

Abstract
The drug cisplatin is widely used to treat a number of tumor types. However, resistance to the drug, which remains poorly understood, limits its usefulness. Previous work using Dictyostelium discoideum as a model for studying drug resistance showed that mutants lacking sphingosine-1-phosphate (S-1-P) lyase, the enzyme that degrades S-1-P, had increased resistance to cisplatin, whereas mutants overexpressing the enzyme were more sensitive to the drug. S-1-P is synthesized from sphingosine and ATP by the enzyme sphingosine kinase. We have identified two sphingosine kinase genes in D. discoideum—sgkA and sgkB—that are homologous to those of other species. The biochemical properties of the SgkA and SgkB enzymes suggest that they are the equivalent of the human Sphk1 and Sphk2 enzymes, respectively. Disruption of the kinases by homologous recombination (both single and double mutants) or overexpression of the sgkA gene resulted in altered growth rates and altered response to cisplatin. The null mutants showed increased sensitivity to cisplatin, whereas mutants overexpressing the sphingosine kinase resulted in increased resistance compared to the parental cells. The results indicate that both the SgkA and the SgkB enzymes function in regulating cisplatin sensitivity. The increase in sensitivity of the sphingosine kinase-null mutants was reversed by the addition of S-1-P, and the increased resistance of the sphingosine kinase overexpressor mutant was reversed by the inhibitor N,N-dimethylsphingosine. Parallel changes in sensitivity of the null mutants are seen with the platinum-based drug carboplatin but not with doxorubicin, 5-fluorouracil, and etoposide. This pattern of specificity is similar to that observed with the S-1-P lyase mutants and should be useful in designing therapeutic schemes involving more than one drug. This study identifies the sphingosine kinases as new drug targets for modulating the sensitivity to platinum-based drugs.
Resistance to the drugs used in chemotherapy is a major obstacle in the treatment of cancer irrespective of their mechanism of cytotoxicity. Cisplatin [cis-diamminedichloroplatinum (II)] is a platinum-based drug that is frequently used in treating a variety of cancers such as small-cell and non-small-cell lung cancer, non-Hodgkin's lymphoma, testicular cancer, ovarian cancer, head and neck cancer, esophageal cancer, and bladder cancer (32). Nevertheless, acquired resistance to the drug often reduces the efficacy of therapy in these cancers, whereas an intrinsic resistance prevents the use of the drug with other cancers. Based on mammalian cell culture studies, several mechanisms of resistance have been proposed, but a good understanding of resistance to this important drug is still lacking (37). The difficulty and cost of bringing new anticancer drugs to market makes it vital to optimize the performance of existing drugs such as cisplatin.
A nonbiased genetic approach using the model organism Dictyostelium discoideum was taken to further our understanding of the molecular and cellular basis of resistance to cisplatin. D. discoideum cells are mitotically dividing single eukaryotic cells with a simple plasma membrane. They are amenable to both random insertional mutagenesis and gene disruption and/or replacement based on homologous recombination (16). Thus, this organism is ideal for identifying specific genes that function in determining drug sensitivity. Using random insertional mutagenesis, five mutants were isolated with increased resistance to cisplatin, and the corresponding genes were identified. These included mutations in genes encoding sphingosine-1-phosphate (S-1-P) lyase, regA cAMP phosphodiesterase, golvesin (a Golgi protein), a P2Y purine receptor homolog, and a CAAX prenyl protease homolog. None of these genes had been previously associated with cisplatin resistance (19).
The S-1-P lyase gene encodes the enzyme that converts S-1-P to phosphoethanolamine and hexadecanal by cleaving the C2-C3 carbon-carbon bond (45) and represents an important step in the regulation of S-1-P levels. There is a great deal of interest in the roles that S-1-P and other sphingolipids play in cell signaling, cancer and chemotherapy and in the cell's response to stress (10, 29, 40, 42). S-1-P has been shown to act as an extracellular signal through specific G-protein-coupled S1P (EDG) receptors as well as an intracellular second messenger. It appears that the balance between S-1-P and ceramide is a crucial determinant, with proportionally greater levels of S-1-P resulting in cell proliferation, while proportionally greater levels of ceramide result in cell death (34, 38). Moreover, numerous reports link S-1-P to various aspects of cell motility, chemotaxis, and metastasis in mammalian and D. discoideum cells (15, 31, 39). S-1-P lyase has been shown to be essential for cell differentiation and morphogenesis in Drosophila melanogaster, Caenorhabditis elegans, and D. discoideum (9, 18, 25).
The increased resistance of D. discoideum cells lacking the S-1-P lyase to cisplatin (19) led to the hypotheses (i) that the S-1-P level regulates the sensitivity to cisplatin and (ii) that, since the S-1-P level can be modulated by its synthetic and degradative enzymes, all of the enzymes in the pathway of sphingomyelin degradation could be potential new drug targets for enhancing the sensitivity of cells to cisplatin.
In a subsequent study (26), we showed that overexpression of the S-1-P lyase had the predicted effect of increasing the sensitivity of the cells to cisplatin (the reverse of the S-1-P lyase-null mutant [sglAΔ]). Exogenously added S-1-P partially blocked the phenotype, whereas N,N-dimethylsphingosine (DMS), an inhibitor of sphingosine kinase (the enzyme that synthesizes S-1-P from sphingosine and ATP) (5), synergistically increased sensitivity to cisplatin. The increase in sensitivity observed in the overexpressing mutants was specific for platinum-based drugs (26). Overall, the present study firmly supported the idea that modulating the levels of sphingolipids in cells can be a powerful way to increase the sensitivity of tumor cells to cisplatin or, alternatively, to increase resistance in cases where the adverse cytotoxic effects of chemotherapy are to be avoided, such as the bone marrow, kidneys, or ovaries (44).
The enzyme sphingosine kinase catalyzes the synthesis of S-1-P from sphingosine and ATP. Mammals, Drosophila, C. elegans, and yeast each have two genes that encode two different sphingosine kinases. The enzymes share five conserved regions (C1 to C5), and mutational analysis has shown that a consensus sequence in region C2, SGDGX17-21K, is required for ATP binding in which the second glycine is critical (14, 33). Sphingosine kinase biochemistry and cell biology are active areas of investigation. Mammalian null mutants are not yet available, but in vitro assays have shown that the two enzymes have different salt and detergent sensitivities and kinetic properties (4, 21). Nuclear export and import signals have been identified in the sphingosine kinase 1 and 2 enzymes, respectively (11, 12), but the precise subcellular localization and tissue distribution remains unclear and may differ between organisms (8, 14, 21, 24, 28). Therefore, the extent to which the two enzymes have distinct cellular functions and regulate the synthesis of S-1-P in different ways remains an open question, and much remains to be learned about how they regulate growth, development, and disease.
