

Significance
Therapies that harness the immune system have recently been approved for cancer treatment. Identification of biomarkers to monitor or predict patients’ responses to immunotherapies would help guide treatment decisions. Herein we analyzed changes in peripheral blood T cells from lung cancer patients receiving immunotherapy blocking the PD-1 inhibitory pathway. We detected CD8 T-cell responses following treatment in most patients. In addition, our data suggest that an increase in proliferation of PD-1+ CD8 T cells in the blood within 4 wk of treatment initiation may be associated with positive clinical outcome. Our analysis provides valuable insights into cancer patients’ responses to PD-1–targeted therapies and warrant further studies on peripheral blood biomarkers.
Keywords: PD-1, cancer immunotherapy, T-cell exhaustion, CD8 T cells, checkpoint inhibition
Abstract
Exhausted T cells in chronic infections and cancer have sustained expression of the inhibitory receptor programmed cell death 1 (PD-1). Therapies that block the PD-1 pathway have shown promising clinical results in a significant number of advanced-stage cancer patients. Nonetheless, a better understanding of the immunological responses induced by PD-1 blockade in cancer patients is lacking. Identification of predictive biomarkers is a priority in the field, but whether peripheral blood analysis can provide biomarkers to monitor or predict patients’ responses to treatment remains to be resolved. In this study, we analyzed longitudinal blood samples from advanced stage non–small cell lung cancer (NSCLC) patients (n = 29) receiving PD-1–targeted therapies. We detected an increase in Ki-67+ PD-1+ CD8 T cells following therapy in ∼70% of patients, and most responses were induced after the first or second treatment cycle. This T-cell activation was not indiscriminate because we observed only minimal effects on EBV-specific CD8 T cells, suggesting that responding cells may be tumor specific. These proliferating CD8 T cells had an effector-like phenotype (HLA-DR+, CD38+, Bcl-2lo), expressed costimulatory molecules (CD28, CD27, ICOS), and had high levels of PD-1 and coexpression of CTLA-4. We found that 70% of patients with disease progression had either a delayed or absent PD-1+ CD8 T-cell response, whereas 80% of patients with clinical benefit exhibited PD-1+ CD8 T-cell responses within 4 wk of treatment initiation. Our results suggest that peripheral blood analysis may provide valuable insights into NSCLC patients’ responses to PD-1–targeted therapies.
High PD-1 expression is a hallmark of exhausted T cells generated in cancer and chronic infections due to persistent antigen stimulation (1–4). PD-1 is induced by T-cell receptor (TCR) signaling, and when PD-l binds to its ligands (PD-L1 or PD-L2), it inhibits TCR/CD28 signaling and T-cell activation (5–7). Blockade of the PD-1 pathway reinvigorates exhausted T cells and can restore antitumor or antivirus immune responses. Antibodies that block the PD-1 pathway have demonstrated antitumor activity in cancer patients, and are now an approved treatment of several different cancers (8).
Although a significant number of cancer patients benefit from PD-1 blockade therapy, many fail to have clinical responses. How PD-1 blockade modulates the immune system in cancer patients is only partially understood, and there is an urgent need to uncover factors that determine clinical responses to this therapy. PD-L1 expression on tumor cells and/or on hematopoietic cells infiltrating the tumor has been associated with clinical responses to PD-1–targeted therapies in different studies (9–11). In advanced melanoma patients, the CD8 T-cell density in tumor biopsies was the best predictor of clinical response to PD-1–targeted therapies, although tumor CD8 T-cell infiltration was also associated with PD-L1 expression in the tumor environment (12). Further analysis of pre- and posttreatment tumor biopsies will certainly reveal additional fundamental aspects about responses to immunotherapies. Nonetheless, there are limitations to tumor site analysis, especially in patients with visceral tumors.
We hypothesized that peripheral blood analysis would provide insights about the immune responses induced by therapies blocking the PD-1 pathway. Peripheral blood analyses are easier to perform, can be repeated at several time points, and may provide a more systemic view of the immune response. In this study we evaluated changes in peripheral blood T cells in 29 non–small cell lung cancer (NSCLC) patients receiving PD-1–targeted therapies.
Results
Proliferation of T Cells in Peripheral Blood of NSCLC Patients After PD-1–Targeted Therapy.
