogen bonds. (Right) Selected region of methyl-TROSY spectra of labeled CikAPsR free (black), or mixed with wild-type KaiB+CImono (blue), or with wild-type KaiB+CImono+wild-type KaiA (red). See fig. S5, E and H, for full spectra. Also see fig. S1 and table S1 for construct details.
To examine the relevance of the CikAPsR-KaiBfs-nmr interface in vivo, we mutated CikAPsR residue C630, which interacts with A41 in KaiBfs-nmr at the center of the interface (Fig. 5A). The C630R substitution eliminated complex formation (fig. S10B) while maintaining structural integrity of CikAPsR (fig. S8, B and C). Cyanobacterial strains that harbored the analogous C644R mutation in cikASe exhibited defects in circadian rhythms and cell division (58) similar to those observed in cikASe knockout strains (Fig. 5, B to D). Thus, in vitro and in vivo experiments demonstrate that the structure of the CikAPsR-KaiBfs-nmr complex accurately reflects interactions between wild-type proteins.
A side-by-side comparison of the parallel β2-β2 CikAPsR-KaiBfs-nmr and antiparallel β6-β2 KaiAcryst-KaiBfs-cryst interfaces revealed that KaiBfs uses the same β2 strand to bind both KaiAcryst and CikAPsR (Fig. 5E). A41D and K43E substitutions in β2 of KaiBfs-cryst, which weakened KaiAΔN-KaiBfs-cryst interactions (fig. S7C), similarly reduced binding of CikAPsR to KaiBfs-nmr (fig. S10C). Consistent with their overlapping binding sites, methyl-TROSY spectra showed that both full-length KaiA and KaiAΔN could displace CikAPsR from a KaiB-CImono complex (Fig. 5E and fig. S5H). Conversely, elevated levels of CikAPsR would be expected to similarly displace KaiA from KaiBfs-KaiC complexes, likely explaining how increased levels of the CikAPsR domain shorten the period of circadian rhythms in vivo (39) and in vitro (60). Notably, although KaiAcryst and CikAPsR bind predominantly to the N-terminal half of KaiBfs, which is conformationally similar in both the ground state (KaiBgs) and fold-switched conformations (KaiBfs) (fig. S11), our studies collectively show that both preferentially bind the fold-switched conformation of KaiB that is stabilized upon binding to the KaiC CI domain. A closer examination of both complexes reveals that the β4 strand and β2-to- β3 loop of the ground-state conformation (KaiBgs) would create putative charge repulsions and steric clashes with KaiAcryst and CikAPsR (Fig. 6). Therefore, by binding to the same β2 strand on KaiBfs, both KaiA and CikAPsR are regulated by the same fold-switch mechanism of KaiB.
Fig. 6. Ground-state KaiB prevents daytime KaiA and CikAPsR recruitment.
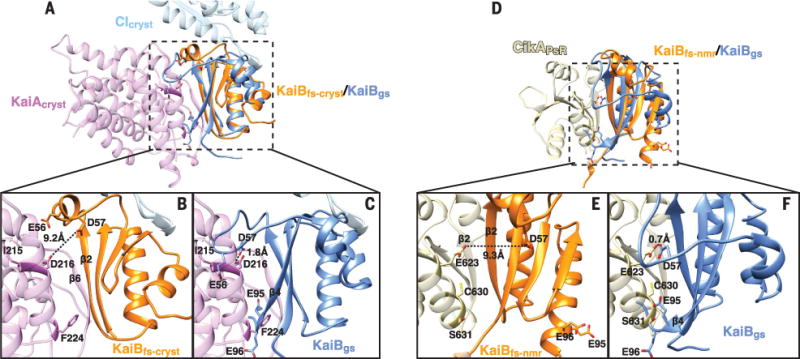
(A) Superposition of the non–fold-switched region (residues 7 to 46) of KaiBgs (blue) (PDB 2QKE, subunit A) with the ternary KaiAcryst-KaiBfs-cryst-CIcryst structure. (B) and (C) are zoomed-in views of the boxed region in (A). Dashed lines, estimated distance between two atoms. (D) Superposition of the non–fold-switched region (residues 7 to 46) of KaiBgs (blue) (PDB 2QKE, subunit A) with the CikAPsR-KaiBfs-nmr structure. (E) and (F) are zoomed-in views of the boxed region in (D). Dashed lines, estimated distance between two atoms.