In the present study we identified two D. discoideum sphingosine kinase genes, sgkA and sgkB, which are homologous to the enzymes of humans and other species. We have constructed null strains for sgkA, sgkB, and an sgkA/sgkB double-null strain, as well as an sgkA overexpressor. We show that altering the expression of the sphingosine kinase genes affects growth rate and sensitivity to cisplatin, with the double null mutant being more sensitive than either of the single null mutants. In contrast, the SgkA overexpressor mutant showed increased resistance to cisplatin. The changes in drug sensitivity of the null mutants are specific to the platinum compounds. The present study further supports D. discoideum as a powerful system for investigating resistance to drugs. More importantly, the present study identifies both of the sphingosine kinases as novel targets for the modulation of platinum based antineoplastic drugs.
MATERIALS AND METHODS
Strains construction, nomenclature, and culture conditions. (i) Sphingosine kinase A null.
The SalI-XbaI fragment containing the sgkA gene from cDNA clone SLG-787 (27) was cloned into pBluescript (Stratagene, La Jolla, Calif.). The EcoRI site within the sgkA gene was replaced with a BamHI site into which the 1.4-kb blasticidin resistance gene (Bsr) cassette was cloned (1). To disrupt the sgkA gene by homologous recombination, D. discoideum Ax4 cells were electroporated with the above vector, linearized with SalI, and selected for blasticidin resistance. The resulting sgkAΔ strain is SA576.
(ii) Sphingosine kinase B null.
The sphingosine kinase B disruption vector was generated by cloning two fragments of the sgkB gene into plasmid pLPBLP on both sides of the Bsr cassette, which was flanked by loxP sites (denoted “floxed-Bsr”) (6). The sgkB fragments were generated by PCR, by using genomic DNA and primers comprised of gene-specific sequences and the restriction sites necessary for cloning. The vector was used to replace the sgkB gene by homologous recombination, producing the sgkBΔ blasticidin-resistant strain HM1080 (clone 1G11). The Bsr cassette was then excised from HM1080 by transient transfection with a G418-resistant plasmid with the nuclear localization signal containing cre recombinase, driven by the actin 15 promoter (pDEX-NLS-cre). Blasticidin-sensitive cre recombinase clones were selected by a protocol similar to that described elsewhere (6). This resulted in the blasticidin-sensitive sgkBΔ strain HM1091.
(iii) Sphingosine kinaseA/sphingosine kinase B double null.
The sphingosine kinase A/B double-null strain was generated by transforming strain HM1091 with a vector containing a floxed-Bsr cassette, flanked by a 5′ and 3′ fragments of the sgkA gene, which were generated by PCR from genomic DNA with primers containing gene-specific sequences and the restriction enzymes needed for cloning. Homologous recombination resulted in the double-null Bsr-resistant strain sgkAΔBΔ strain HM1093 (clone D8).
(iv) SgkA overexpressor.
An sgkA overexpression vector was generated by cloning a reverse transcription-PCR product of the sgkA gene into the pDXA3C vector (23), in which the C-terminal myc-tag had been replaced by a FLAG tag. The stable G418-resistant sgkA overexpressing (sgkAOE) strain, SA604, was generated by transforming Ax3-ORF cells (containing the transacting origin of replication recognition factor) (23) with the above vector and selecting for G418 resistance.
All strains were stored as frozen or desiccated spores and fresh cultures were started monthly in liquid HL-5 medium (41). Cell concentrations in HL5 liquid medium were determined by using a Coulter Z1 Particle Counter.
Molecular biology techniques.
Cloning and Southern analyses were performed according to standard published techniques (26, 36). Western analyses were performed according to the manufactures instructions with anti-FLAG monoclonal antibody (MAb) M2 at a dilution of 1:2,000 (Sigma Chemical Co., St. Louis, Mo.).
Sphingosine kinase assay.
Sphingosine kinase enzyme activity was assayed as described previously (30). Frozen pellets of 108 D. discoideum cells were lysed in 500 μl of SK buffer with 0.2 M KCl, followed by six cycles of freezing and thawing, and centrifugation for 30 min at 150,000 × g at 4°C (Beckman TL-100). Supernatants were transferred to clean tubes, and the protein concentration was determined by BCA (Pierce Chemical Co., Rockford, Ill.). A total of 50 to 150 μg of protein extract was brought to 180 μl with SK buffer containing either 0.2 or 1.0 M KCl and incubated with 10 μl of 1 mM sphingosine (delivered in 5% Triton X-100) and 10 μl of ATP mix (9 μl of 20 mM ATP in 200 mM MgCl2 plus 1 μl of [γ-32P]ATP (3,000 Ci/mmol; New England Nuclear, Boston, Mass.) for 90 min at 28°C. The reaction was stopped with 20 μl of 1 N HCl and 800 μl of chloroform-methanol-HCl (50:100:1) for 10 min at room temperature. We then added 250 μl of chloroform and 250 μl of 2 M KCl, and the samples were mixed and centrifuged to separate the phases. The top aqueous layer was aspirated, and 100-μl aliquots of the organic phase were spotted onto thin-layer chromatography plates (Silica Gel 60; Merck). The plates were developed in chloroform-acetone-methanol-acetic acid-H2O (10:4:3:2:1) and visualized on a Fuji phosphorimager. Spots were then scraped from the plates and quantitated in a scintillation counter. One unit activity was defined as the number of picomoles of S-1-P generated per minute per milligram of protein extract.
Reagents.
Sphingosine, DMS, and S-1-P were from BIOMOL (Plymouth Meeting, Pa.).
Immunofluorescence microscopy.
sgkAOE cells were harvested from HL5 medium and washed twice in LPS buffer (20 mM KCl, 2.5 mM MgCl2, 40 mM K-PO4, pH 6.5, containing 0.5 mg of streptomycin sulfate/ml) in a clinical centrifuge. A total of 4 × 104 cells in 80 μl of LPS were allowed to settle and adhere to glass coverslips. The cells were fixed and permeabilized as described previously (3). The cells were incubated with anti-FLAG MAb (1:500), followed by rhodamine-conjugated goat anti-mouse immunoglobulin G (1:200; Cappel). The cells were photographed by using a Zeiss IM microscope with a ×100 Neofluar lens.
Drug sensitivity.
Viable D. discoideum cells grow and form distinct clear clonal plaques when coplated on lawns of Klebsiella aerogenes (41). Cell viability after drug treatment was measured by counting plaques, each representing a surviving cell, by using a rapid plaque assay in 24-well plates (2). Threefold serial dilutions of cells were performed in 96-well plates by mixing 100 μl with 200 μl of SS (0.6 g of NaCl, 0.75 g of KCl, and 0.4 g of CaCl2 per liter) by using an automatic repeating pipettor that automatically mixes dilutions (Matrix Technologies Corp., Hudson, N.H.). A total of 15 μl of each diluted sample was added to 20 μl of K. aerogenes food source in 24-well plates containing 2 ml of SM agar per well (41). The plates were placed on a rotating shaker for 10 min to ensure even distribution and then incubated at 22°C for 3 days when plaques began to appear. Plates were scanned twice a day and plaques were counted. Each experiment was performed in duplicate, and survival was calculated as the average percent survival relative to the untreated culture. In experiments in which cells were cotreated with cisplatin and another inhibitor or activator, the second compound was added 1 h prior to the addition of cisplatin. The fold change in resistance or sensitivity was calculated as the ratio of the survival after combination treatment to the expected survival if each of the compounds acted independently as follows: (survival after DMS alone × survival after cisplatin alone)/observed survival after cotreatment with both drugs. Standard error and P values were calculated by using the statistical tool in Microsoft Excel.