To have a better understanding of the mechanisms involved in clinical responses to PD-1 pathway blockade in cancer patients, we monitored T-cell activation in the blood of advanced-stage NSCLC patients receiving PD-1–targeted therapies. We analyzed 29 NSCLC patients that received either anti–PD-L1 or anti–PD-1 blocking antibodies. A summary of the patients’ characteristics is described in Table S1. To monitor T-cell responses to PD-1–targeted therapies, we isolated peripheral blood mononuclear cells (PBMCs), and T cells were analyzed by flow cytometry. Blood samples were obtained at baseline (before treatment initiation) and before each treatment cycle (Fig. 1A). Treatment cycles were scheduled according to the clinical trial, approved drug interval, or health condition of each patient. We analyzed CD8 and CD4 T cells, and intracellular Foxp3 expression was used to discriminate between conventional CD4 T cells (CD4 T cells) and regulatory CD4 T cells (Treg cells; Fig. 1B). To assess T-cell activation, we examined intracellular expression of Ki-67, a cell-cycle marker expressed by cycling or recently divided cells (Fig. 1C) (13). Similar to healthy subjects, Treg cells presented the highest frequency of Ki-67+ cells compared with conventional CD4 T cells or CD8 T cells in baseline samples from NSCLC patients. Because the baseline frequency of Ki-67+ T cells was variable between patients and T-cell subsets, to study the effect of PD-1–targeted therapy on NSCLC patients’ T cells, we calculated the fold increase from baseline in the frequency of Ki-67+ T cells at all posttreatment time points available (2–12 wk post treatment initiation). The highest fold increase from baseline is shown for each patient in Fig. 1D. With this analysis it became evident that the highest fold increase in Ki-67+ T cells occurred among CD8 T cells. As a control, we analyzed the degree of change in the proliferation of T cells in blood samples collected in 2-wk intervals from healthy volunteers, and only minimal differences were detected (Fig. S1). We conclude that the increase in Ki-67+ T cells in peripheral blood can be used to identify patients with CD8 T-cell responses elicited by PD-1–targeted therapy, and this induction of CD8 T-cell proliferation occurs in a large fraction of NSCLC patients (∼70% in this study).
Table S1.
Demographic and clinical characteristics of NSCLC patients on this study
| Patient characteristics | n = 29 |
| Median age (range) | 66 (48–76) |
| Sex, n (%) | |
| Male | 20 (69) |
| Female | 9 (31) |
| Race, n (%) | |
| Caucasian | 18 (62) |
| African-American | 11 (38) |
| Smoking status, n (%) | |
| Former | 23 (79) |
| Current | 2 (7) |
| Never | 4 (14) |
| Histology, n (%) | |
| Adenocarcinoma | 20 (69) |
| Squamous cell carcinoma | 7 (24) |
| Poorly differentiated | 1 (3.5) |
| Adenosquamous | 1 (3.5) |
| Stage at diagnosis, n (%) | |
| I–III | 10 (35) |
| IV | 19 (65) |
| Prior chemotherapy, n (%) | |
| One line | 13 (45) |
| Two lines | 8 (28) |
| Three or more lines | 4 (14) |
| Prior radiation, n (%) | 25 (86) |
| Prior surgery, n (%) | 11 (38) |
| Study drug, n (%) | |
| Nivolumab | 21 (73) |
| Pembrolizumab | 5 (17) |
| Atezolizumab | 3 (10) |
| Best disease response, n (%) | |
| Partial response (PR) | 12 (41) |
| Stable disease (SD) | 7 (24) |
| Progressive disease (PD) | 10 (35) |
Fig. 1.

Proliferation of CD8 T cells can be detected in the blood of NSCLC patients after PD-1–targeted therapies. (A) Study design. (B) Dot plots show summary of gating strategy. (C) Dot plots show frequency of Ki-67+ cells among CD8 T cells, Foxp3neg CD4 T cells, and Foxp3+ CD4 Treg cells at baseline (pre) and 3 wk after treatment initiation (post) for one representative patient. Gates were determined based on naïve T cells. (D) Graph shows best fold increase in Ki-67+ cells among different T-cell populations (post PD-1 therapy compared with baseline). A 1.5-fold threshold is shown as gray shaded area. Lines represent the median fold increase for each population; n = 29. Patients with ≥1.5-fold increase in Ki-67+ CD8 T cells after PD-1–targeted therapy are indicated by dashed red rectangle.
Fig. S1.