Discussion
Here, we presented four structures assembled by the KaiBfs-KaiC hub, all of which are obligately dependent on the rare, metamorphic fold-switching of KaiB and ATP hydrolysis by the KaiC CI domain. Together, they reveal how this posttranslational circadian clock transitions from day to night and provide the basis for a revised mechanistic model of cyanobacterial circadian rhythms (Fig. 1). Additionally, structural analysis revealed that KaiA exists in dynamic equilibrium between active and inactive states, the latter of which is stabilized by interaction with the KaiBfs-KaiC hub. Because KaiA and CikA compete for overlapping binding sites, as do KaiB and SasA, regulation of the oscillator and output signal transduction are tightly coupled. Although no metamorphic proteins have been reported in the transcription-based vertebrate circadian clock, several clock protein interactions depend similarly on competition for mutually exclusive binding events (61), suggesting that some parallels in timekeeping strategies may exist in the network architectures of diverse circadian clocks.
Supplementary Material
Acknowledgments
We thank staff at the 8.3.1 beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory, and the 23-ID-B beamline at the Advanced Photon Source, Argonne National Laboratory for help with data collection. Access to the 8.3.1 beamline was provided by the University of California Office of the President Multicampus Research Programs and Initiatives grant MR-15-328599 and the Program for Breakthrough Biomedical Research, which is partly funded by the Sandler Foundation. The Advanced Light Source (contract DE-AC02-05CH11231) and Advanced Photon Source (contract DE-AC02-06CH11357) are supported by the U.S. Department of Energy. This work was supported by grants from the Air Force Office of Scientific Research (FA9550-13-1-0154 to A.L.), U.S. National Institutes of Health (R01GM107521 to A.L., R01GM107069 to C.L.P., F31CA189660 to A.K.M., and R35GM118290 to S.S.G.). R.T. was supported by the National Science Foundation Graduate Research Fellowship, and S.E.C. was supported by the American Cancer Society Postdoctoral Fellowship PF-12-262-01-MPC. S.L. and the Biomolecular/Proteomics Mass Spectrometry Facility are supported by the U.S. National Institutes of Health (1U19AI117905, R01GM020501, and R01AI101436). We thank M. Lutz for facility support; S. Grimaldi for maintaining the NMR cryoplatform; D. Rice for maintaining the NMR Facility; D. Garrett at the U.S. National Institutes of Health for providing NMR software XIPP; C. Schwieters for Xplor-NIH support; and National Magnetic Resonance Facility at Madison (NMRFAM) Advanced Workshop instructors M. Westler, G. Cornilescu, V. Chen, and W. Lee at the NMRFAM. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). We thank L. Zhang for RDC NMR guidance and R. Peterson, F. Delaglio, M. Paddock, and J. Boyd for helpful discussions. C.L.P., A.L., S.S.G., S.L., R.T., A.C., and S.E.C. designed the experiments and contributed reagents. R.T., N.F.G., A.C., J.L., S.E.C., Y.-G.C., J.H., S.L., and S.T. performed the experiments. Data were analyzed by R.T., N.F.G, A.C., J.L., S.E.C., Y.-G.C., J.H., S.L., S.T., C.L.P., A.L., and S.S.G. A.K.M. contributed to some experiments and discussions. R.T., A.L., and C.L.P wrote the paper. All authors approve of the conclusions and final version of the paper. The atomic coordinates and structure factors for KaiBfs-cryst-CIcryst, KaiBfs-cryst*-KaiCS431E, KaiAcryst-KaiBfs-cryst-CIcryst, and CikAPsR-KaiBfs-nmr complexes have been deposited in the Protein Data Bank under accession codes 5JWO, 5JWQ, 5JWR, and 5JYV, respectively. Free CikAPsR and KaiBfs-nmr structures are deposited under 5JYU and 5JYT, respectively. Structural statistics are provided in the supplementary materials (tables S2 and S3).