Cisplatin uptake.
Cultures (5 ml at 2 × 106 cell/ml) were treated with the indicated amounts of cisplatin, harvested, and washed twice in ice-cold phosphate-buffered saline by centrifugation. The cell pellets were incubated for 2 h in 150 μl of 6 N HNO3 at 85 to 90°C. A total of 540 μl of 1.3% Triton X-100 was added to each sample (to a final concentration of 1.3 N HNO3 and 1% Triton X-100), and the concentration of platinum was determined by flameless atomic absorption spectrometry with a Varian SPECTRAA-220Z graphite furnace double-beam atomic spectrophotometer (Houston, Tex.) with Zeeman background correction. The platinum standard (Buck Scientific, East Norwalk, Conn.) used for calibration included equivalent amounts of HNO3 and Triton X-100 as the samples. Use of a flameless spectrophotometer provides the sensitivity necessary to measure intracellular cisplatin when it was delivered to the cells at moderate cytotoxic concentrations (i.e., <100% cytotoxic). Nonspecific adsorption of cisplatin to cells was monitored by treating cells with cisplatin at 4°C and was minimal in all cases.
RESULTS
D. discoideum encodes two sphingosine kinase enzymes.
D. discoideum has two genes with homology to the human sphingosine kinases 1 and 2 (Fig. 1A), which have been named sgkA (AY283053) and sgkB (AY679519). They encode predicted proteins of 70.1 and 83.4 kDa. The Dictyostelium SgkA and SgkB proteins share 50% identity and are highly homologous to sphingosine kinases in humans, mice, Drosophila, yeast, and Arabidopsis. Figure 1B presents an expanded view of the five conserved domains found in the sphingosine kinases of the above species (20). Both the SgkA and the SgkB proteins have the typical nucleotide binding motif in region C2. Human sphingosine kinase 2 has a 9-amino-acid motif similar to that in BH3-only proteins (22), and motifs with significant homology are also seen in both SgkA and SgkB.
FIG. 1.

Homology of the D. discoideum SgkA and SgkB proteins with sphingosine kinases of other species. (A) Homology between the human and D. discoideum enzymes. (B) Homology of the sphingosine kinase conserved regions (C1 to C5) among humans, mice, Drosophila, D. discoideum, yeast, and Arabidopsis. Accession numbers used for alignments are: hSPHK1, AAG01980; hSPHK2, AAH06161; mSPHK1, AAC61697; mSPHK2, AAF74125; LCB4, NP_014814; LCB5, NP_013361; Drosophila sp. strain SK1, AAF48045; Drosophila sp. strain SK2, AAF47706; and AtLCBK1, BAB07787. It should be noted that the D. discoideum SgkB protein uniquely contains stretches of 5, 13, 26, and 7 asparagine residues, a common feature in D. discoideum proteins.
Generation of sphingosine kinase-null mutants.
Sphingosine kinase-null mutants were generated by homologous recombination as described in Materials and Methods. Figure 2A to C shows the structures of the homologous recombination vectors used to make the sgkAΔ, sgkBΔ, and sgkAΔBΔ mutants. The PCR primers used to verify the gene disruptions in the mutant strains correspond to genomic sequences outside those in the disruption vectors. The PCR results are presented for each mutant (Fig. 2D to F). Supporting the PCR analyses, Southern analyses of each mutant are shown in Fig. 2G to I. Both analyses confirm that the genes were successfully disrupted.
FIG. 2.

Construction and verification of sgk-null strains. (A to C) Schematic presentations of the construction vectors used for homologous disruption of the genes (not drawn to scale). Arrows represent the oligonucleotide primers used for verification of the gene disruptions by PCR. The PCR primers were outside the regions of the genes used for the construction vectors. Abbreviations: B, BamHI; H, HindIII; Nd, NdeI; No, NotI; S, SacII; A, ApaI. (D to F) PCR verification of gene disruptions. (G to I) Southern analyses of the sgk single and double mutants. sgkAΔ (A and D) primers 1 and 2 are sgkA-specific primers and yield the predicted 2-kb fragment from the AX4 parent strain. Primers 3 and 4 are Bsr-specific primers. Primer combinations 1 and 4 (1/4) and 3/2 generate the predicted 2.1- and 1.7-kb PCR products, respectively, from the sgkAΔ strain. (G) Southern analysis with an sgkA-specific probe confirms the insertion of a 1.4-kb cassette into the 5-kb sgkA NdeI fragment. sgkBΔ (B and E) primers 5 and 6 are sgkB-specific primers and generated the predicted 2.5-kb fragment from the Ax4 parent and a 3.4-kb fragment from the disrupted Bsr-containing gene. The cre recombinase product was verified with the same primers to generate a 2.0-kb PCR fragment(representing the loss of the Bsr cassette and part of the sgkB gene). (H) Southern analysis with the sgkB-specific probe confirms the disruption of the parental copy of the gene. sgkAΔBΔ (C and F) The disruption was verified by PCR with sgkA-specific primers 7 and 8, which generate a 2.5-kb fragment in the parental strain (Ax4) and in the sgkBΔ (where sgkA is unaffected) and a 3.3-kb fragment in the double mutant, where the Bsr cassette is inserted into the sgkA gene. (I) Southern analyses of the double mutant with an sgkA-specific probe confirms the disruption of the parental 5-kb NdeI fragment and the generation of 2.9-kb NdeI-HindIII, 1.7-kb NdeI, and 1.4-kb HindIII-NdeI fragments in the mutant strain.
Sphingosine kinase-null mutants have reduced enzyme activity.
The level of sphingosine kinase activity was measured in the sphingosine kinase single and double mutants. The assays were performed at 28°C, at which temperature the activity is higher than at 37°C, and remains linear for a longer period of time (unpublished). This may reflect the fact that D. discoideum grows optimally at 22°C. Studies with human sphingosine kinase have shown that the two enzymes have dramatically different sensitivities to KCl, with human sphingosine kinase 2 being activated in 1 M KCl. Figure 3A shows that the D. discoideum SgkB enzyme, which is active in the sgkAΔ cells, is activated 35-fold in 1 M KCl compared to its activity in 200 mM KCl, whereas the activity of the SgkA enzyme, which is active in the sgkBΔ cells, is activated only 2.4-fold in high salt and constitutes a much lower level of activity than SgkB. On the basis of this salt specificity, we suggest that the D. discoideum SgkB is the ortholog of the human Sphk2 enzyme.
FIG. 3.