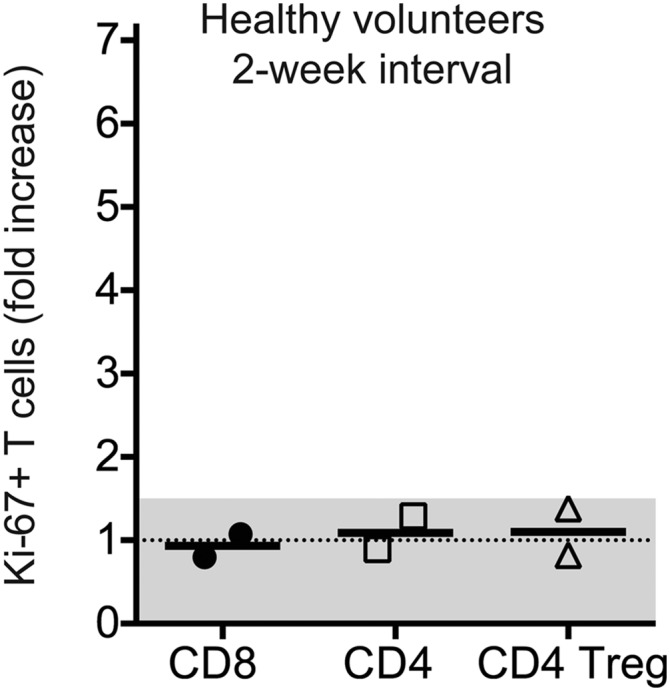
Minimal changes on Ki-67+ T cells in the peripheral blood of healthy volunteers. PBMCs were isolated consecutively from two healthy subjects recruited through Emory Vaccine Center. The frequency of Ki-67+ cells among CD8 T cells, Foxp3neg CD4 T cells, and CD4 Treg cells was assessed at baseline and after 2 wk. Graph shows the fold increase in Ki-67+ cells among the different T-cell populations. Lines represent the median fold increase for each population. A 1.5-fold threshold is shown as a gray shaded area.
It is interesting to note that overall there were no significant changes in the absolute count of white blood cells, lymphocytes, or neutrophils after PD-1–targeted therapies (Fig. S2). The lymphocyte counts were, in general, lower in NSCLC patients compared with hospital reference values. In addition, NSCLC patients had a lower frequency of naïve CD8 T cells compared with healthy subjects (Fig. S3A). These differences may be due to age or prior chemotherapy and/or radiotherapy treatment of NSCLC patients (14). However, PD-1 expression within different CD8 and CD4 T-cell subsets from NSCLC patients before PD-1–targeted therapy was comparable to healthy subjects (Fig. S3B) (15). We propose that peripheral blood monitoring can be used to study responses to PD-1–targeted therapies; however, it is imperative to perform a careful analysis with adequate markers to identify T-cell populations of interest.
Fig. S2.

Complete blood counts are not affected by PD-1–targeted therapy. (A) Graphs show white blood cell (WBC) counts, absolute neutrophil counts (ANCs), and absolute lymphocyte counts (ALCs) at baseline. Patients were grouped according to clinical outcome. Yellow shaded areas indicate range of reference values. (B) Graphs show changes in complete blood counts (WBC, ANC, and ALC) as fold change (posttreatment/pretreatment). Each line represents one patient, and colors indicate clinical outcomes (blue, partial response; gray, stable disease; red, disease progression). Shaded gray areas indicate the mean fold increase for all time points. P, patient.
Fig. S3.

Distribution of T-cell subsets and PD-1 expression on NSCLC patients and healthy subjects. Analysis was performed in whole blood obtained from NSCLC patients before PD-1–targeted therapy initiation. Healthy volunteers were recruited through Emory Vaccine Center. (A) Graphs show frequency of CCR7+CD45RA+ naïve (N), CCR7+CD45RAneg central memory (TCM), CCR7negCD45RAneg effector memory (TEM), and CCR7negCD45RA+ effector memory RA (TEMRA) on CD8 T cells (Left) and CD4 T cells (Right). (B) Graphs show frequency of PD-1+ T cells among the different subsets described in A, on CD8 T cells (Left) and CD4 T cells (Right). Gates were established based on naïve T cells. Lines represent the median. Unpaired Student t test.
CD8 T Cells That Proliferate After PD-1–Targeted Therapies Have an Activated Effector-Like Phenotype and Express PD-1.