Footnotes
The authors declare no competing financial interests.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/355/6330/1174/suppl/DC1
Materials and Methods
References (62–82)
REFERENCES AND NOTES
- 1.Whitehead K, Pan M, Masumura K, Bonneau R, Baliga NS. PLOS ONE. 2009;4:e5485. doi: 10.1371/journal.pone.0005485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edgar RS, et al. Nature. 2012;485:459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondo T, et al. Proc Natl Acad Sci USA. 1993;90:5672–5676. doi: 10.1073/pnas.90.12.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pittendrigh CS. Proc Natl Acad Sci USA. 1954;40:1018–1029. doi: 10.1073/pnas.40.10.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konopka RJ, Benzer S. Proc Natl Acad Sci USA. 1971;68:2112–2116. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell-Pedersen D, et al. Nat Rev Genet. 2005;6:544–556. doi: 10.1038/nrg1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunlap JC, Loros JJ, DeCoursey PJ. Chronobiology: Biological Timekeeping. Sinauer Associates, Inc; Sunderland, MA: 2004. [Google Scholar]
- 8.Papazyan R, Zhang Y, Lazar MA. Nat Struct Mol Biol. 2016;23:1045–1052. doi: 10.1038/nsmb.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panda S. Science. 2016;354:1008–1015. doi: 10.1126/science.aah4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musiek ES, Holtzman DM. Science. 2016;354:1004–1008. doi: 10.1126/science.aah4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bass J, Lazar MA. Science. 2016;354:994–999. doi: 10.1126/science.aah4965. [DOI] [PubMed] [Google Scholar]
- 12.Vijayan V, Zuzow R, O’Shea EK. Proc Natl Acad Sci USA. 2009;106:22564–22568. doi: 10.1073/pnas.0912673106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woelfle MA, Ouyang Y, Phanvijhitsiri K, Johnson CH. Curr Biol. 2004;14:1481–1486. doi: 10.1016/j.cub.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 14.Lambert G, Chew J, Rust MJ. Biophys J. 2016;111:883–891. doi: 10.1016/j.bpj.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diamond S, Jun D, Rubin BE, Golden SS. Proc Natl Acad Sci USA. 2015;112:E1916–E1925. doi: 10.1073/pnas.1504576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishiura M, et al. Science. 1998;281:1519–1523. doi: 10.1126/science.281.5382.1519. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima M, et al. Science. 2005;308:414–415. doi: 10.1126/science.1108451. [DOI] [PubMed] [Google Scholar]
- 18.Iwasaki H, et al. Cell. 2000;101:223–233. doi: 10.1016/S0092-8674(00)80832-6. [DOI] [PubMed] [Google Scholar]
- 19.Schmitz O, Katayama M, Williams SB, Kondo T, Golden SS. Science. 2000;289:765–768. doi: 10.1126/science.289.5480.765. [DOI] [PubMed] [Google Scholar]
- 20.Cohen SE, Golden SS. Microbiol Mol Biol Rev. 2015;79:373–385. doi: 10.1128/MMBR.00036-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutu A, O’Shea EK. Mol Cell. 2013;50:288–294. doi: 10.1016/j.molcel.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe J, et al. Science. 2015;349:312–316. doi: 10.1126/science.1261040. [DOI] [PubMed] [Google Scholar]
- 23.Wang J. J Struct Biol. 2004;148:259–267. doi: 10.1016/j.jsb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Nishiwaki T, Iwasaki H, Ishiura M, Kondo T. Proc Natl Acad Sci USA. 2000;97:495–499. doi: 10.1073/pnas.97.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egli M, et al. Biochemistry. 2012;51:1547–1558. doi: 10.1021/bi201525n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishiwaki T, Kondo T. J Biol Chem. 2012;287:18030–18035. doi: 10.1074/jbc.M112.350660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang YG, Kuo NW, Tseng R, LiWang A. Proc Natl Acad Sci USA. 2011;108:14431–14436. doi: 10.1073/pnas.1104221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng R, et al. J Mol Biol. 2014;426:389–402. doi: 10.1016/j.jmb.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang YG, Tseng R, Kuo NW, LiWang A. Proc Natl Acad Sci USA. 2012;109:16847–16851. doi: 10.1073/pnas.1211508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin J, Chew J, Chockanathan U, Rust MJ. Proc Natl Acad Sci USA. 2014;111:E3937–E3945. doi: 10.1073/pnas.1408692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishiwaki T, et al. EMBO J. 2007;26:4029–4037. doi: 10.1038/sj.emboj.7601832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rust MJ, Markson JS, Lane WS, Fisher DS, O’Shea EK. Science. 2007;318:809–812. doi: 10.1126/science.1148596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vakonakis I, et al. Proc Natl Acad Sci USA. 2004;101:1479–1484. doi: 10.1073/pnas.0305516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye S, Vakonakis I, Ioerger TR, LiWang AC, Sacchettini JC. J Biol Chem. 2004;279:20511–20518. doi: 10.1074/jbc.M400077200. [DOI] [PubMed] [Google Scholar]