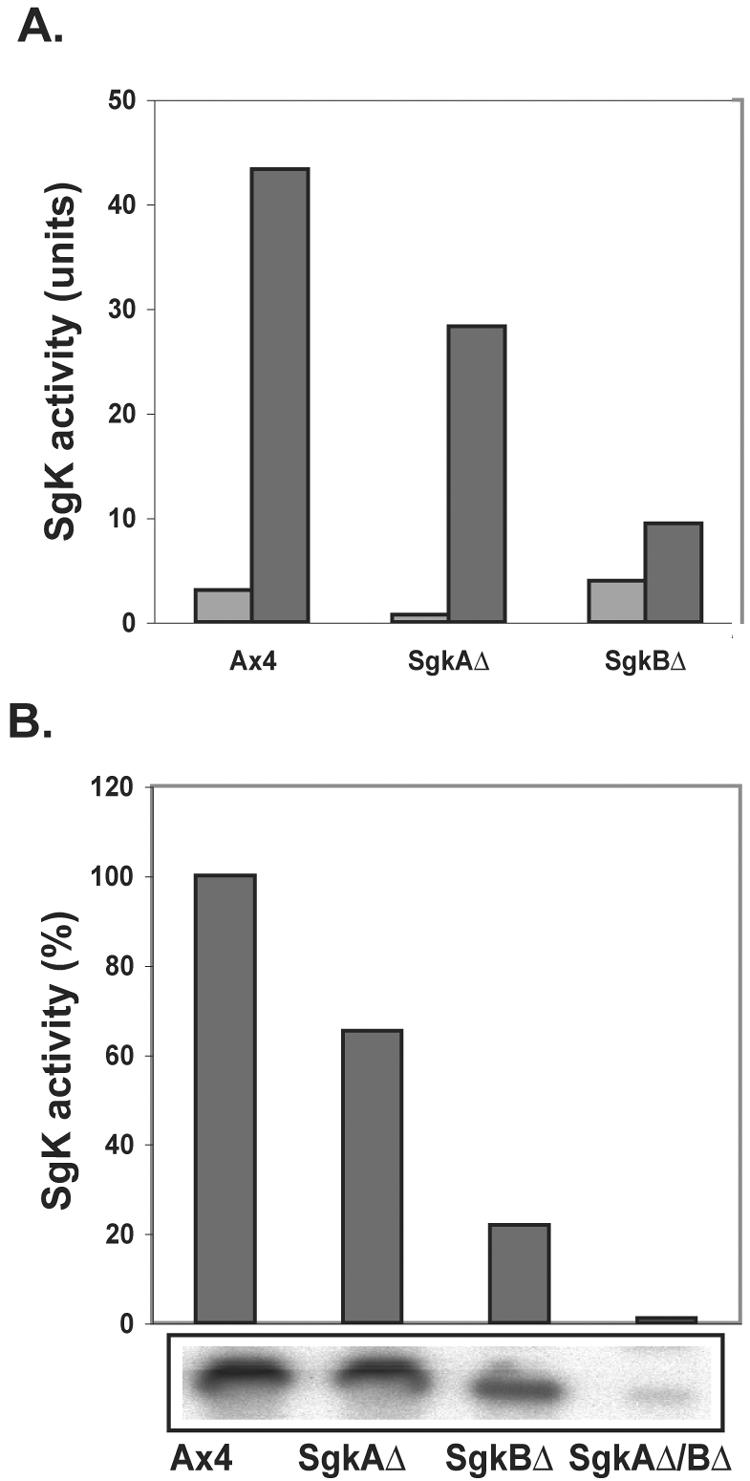
Sphingosine kinase activity in sgk-null mutants. (A) Sphingosine kinase when assayed in low (0.2 M) KCl (light gray bars) and high (1.0 M) KCl (dark gray bars). (B) Comparison of sphingosine kinase activity in the parental and sgk-null mutants when assayed in high salt. The lower box shows the phosphorimager picture of the S-1-P bands on the thin-layer chromatography plate from which the quantitation was made.
Figure 3B presents the sphingosine kinase activity levels for the parent Ax4 and the three null mutants. The sgkAΔ mutant has decreased activity compared to the parent but higher activity than the sgkBΔ mutant, suggesting that SgkB is the predominant activity in mitotically dividing D. discoideum cells. The sgkAΔBΔ double mutant has essentially no sphingosine kinase activity. The results are typical of three independent experiments.
Characterization of the sgkAOE strain.
Figure 4A demonstrates expression of the SgkA FLAG-tagged fusion protein in the sgkAOE strain, as assayed by Western analysis with anti-FLAG MAb. The sgkA gene in this construct is under the control of the strong actin 15 promoter, which results in significant overexpression and, indeed, the level of SgkA enzyme activity in the stable sgkAOE strain is 13-fold higher than in the parental cells (Fig. 4B). Figure 4C shows the localization of the overexpressed FLAG-tagged protein in mitotically dividing D. discoideum cells. The sgkAOE strain has somewhat higher percentage of multinucleate cells than the parental strain. The protein is diffusely distributed throughout the cytosol and appears to be enriched within the nuclei (arrows) and in some pseudopodia (p). This is true for mononucleate cells, as well as for multinucleate cells (shown) which, because of their large size, are easier to visualize. These results are similar to what has been observed when sphingosine kinase was overexpressed in human cells (12).
FIG.4.

Characterization of the sgkA overexpressor mutant. (A) Western blot analysis of the SgkA fusion protein in the sgkAOE strain and in a vector control with anti-FLAG MAb. (B) Sphingosine kinase activity in the sgkAOE strain. Protein extracts from the overexpressor and the parent strain were assayed in SK buffer plus 1 M KCl and separated on thin-layer chromatography plates (lower panel). The spots were excised and quantitated by liquid scintillation counting. Bars: sgkAOE, pink bar; Ax4 parent strain, red bar. (C) Three examples of immunofluorescent localization with anti-FLAG MAb of the Sgk fusion protein in the sgkAOE strain. Image type: A, C, and E, fluorescence; B, D, and F, phase contrast. Arrows point to the position of the nuclei which contain the characteristic three nucleoli (phase images). Pseudopodia are indicated by “p.” No fluorescence was observed when the primary anti-FLAG antibody was omitted.
Altering the levels of sphingosine kinase enzymes results in altered growth rates.
In previous studies (26), it was shown that overexpression of the S-1-P lyase gene resulted in a decrease in growth rate and that the decrease in rate reflected the level of SglA enzyme expression. Based on this we predicted that disrupting the sgk genes also would result in a decrease in growth rate. Figure 5 confirms this prediction. Figures 5A and B show that the plaque size decreases in the order of Ax4 (parent) >sgkAΔ > sgkBΔ > sgkAΔBΔ. The effect is proportional to the kinase levels in the mutants (Fig. 3B). Conversely, overexpression of the sgkA gene (Fig. 4B) results in an increase in plaque size compared to the parent cells. The difference in growth rate of the mutants is less pronounced when the cells are growing axenically in liquid medium. Nevertheless, even under these conditions the sgk-null mutants have a consistently slower growth rate, whereas the sgkA overexpressor has a more rapid growth rate (Fig. 5C). In summary, both assays indicate that sphingosine kinase has a function in controlling growth in D. discoideum.