We next sought to characterize posttreatment Ki-67+ CD8 T cells in patients that demonstrated ≥1.5–fold increase in CD8 T-cell proliferation following treatment initiation (n = 20, immunologically responsive patients highlighted by dashed red rectangle in Fig. 1D). Therapeutic anti–PD-1 administration hampers detection of PD-1 expression on peripheral blood cells by commercially available antibodies. Because both pembrolizumab and nivolumab are human IgG4 anti–PD-1, to detect PD-1–expressing cells in patients that received anti–PD-1 therapeutic antibodies, we performed an indirect anti–human IgG4 staining as previously described (9) (Fig. S4). In most patients, Ki-67+ CD8 T cells elicited after PD-1–targeted therapies were predominantly PD-1+ cells (Fig. 2 A and B). This result suggests that the increase in T-cell proliferation is a direct effect of blockade of the PD-1 pathway on PD-1–expressing CD8 T cells.
Fig. S4.

Detection of therapeutic anti–PD-1 antibodies by indirect staining. Dot plots show staining of PBMCs isolated from NSCLC P20 at baseline (pre) and 2 wk after the second infusion with nivolumab, a human IgG4 anti–PD-1 (post). Dot plots show CD8 and PD-1 (anti–PD-1, anti–human-IgG4, or anti–human-IgG4 + anti–PD-1) expression on CD3+ live cells. Combined anti–human-IgG4 + anti–PD-1 staining was used to analyze all posttreatment PBMC samples from NSCLC patients receiving therapeutic anti–PD-1. Data are representative of NSCLC patients receiving nivolumab or pembrolizumab.
Fig. 2.

Blockade of the PD-1 pathway induces proliferation of effector-like PD-1+ CD8 T cells. Analysis was performed at the best CD8 T-cell response time point on NSCLC patients with ≥1.5-fold increase in Ki-67+ CD8 T cells after treatment (n = 20). Gates were determined based on naïve CD8 T cells. (A) Graph shows frequency of PD-1+ cells among Ki-67+ CD8 T cells. Patients whose responding Ki-67+ CD8 T cells were mostly PD-1neg are indicated by gray circles. P, patient. (B) Dot plots show proliferation and PD-1 expression on CD8 T cells, on two representative patients with PD-1+ CD8 T-cell responses. (C) As in B, but depicted are two representative patients with PD-1neg CD8 T-cell responses. (D) Graph shows frequency of Bcl-2lo cells. (E) Dot plots show proliferation and Bcl-2 expression on responding PD-1+ Ki-67+ T cells (red dots) over the contour plot for total CD8 T cells on two representative patients. (F) Graph shows frequency of HLA-DR+CD38+ cells. (G) Dot plots show HLA-DR and CD38 expression as in E. (H) Graph shows frequency of CCR7neg CD45RAneg cells. (I) Dot plots show CCR7 and CD45RA expression as in E. (J) Graph shows frequency of granzyme B+ cells. (K) Dot plots show Ki-67 and granzyme B expression as in E. P, patient; C, treatment cycle.
Among patients with CD8 T-cell responses following PD-1–targeted therapy (n = 20), two patients demonstrated CD8 T-cell proliferation mostly among PD-1neg cells (P32 and P47) and three patients demonstrated a mixed response comprised of PD-1+ and PD-1neg proliferating CD8 T cells (Fig. 2 A and C). We next assessed phenotypic differences between PD-1+ and PD-1neg proliferating CD8 T cells. Several markers can be used to identify effector T cells that expand during immune responses—for example, yellow fever virus-specific effector CD8 T cells induced by vaccination are human leukocyte antigen–antigen D-related positive (HLA-DR+) CD38+ Ki-67+ Bcl-2lo (16, 17) (Fig. S5). To compare the phenotype of Ki-67+PD-1+ and Ki-67+PD-1neg CD8 T cells induced by PD-1–targeted therapy in NSCLC patients, we analyzed Bcl-2, HLA-DR, and CD38 expression. Bcl-2 is an antiapoptotic protein, highly expressed on naïve and memory T cells, but down-regulated in effector cells. PD-1+ Ki-67+ CD8 T cells had lower Bcl-2 expression than PD-1neg Ki-67+ counterparts (Fig. S6A). All responding Ki-67+PD-1+ CD8 T cells down-regulated Bcl-2 compared with naïve CD8 T cells (Fig. 2 D and E). Bcl-2 down-regulation is indicative of TCR engagement and effector cell differentiation on PD-1+ Ki-67+ CD8 T cells. Furthermore, expression of the activation markers CD38 and HLA-DR was more pronounced among PD-1+ Ki-67+ CD8 T cells than PD-1neg Ki-67+ counterparts (Fig. S6B). And most PD-1+ Ki-67+ CD8 T cells in patients with PD-1+ CD8 T-cell responses were positive for both CD38 and HLA-DR (Fig. 2 F and G). Consistent with effector phenotype, responding PD-1+Ki-67+ CD8 T cells lacked CD45RA and CCR7 expression (Fig. 2 H and I). To examine the cytotoxic potential of responding CD8 T cells, we analyzed intracellular granzyme B expression. Granzyme B was expressed on 25–80% of PD-1+ Ki-67+ responding CD8 T cells (Fig. 2 J and K), similar to PD-1+Ki-67neg or PD-1negKi-67+ CD8 T cells (Fig. S6C). Hence, PD-1–targeted therapies induce proliferation of effector-like PD-1+ CD8 T cells with the potential to exert cytotoxic activity.