- 35.Williams SB, Vakonakis I, Golden SS, LiWang AC. Proc Natl Acad Sci USA. 2002;99:15357–15362. doi: 10.1073/pnas.232517099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwasaki H, Nishiwaki T, Kitayama Y, Nakajima M, Kondo T. Proc Natl Acad Sci USA. 2002;99:15788–15793. doi: 10.1073/pnas.222467299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim YI, Dong G, Carruthers CW, Jr, Golden SS, LiWang A. Proc Natl Acad Sci USA. 2008;105:12825–12830. doi: 10.1073/pnas.0800526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vakonakis I, LiWang AC. Proc Natl Acad Sci USA. 2004;101:10925–10930. doi: 10.1073/pnas.0403037101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang YG, et al. Science. 2015;349:324–328. doi: 10.1126/science.1260031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takai N, et al. Proc Natl Acad Sci USA. 2006;103:12109–12114. doi: 10.1073/pnas.0602955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitayama Y, Iwasaki H, Nishiwaki T, Kondo T. EMBO J. 2003;22:2127–2134. doi: 10.1093/emboj/cdg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, Mori T, Johnson CH. EMBO J. 2003;22:2117–2126. doi: 10.1093/emboj/cdg168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murzin AG. Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- 44.Phong C, Markson JS, Wilhoite CM, Rust MJ. Proc Natl Acad Sci USA. 2013;110:1124–1129. doi: 10.1073/pnas.1212113110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami R, Mutoh R, Ishii K, Ishiura M. Genes Cells. 2016;21:890–900. doi: 10.1111/gtc.12392. [DOI] [PubMed] [Google Scholar]
- 46.Murakami R, et al. J Biol Chem. 2012;287:29506–29515. doi: 10.1074/jbc.M112.349092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pattanayek R, et al. PLOS ONE. 2011;6:e23697. doi: 10.1371/journal.pone.0023697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mutoh R, Nishimura A, Yasui S, Onai K, Ishiura M. PLOS ONE. 2013;8:e80200. doi: 10.1371/journal.pone.0080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paddock ML, Boyd JS, Adin DM, Golden SS. Proc Natl Acad Sci USA. 2013;110:E3849–E3857. doi: 10.1073/pnas.1315170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snijder J, et al. Proc Natl Acad Sci USA. 2014;111:1379–1384. doi: 10.1073/pnas.1314326111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villarreal SA, et al. J Mol Biol. 2013;425:3311–3324. doi: 10.1016/j.jmb.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pattanayek R, et al. PLOS ONE. 2009;4:e7529. doi: 10.1371/journal.pone.0007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugiyama M, et al. Sci Rep. 2016;6:35567. doi: 10.1038/srep35567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garces RG, Wu N, Gillon W, Pai EF. EMBO J. 2004;23:1688–1698. doi: 10.1038/sj.emboj.7600190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kageyama H, et al. Mol Cell. 2006;23:161–171. doi: 10.1016/j.molcel.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 56.Vakonakis I, et al. J Biomol NMR. 2001;21:179–180. doi: 10.1023/a:1012478912174. [DOI] [PubMed] [Google Scholar]
- 57.Pattanayek R, Egli M. Biochemistry. 2015;54:4575–4578. doi: 10.1021/acs.biochem.5b00694. [DOI] [PubMed] [Google Scholar]
- 58.Dong G, et al. Cell. 2010;140:529–539. doi: 10.1016/j.cell.2009.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mori T, et al. PLOS Biol. 2007;5:e93. doi: 10.1371/journal.pbio.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, Dong G, Golden SS. Mol Microbiol. 2006;60:658–668. doi: 10.1111/j.1365-2958.2006.05138.x. [DOI] [PubMed] [Google Scholar]
- 61.Xu H, et al. Nat Struct Mol Biol. 2015;22:476–484. doi: 10.1038/nsmb.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.