FIG. 5.

sgk mutants have altered growth rates. (A) Clonal plaques of parental and sgk mutant strains. (B) Size measurements of plaques from panel A. The inset zooms in on differences at day 4, when plaques were very small. (C) Growth of parental and sgk mutant cells in liquid HL5 medium was monitored daily for 10 days. The data are the average of duplicate experiments. The error bars have been omitted to avoid masking the symbols. Symbols and their respective strains are as defined at the bottom of the figure.
Sphingosine kinase mutants have altered sensitivity to cisplatin and carboplatin.
Based on previous work on D. discoideum mutants either overexpressing or lacking the S-1-P lyase gene (19, 26), we hypothesized that altering the levels of sphingosine kinase should also alter sensitivity to cisplatin and the related platinum-based drug carboplatin. Specifically, it was predicted that deleting the sphingosine kinase genes would have a similar effect to overexpressing the S-1-P lyase (increased drug sensitivity), whereas overexpressing the sphingosine kinase genes would mimic the S-1-P lyase-null mutant (increased drug resistance). Figure 6A shows the sensitivity of the sgkAΔ, sgkBΔ, and sgkAΔBΔ mutants to a range of cisplatin concentrations. The parent cells showed a typical response as seen previously (26). The disruption of the sgkA and sgkB genes resulted in increased sensitivity, with sgkBΔ being slightly more sensitive than sgkAΔ. The double mutant was more sensitive to cisplatin than either of the single mutants. More cytotoxicity was seen after 24 h compared to the 5 h treatment (Fig. 6B). Similar results were observed when the mutants were tested for sensitivity to carboplatin (Fig. 6C), although cells are generally less sensitive to carboplatin, as observed previously (26). Increasing the treatment with carboplatin to 24 h did not significantly increase cytotoxicity (Fig. 6D). In contrast to the sgk-null mutants, overexpression of SgkA resulted in an increase in resistance to both cisplatin and carboplatin. The results confirm the predictions and clearly show that both D. discoideum sphingosine kinase enzymes function in regulating sensitivity to both cisplatin and carboplatin.
FIG. 6.

sgk mutants are altered in sensitivity to cisplatin and carboplatin. (A and B) Sensitivity of parental and sgk mutant strains to increasing concentrations of cisplatin at 5 and 24 h, respectively. (C and D) Sensitivity of the parental and sgk-null mutant strains to increasing concentrations of carboplatin at 5 and 24 h, respectively. Symbols and their respective strains are as defined at the bottom of the figure.
Sensitivity to other drugs is not altered in the sphingosine kinase-null mutants.
Altering the levels of the S-1-P lyase had the effect of specifically modulating the sensitivity of cells to the platinum-based drugs, cisplatin and carboplatin, but not to the other DNA-damaging drugs tested (26). This suggested that a similar profile of sensitivity would be seen after altering the expression of the sphingosine kinases. The data in Fig. 7 show that the sensitivity of the sgk-null mutants to cisplatin and carboplatin is inversely proportional to the level of total sphingosine kinase activity in the mutants (see also Fig. 6), whereas there is no significant alteration in sensitivity to doxorubicin, 5-flurouracil, and etoposide. Overexpression of the kinase resulted in increased resistance to the platinum based drugs and, to a lesser extent, to the other drugs.
FIG. 7.

Sensitivity of parental and sgk mutant strains to a variety of DNA-damaging agents tested after 24 h of treatment. Symbols are defined at the bottom of the figure. The results are the averages of duplicate experiments, and the symbols above the bars indicate that they are significantly different than the control as follows: *, < 0.05; and **, < 0.01.
Uptake of cisplatin is not altered in sphingosine kinase mutants.
The accumulating data suggested that the effect of modulating the sphingosine kinase and S-1-P lyase enzymes on cisplatin sensitivity in D. discoideum was due to an effect on S-1-P-mediated intracellular signaling. However, even though S-1-P is not a component of the plasma membrane, it was possible that the proximal cause of the alterations in cisplatin sensitivity was due to changes in cisplatin uptake. Therefore, atomic absorption spectroscopy was used to measure the cisplatin uptake at moderate cytotoxic levels (150 μM) in mutant and parental cells. Figure 8A and B shows that there is no significant difference in accumulation between the parental and sphingosine kinase mutant cells as a function of either cisplatin concentration or time. Likewise, Fig. 8C and D show that there was no significant difference in accumulation between the parental and mutant S-1-P lyase mutant cells. Although altered uptake remains a possible proximal cause for the changes in drug sensitivity in the other mutants that we identified in the original genetic screen (19), we believe that the role of sphingosine kinase and S-1-P lyase in the response to cisplatin is not through altering the uptake of the drug.
FIG. 8.

Cisplatin accumulation is not altered in the sgk mutants. (A and B) Cisplatin accumulation in the parental and sgk (sphingosine kinase) mutant cells as a function of cisplatin concentration and time, respectively. (C and D) Cisplatin accumulation in the parental and sgl (sphingosine lyase) mutant cells as a function of cisplatin concentration and time, respectively. The concentration dependence assays were incubated for 30 min. Cellular levels of platinum were measured by flameless atomic absorption spectrometry, and cisplatin levels were extrapolated from standard curves by using a platinum standard solution.
DMS sensitizes cells to cisplatin.
Figures 6 and 7 demonstrate that decreasing sphingosine kinase activity increases sensitivity to cisplatin, whereas increasing sphingosine kinase activity increases resistance relative to the parental cells. This increase in resistance can be reversed pharmacologically by treatment of the sgkAOE mutant with the sphingosine kinase inhibitor DMS. Parental Ax4 and sgkAOE cells were treated with increasing levels (0 to 10 μM) of DMS in the presence or absence of cisplatin for 5 and 24 h.
Parental Ax4 D. discoideum cells are killed by DMS in a time- and dose-dependent manner (Fig. 9A and B, open circles). Concentrations of 2.5, 5, and 10 μM result in 102, 78, and 28% survival at 5 h and in 77, 38, and 27% survival at 24 h, respectively. Treatment of parallel cultures with 150 μM cisplatin alone reduced viability to 66% after 5 h and 29% after 24 h. Cotreatment of the cells with both DMS and 150 μM cisplatin resulted in a synergistic increase in drug sensitivity (Fig. 9A and B, closed circles). For example, at 2.5 and 5 μM DMS, sensitivity to cisplatin after 5 h increased 1.5- and 2.3-fold, respectively, and after 24 h it increased 1.5- and 1.5-fold, respectively, over what would be predicted if the two drugs were working independently.
FIG. 9.

Pretreatment with DMS increases the sensitivity of cells to cisplatin. Symbols: ○, DMS alone was added at the indicated concentrations to growing cultures; •, DMS at the indicated concentrations was added to growing cultures 1 h before addition of 150 μΜ cisplatin. The closed circle points at zero DMS represent survival with 150 μM cisplatin alone and were done in triplicate. Parental Ax4 cells (A and B) or the sglAOE mutant cells (C and D). Cell viability was assayed at 5 h (A and C) and 24 h (B and D).