Fig. S5.

Phenotype of effector CD8 T cells induced by vaccination with live attenuated virus. (A) Dot plot shows gating of yellow fever (YF)-specific CD8 T cells by tetramer staining 14 d after s.c. administration of 17D live-attenuated yellow fever vaccine strain. (B) Dot plots show phenotype of YF-specific CD8 T cells (blue dots). Underlying contour plots show phenotype of total CD8 T cells.
Fig. S6.

Characterization of peripheral blood CD8 T-cell following PD-1–targeted therapy of NSCLC patients. (A) Graph shows Bcl-2 expression levels on Ki-67+ CD8 T cells (comparison between PD-1neg and PD-1+). Paired Student t test. (B) Graph shows frequency of Ki-67+ CD8 T cells that coexpress HLA-DR and CD38 (comparison between PD-1neg and PD-1+). Paired Student t test. (C) Graph shows frequency of granzyme B+ cells among the indicated CD8 T-cell populations. Gates were determined based on naïve T cells. Repeated-measures ANOVA and Tukey’s multiple comparisons test. MFI, mean fluorescence intensity.
CD8 T Cells Responding to PD-1–Targeted Therapies Express Costimulatory Molecules and Coinhibitory Receptors.
We next assessed the expression of costimulatory molecules on peripheral blood CD8 T cells responding to PD-1–targeted therapy. Our previous study shows that in mice receiving PD-1 pathway blockade, expansion of PD-1+ CD8 T cells is contingent on CD28 expression and engagement. We also reported that PD-1+ CD8 T cells that proliferate in the blood of NSCLC patients following PD-1–targeted therapy expressed CD28 (18). Here we show that PD-1+ Ki-67+ responding CD8 T cells were not only CD28+, but also CD27+ (Fig. 3 A–C). In addition, inducible costimulator (ICOS) expression was also increased in PD-1+ Ki-67+ CD8 T cells relative to naïve (Fig. 3D). ICOS levels were higher on responding PD-1+ Ki-67+ CD8 T cells than PD-1+Ki-67neg or PD-1negKi-67+ CD8 T cells (Fig. 3 E and F). In summary, our data show that PD-1–targeted therapies in NCSLC patients induce proliferation of peripheral blood PD-1+ CD8 T cells with effector-like features, and these cells also express costimulatory molecules.
Fig. 3.

PD-1+ Ki-67+ CD8 T cells responding to PD-1–targeted therapies express costimulatory molecules. Analysis was performed at the best CD8 T-cell response time point, on NSCLC patients with ≥1.5-fold increase in Ki-67+ CD8 T cells after treatment; n = 18. Gates were determined based on naïve CD8 T cells. (A) Graph shows frequency of CD28+ cells. (B) Graph shows frequency of CD27+ cells. (C) Dot plots show CD28 and CD27 expression on responding PD-1+ Ki-67+ T cells (red dots) over the contour plot for total CD8 T cells on two representative patients. (D) Graph shows frequency of ICOS+ cells. (E) Graph shows ICOS mean fluorescence intensity (MFI) on the indicated CD8 T-cell populations. Repeated-measures ANOVA and Tukey’s multiple-comparisons test. (F) Dot plots show proliferation and ICOS expression as in C. P, patient; C, treatment cycle.