The sgkAOE mutant cells are also affected by DMS in a dose- and time-dependent manner but to a lesser extent. The cells are more resistant to DMS than the parental cells (Fig. 9C and D). As demonstrated above, these cells are initially more resistant to cisplatin than the parental cells. Similar to the results described for the parent cells, cotreatment with DMS and cisplatin resulted in a synergistic reduction in viability, e.g., at 2.5 and 5 μM DMS sensitivity after 24 h was increased 1.9- and 1.5-fold, respectively. The lower fold-increases after 5 h (1.1- and 1.2, respectively) probably reflect the higher level of SgkA activity in these cells.
Overall, these data confirm the findings of our previous study showing that DMS can potentiate the efficacy of cisplatin (26) and demonstrate that the increase in resistance in this sgkAOE mutant was due to the increase in sphingosine kinase enzyme activity per se rather than some other function of the SgkA protein.
Exogenous S-1-P counteracts cisplatin sensitivity in sphingosine kinase-null mutants.
Figure 10 demonstrates that adding S-1-P to either parental or sgk-null mutant cells reverses their sensitivity to cisplatin. The mutants show the same relative sensitivities to cisplatin that were observed in the previous experiments. S-1-P has little effect on the cell viability at any of the concentrations used. The effect of S-1-P was observed with 5 μM S-1-P. These data are consistent with previous work showing that exogenously added S-1-P reversed the increased cisplatin resistance of mutants overexpressing the S-1-P lyase.
FIG. 10.

S-1-P increases resistance of the sgk-null mutants to cisplatin. S-1-P at the indicated concentrations was added to growing cultures of the parental Ax4 cells and the sgk-null mutant cells 1 h prior to the addition of 150 μM cisplatin. The cells were harvested after 5 h and plated for viability. Symbols: ○, S-1-P alone; •, S-1-P at the indicated concentrations plus 150 μM cisplatin.
DISCUSSION
Over the past several years there has been a surge in interest in the roles of sphingolipids in cell function. It is now clear that sphingolipids play pivotal roles in the signaling pathways that affect growth, developmental programs, stress response, and disease (17, 29). In particular, a substantial body of work now exists supporting the idea that the balance between S-1-P and ceramide (or S-1-P and sphingosine) plays a central role in whether a cell proliferates or dies in response to extracellular signals and stress, where S-1-P is “progrowth” and ceramide is “proapoptosis.” Thus, the discovery of an S-1-P lyase-null mutant in a genetic selection for cisplatin-resistant mutants suggested that (i) the absence in S-1-P lyase in the cells leads to an increase in S-1-P, which in turn leads to increased cell survival and proliferation even in the presence of the cytotoxic levels of cisplatin, and (ii) that the enzymes that control the level of S-1-P could be used as therapeutic targets to increase the efficacy of cisplatin.
Resistance to cisplatin is a common problem in cancer therapy, and increasing the sensitivity of tumors to drugs is an important goal. Conversely, in some cases it is desired to be able to reduce the cytotoxicity of cisplatin to certain cells and organs, such as the kidney, where cisplatin is highly nephrotoxic (47), or in the ovaries (44). Thus, being able to alter the cells response to the drug could be very beneficial in cancer therapy.
D. discoideum affords an excellent system for testing these ideas because both null and overexpressor mutants can be constructed and tested for drug sensitivity. In this regard we showed that overexpression of S-1-P lyase increased the sensitivity of cells to cisplatin and carboplatin, a related platinum compound used in therapy. Interestingly, the increased S-1-P lyase gene expression did not sensitize the cells to other DNA-damaging drugs that exert their cytotoxicity by other mechanisms (i.e., not cross-linking purines).
Sphingosine kinase enzymes function upstream of the S-1-P lyase, in the synthesis of S-1-P, and are important regulators of S-1-P levels (see reference 17). Eukaryotes studied to date have two genes encoding homologous sphingosine kinases, which are characterized by five conserved domains. The sphingosine kinases are, in turn, regulated by a variety of agonists of receptor tyrosine kinases and G-protein-coupled receptors. The activation of the sphingosine kinases is at least partially regulated by interaction with other proteins and by phosphorylation. Aspects of many biological processes, including Ca2+ homeostasis, cell motility, inflammation, and immunity, are regulated by sphingosine kinase. However, much remains to be learned about the cellular functions of these enzymes.
In the present study we identified two D. discoideum sphingosine kinases, sgkA and sgkB, which are homologous to the human Sphk1 and Sphk2 enzymes. Disrupting and overexpressing the sphingosine kinase enzymes in D. discoideum allowed us to further characterize these enzymes and to demonstrate their involvement in the cellular response to cisplatin. The D. discoideum enzymes exhibit a differential sensitivity to salt similar to that of the human enzyme (4, 21). On the basis of the salt sensitivity, we suggest that the D. discoideum SgkB is the ortholog of the mammalian Sphk2. The SgkA protein in the sgkAOE mutant is distributed throughout the cytosol, with some enrichment in the nuclei, reminiscent of the localization when Sphk1 is overexpressed in human cells (12).
The sphingosine kinase mutant strains show changes in growth rate consistent with changes that were seen previously with strains overexpressing different levels of S-1-P lyase. It also was previously shown that DMS slows the growth rate in D. discoideum (26). These growth phenotypes are consistent with those observed in HEK cells overexpressing the human S-1-P lyase gene (35). Yeast strains lacking sphingosine kinase activity also showed attenuated growth (13). Indeed, it has been suggested that human sphingosine kinase is an oncogene that functions through the H-ras pathway (46).
We show here that sphingosine kinase mutants have altered sensitivity to platinum-based drugs. The sgk-null mutants were more sensitive to cisplatin than the parent, and the sgkAOE cells were more resistant. This was not due to altered drug uptake in the different strains. These data are consistent with data from the S-1-P lyase mutants (19, 26). The sgkAΔBΔ mutant was more sensitive than either of the single mutants, but sgkBΔ was more sensitive than sgkAΔ, suggesting that the SgkB enzyme plays the predominant role in determining drug sensitivity either due to its relative activity or intracellular localization or both. Nevertheless, it is clear that both enzymes contribute to the overall cisplatin sensitivity of the cells. As with the S-1-P lyase-overexpressing cells, the sgk-null cells show parallel changes in drug sensitivity to carboplatin, another platinum-based drug, but not with DNA-damaging drugs with other mechanisms of action. However, the sgkAOE cells also showed a limited increased resistance to the other drugs, a finding different from what was observed previously with the S-1-P lyase-null cells (26). This could be due to a high level of S-1-P in the sgkAOE cells, which results in the simultaneous activation of several survival pathways such as the extracellular signal-regulated kinase mitogen-activated protein kinase pathway and the phosphatidylinositol 3-kinase pathway. Alternatively, in contrast to the S-1-P lyase-null cells, high levels of sphingosine kinase could cause a reduction in sphingosine and/or ceramide levels in the sgkAOE cells. To this end, direct measurements of S-1-P levels in D. discoideum have proven to be challenging. We are now working on methods to measure the relative levels of S-1-P, sphingosine, and ceramide in the mutants and in response to cisplatin. It also will be interesting to modulate the enzymes that alter ceramide levels, such as ceramide synthase and serine palmitoyltransferase, and to assess the effect on cisplatin sensitivity and specificity.
Overexpression of the sgkA gene results in increased resistance similar to the disruption of the S-1-P lyase, whereas disruption of the kinases results in increased sensitivity similar to the overexpression of the S-1-P lyase. These data strongly support the idea that it is the increase in S-1-P that leads to drug resistance in the S-1-P lyase-null mutant rather than the lack of the products of the S-1-P lyase. Consistent with this is a recent report demonstrating degradation of the human sphingosine kinase 1 enzyme and an increase in apoptosis after treatment with actinomycin D (43).
The identification of the S-1-P lyase-null mutant in the initial genetic selection has led to multiple protein targets for drug therapy that can sensitize cells to platinum drugs (i.e., the two sphingosine kinases and the S-1-P lyase). The demonstration of multiple targets is important because it may not be possible to develop appropriate compounds to modulate a particular target. To this end, the sphingosine kinase inhibitor DMS was readily available, and it was demonstrated that it increases the sensitivity of cells to cisplatin (26; the present study). Additional sphingosine kinase inhibitors have been developed recently and need to be tested for their ability to increase sensitivity to cisplatin (7).
Acknowledgments
This study was supported by NIH grants GM53929, CA95872 (S.A. and H.A.), and CA57530 (M.H.H.).
D.T. thanks Rob Kay for his support during the course of this work. We thank Christopher Foote for assistance and Alan Kimmel and Lisa Kreppel for the Cre-loxP system plasmids.
REFERENCES
- 1.Adachi, H., T. Hasebe, K. Yoshinaga, T. Ohta, and K. Sutoh. 1994. Isolation of Dictyostelium discoideum cytokinesis mutants by restriction enzyme-mediated integration of the blasticidin S resistance marker. Biochem. Biophys. Res. Commun. 205:1808-1814. [DOI] [PubMed] [Google Scholar]
- 2.Alexander, H., A. N. Vomund, and S. Alexander. 2003. Viability assay for Dictyostelium for use in drug studies. BioTechniques 35:464-470. [DOI] [PubMed] [Google Scholar]
- 3.Alexander, S., L. M. Sydow, D. Wessels, and D. R. Soll. 1992. Discoidin proteins of Dictyostelium are necessary for normal cytoskeletal organization and cellular morphology during aggregation. Differentiation 51:149-161. [DOI] [PubMed] [Google Scholar]
- 4.Billich, A., F. Bornancin, P. Devay, D. Mechtcheriakova, N. Urtz, and T. Baumruker. 2003. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J. Biol. Chem. 278:47408-47415. [DOI] [PubMed] [Google Scholar]
- 5.Edsall, L. C., J. R. Van Brocklyn, O. Cuvillier, B. Kleuser, and S. Spiegel. 1998. N,N-Dimethylsphingosine is a potent competitive inhibitor of sphingosine kinase but not of protein kinase C: modulation of cellular levels of sphingosine 1-phosphate and ceramide. Biochemistry 37:12892-12898. [DOI] [PubMed] [Google Scholar]
- 6.Faix, J., L. Kreppel, G. Shaulsky, M. Schleicher, and A. R. Kimmel. 2004. A rapid and efficient method to generate multiple gene disruptions in Dictyostelium using a single selectable marker and the cre-loxP system. Nucleic Acids Res. 32:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.French, K. J., R. S. Schrecengost, B. D. Lee, Y. Zhuang, S. N. Smith, J. L. Eberly, J. K. Yun, and C. D. Smith. 2003. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 63:5962-5969. [PubMed] [Google Scholar]
- 8.Gijsbers, S., G. Van der Hoeven, and P. P. Van Veldhoven. 2001. Subcellular study of sphingoid base phosphorylation in rat tissues: evidence for multiple sphingosine kinases. Biochim. Biophys. Acta 1532:37-50. [DOI] [PubMed] [Google Scholar]
- 9.Herr, D. R., H. Fyrst, V. Phan, K. Heinecke, R. Georges, G. L. Harris, and J. D. Saba. 2003. Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development 130:2443-2453. [DOI] [PubMed] [Google Scholar]
- 10.Hla, T. 2004. Physiological and pathological actions of sphingosine 1-phosphate. Semin. Cell Dev. Biol. 15:513-520. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi, N., T. Okada, S. Hayashi, T. Fujita, S. Jahangeer, and S. Nakamura. 2003. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 278:46832-46839. [DOI] [PubMed] [Google Scholar]
- 12.Inagaki, Y., P. Y. Li, A. Wada, S. Mitsutake, and Y. Igarashi. 2003. Identification of functional nuclear export sequences in human sphingosine kinase 1. Biochem. Biophys. Res. Com. 311:168-173. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins, G. M., and Y. A. Hannun. 2001. Role for de novo sphingoid base biosynthesis in the heat-induced transient cell cycle arrest of Saccharomyces cerevisiae. J. Biol. Chem. 276:8574-8581. [DOI] [PubMed] [Google Scholar]
- 14.Kohama, T., A. Olivera, L. Edsall, M. M. Nagiec, R. Dickson, and S. Spiegel. 1998. Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 273:23722-23728. [DOI] [PubMed] [Google Scholar]
- 15.Kumar, A., A. Wessels, K. Daniels, H. Alexander, S. Alexander, and D. R. Soll. 2004. Sphingosine-1-phosphate plays a role in the suppression of lateral pseudopod formation during Dictyostelium discoideum cell migration and chemotaxis. Cell Motil. Cytoskeleton 59:227-241. [DOI] [PubMed] [Google Scholar]
- 16.Kuspa, A., and W. F. Loomis. 1992. Tagging developmental genes in Dictyostelium by restriction enzyme-mediated integration of plasmid DNA. Proc. Natl. Acad. Sci. USA 89:8803-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Stunff, H., S. Coursol, S. Milstien, and S. Spiegel. 2004. Sphingosine-1-phosphate metabolism in mammalian cell signaling. Lipid Metab. Membr. Biogen. 6:313-335. [Google Scholar]
- 18.Li, G., C. Foote, S. Alexander, and H. Alexander. 2001. Sphingosine-1-phosphate lyase has a central role in the development of Dictyostelium discoideum. Development 128:3473-3483. [DOI] [PubMed] [Google Scholar]
- 19.Li, G. C., H. Alexander, N. Schneider, and S. Alexander. 2000. Molecular basis for resistance to the anticancer drug cisplatin in Dictyostelium. Microbiology 146:2219-2227. [DOI] [PubMed] [Google Scholar]
- 20.Liu, H., D. Chakravarty, M. Maceyka, S. Milstien, and S. Spiegel. 2002. Sphingosine kinases: a novel family of lipid kinases. Prog. Nucleic Acids Res. Mol. Biol. 71:493-511. [DOI] [PubMed] [Google Scholar]
- 21.Liu, H., M. Sugiura, V. E. Nava, L. C. Edsall, K. Kono, S. Poulton, S. Milstien, T. Kohama, and S. Spiegel. 2000. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275:19513-19520. [DOI] [PubMed] [Google Scholar]
- 22.Liu, H., R. E. Toman, S. K. Goparaju, M. Maceyka, V. E. Nava, H. Sankala, S. G. Payne, M. Bektas, I. Ishii, J. Chun, S. Milstien, and S. Spiegel. 2003. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 278:40330-40336. [DOI] [PubMed] [Google Scholar]
- 23.Manstein, D. J., H. P. Schuster, P. Morandini, and D. M. Hunt. 1995. Cloning vectors for the production of proteins in Dictyostelium discoideum. Gene 162:129-134. [DOI] [PubMed] [Google Scholar]
- 24.Melendez, A. J., E. Carlos-Dias, M. Gosink, J. M. Allen, and L. Takacs. 2000. Human sphingosine kinase: molecular cloning, functional characterization and tissue distribution. Gene 251:19-26. [DOI] [PubMed] [Google Scholar]
- 25.Mendel, J., K. Heinecke, H. Fyrst, and J. D. Saba. 2003. Sphingosine phosphate lyase expression is essential for normal development in Caenorhabditis elegans. J. Biol. Chem. 278:22341-22349. [DOI] [PubMed] [Google Scholar]
- 26.Min, J., A. Stegner, H. Alexander, and S. Alexander. 2004. Overexpression of sphingosine-1-phosphate lyase or inhibition of sphingosine kinase in Dictyostelium discoideum results in a selective increase in sensitivity to platinum based chemotherapy drugs. Eukaryot. Cell 3:795-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morio, T., H. Urushihara, T. Saito, Y. Ugawa, H. Mizuno, M. Yoshida, R. Yoshino, B. N. Mitra, M. Pi, T. Sato, K. Takemoto, H. Yasukawa, J. Williams, M. Maeda, I. Takeuchi, H. Ochiai, and Y. Tanaka. 1998. The Dictyostelium developmental cDNA project: generation and analysis of expressed sequence tags from the first-finger stage of development. DNA Res. 5:335-340. [DOI] [PubMed] [Google Scholar]
- 28.Nava, V. E., E. Lacana, S. Poulton, H. Liu, M. Sugiura, K. Kono, S. Milstien, T. Kohama, and S. Spiegel. 2000. Functional characterization of human sphingosine kinase-1. FEBS Lett. 473:81-84. [DOI] [PubMed] [Google Scholar]
- 29.Ogretmen, B., and Y. A. Hannun. 2004. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 4:604-616. [DOI] [PubMed] [Google Scholar]
- 30.Olivera, A., K. Barlow, and S. Spiegel. 1999. Assaying sphingosine kinase activity. Methods Enzymol. 311:215-223. [DOI] [PubMed] [Google Scholar]
- 31.Oskouian, B., and J. D. Saba. 2004. Death and taxis: what non-mammalian models tell us about sphingosine-1-phosphate. Semin. Cell Dev. Biol. 15:529-540. [DOI] [PubMed] [Google Scholar]
- 32.Perry, M. C. (ed.). 1997. The chemotherapy source book, 2nd ed. The Williams & Wilkins Co., Baltimore, Md.
- 33.Pitson, S. M., P. A. Moretti, J. R. Zebol, R. Zareie, C. K. Derian, A. L. Darrow, J. Qi, R. J. D'Andrea, C. J. Bagley, M. A. Vadas, and B. W. Wattenberg. 2002. The nucleotide-binding site of human sphingosine kinase 1. J. Biol. Chem. 277:49545-49553. [DOI] [PubMed] [Google Scholar]
- 34.Pyne, S. 2002. Cellular signaling by sphingosine and sphingosine 1-phosphate: their opposing roles in apoptosis, p. 245-268. In A. Quinn and A. Kagan (ed.), Phospholipid metabolism in apoptosis, vol. 36. Kluwer Academic/Plenum, New York, N.Y. [PubMed]
- 35.Reiss, U., B. Oskouian, J. Zhou, V. Gupta, P. Sooriyakumaran, S. Kelly, E. Wang, A. H. Merrill, Jr., and J. D. Saba. 2004. Sphingosine-phosphate lyase enhances stress-induced ceramide generation and apoptosis. J. Biol. Chem. 279:1281-1290. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 37.Siddik, Z. H. 2003. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22:7265-7279. [DOI] [PubMed] [Google Scholar]
- 38.Spiegel, S. 1999. Sphingosine 1-phosphate: a prototype of a new class of second messengers. J. Leukoc. Biol. 65:341-344. [DOI] [PubMed] [Google Scholar]
- 39.Spiegel, S., D. English, and S. Milstien. 2002. Sphingosine 1-phosphate signaling: providing cells with a sense of direction. Trends Cell Biol. 12:236-242. [DOI] [PubMed] [Google Scholar]
- 40.Spiegel, S., and S. Milstien. 2002. Sphingosine 1-phosphate, a key cell signaling molecule. J. Biol. Chem. 277:25851-25854. [DOI] [PubMed] [Google Scholar]
- 41.Sussman, M. 1987. Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Methods Cell Biol. 28:9-29. [DOI] [PubMed] [Google Scholar]
- 42.Taha, T. A., K. M. Argraves, and L. M. Obeid. 2004. Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1682:48-55. [DOI] [PubMed] [Google Scholar]
- 43.Taha, T. A., W. Osta, L. Kozhaya, J. Bielawski, K. R. Johnson, W. E. Gillanders, G. S. Dbaibo, Y. A. Hannun, and L. M. Obeid. 2004. Downregulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53. J. Biol. Chem. 279:20546-20554. [DOI] [PubMed] [Google Scholar]
- 44.Tilly, J. L., and R. N. Kolesnick. 2002. Sphingolipids, apoptosis, cancer treatments, and the ovary: investigating a crime against female fertility. Biochim. Biophys. Acta 1585:135-138. [DOI] [PubMed] [Google Scholar]
- 45.van Veldhoven, P. P., and G. P. Mannaerts. 1993. Sphingosine-phosphate lyase. Adv. Lipid Res. 26:69-98. [PubMed] [Google Scholar]
- 46.Xia, P., J. R. Gamble, L. Wang, S. M. Pitson, P. A. Moretti, B. W. Wattenberg, R. J. D'Andrea, and M. A. Vadas. 2000. An oncogenic role of sphingosine kinase. Curr. Biol. 10:1527-1530. [DOI] [PubMed] [Google Scholar]
- 47.Zhang, L., and M. H. Hanigan. 2003. Role of cysteine S-conjugate beta-lyase in the metabolism of cisplatin. J. Pharmacol. Exp. Ther. 306:988-994. [DOI] [PubMed] [Google Scholar]