We also noticed that after treatment, PD-1+ Ki-67+ CD8 T cells expressed higher levels of PD-1 compared with nonproliferating PD-1+ Ki-67neg CD8 T cells (Fig. 4A). To further evaluate the expression of inhibitory molecules, we analyzed intracellular expression of cytotoxic T lymphocyte-associated protein 4 (CTLA-4). CTLA-4 is expressed by PD-1hi tumor infiltrating lymphocytes (TILs), and higher frequency of CTLA-4+ CD8 TILs has been recently associated with response to PD-1–targeted therapy in melanoma patients (19). As previously reported, unlike TILs, CTLA-4 can be hardly detected on peripheral blood total CD8 T cells (20), but there was significant CTLA-4 expression on PD-1+ Ki-67+ CD8 T cells after PD-1 blockade compared with naïve, nondividing PD-1+ or PD-1neg Ki-67+ CD8 T cells (Fig. 4 B–D). Up to 50% of responding CD8 T cells expressed CTLA-4, and it was most evident among cells with highest Ki-67 expression. High levels of PD-1 and increased CTLA-4 expression likely reflect increased TCR signaling due to the blockade of inhibitory PD-1 signals. Therefore, after PD-1–targeted therapy of lung cancer patients, in most subjects we can detect activated CD8 T cells in peripheral blood that have effector-like features and express both coinhibitory and costimulatory molecules.
Fig. 4.

Increased expression of PD-1 and CTLA-4 on PD-1+ Ki-67+ CD8 T cells responding to PD-1–targeted therapies. Analysis was performed at the best CD8 T-cell response time point, on NSCLC patients with ≥1.5-fold increase in Ki-67+ CD8 T cells after treatment; n = 18. Gates were determined based on naïve CD8 T cells. (A) Graph shows PD-1 MFI on PD-1+ CD8 T cells (Ki-67neg and Ki-67+). Paired Student t test. (B) Graph shows CTLA-4 MFI on the indicated CD8 T-cell populations. Repeated-measures ANOVA and Tukey’s multiple-comparisons test. (C) Graph shows frequency of CTLA-4+. (D) Dot plots show proliferation and CTLA-4 expression on responding PD-1+ Ki-67+ T cells (red dots) over the contour plot for total CD8 T cells on two representative patients. P, patient; C, treatment cycle.
Minimal Effects on EBV-Specific CD8 T Cells After PD-1–Targeted Therapy of NSCLC Patients.
There are no known antigens specific to NSCLC, and several reports now suggest that immunotherapies (blockade of CTLA-4 or PD-1 pathway) may rescue CD8 T cells recognizing mutation-derived neoantigens unique to each patient (21–25). Having established that PD-1–targeted therapies induce proliferation of PD-1–expressing CD8 T cells, we sought to evaluate whether PD-1–targeted therapies would activate PD-1+ CD8 T cells indiscriminately. To address this question, we assessed changes in EBV-specific PD-1+ CD8 T cells.
NSCLC patients with detectable EBV-specific CD8 T cells at baseline and presenting CD8 T-cell responses following PD-1–targeted therapy were further analyzed. EBV-specific PD-1+ CD8 T cells were present in patient 2 at baseline, and their frequency remained unchanged upon blockade of the PD-1 pathway (Fig. S7A, dot plots on the left). Intracellular Ki-67 staining confirmed that in contrast to EBV-tetramerneg PD-1+ CD8 T cells, EBV-specific CD8 T cells did not enter cell cycle (Fig. S7A, dot plots on the right). Likewise, the frequency of EBV-specific CD8 T cells was unaltered upon blockade of the PD-1 pathway in three other NSCLC patients (Fig. S7B). After PD-1–targeted therapy, despite activation of total CD8 T cells, there was minimal activation of EBV-specific CD8 T cells as assessed by HLA-DR and CD38 coexpression (Fig. S7C). Higher levels of activation (25% CD38/HLA-DR+) were observed in the patient with the highest levels of PD-1 expression among EBV-specific CD8 T cells (Fig. S7D). Thus, we conclude that PD-1–targeted therapies do not activate all PD-1–expressing cells in a similar manner. In this study, we did not discern potential differences among EBV-specific CD8 T cells that recognize latent or lytic epitopes, and we did not evaluate expression of EBV antigens in NSCLC patients. Cognate antigen levels, which govern PD-1 expression levels, most likely regulate T-cell activation by PD-1–targeted therapies; thus, the minimal effects observed on EBV-specific CD8 T cells on NSCLC patients may reflect low levels of EBV antigen expression. Uncovering factors that determine responsiveness of PD-1+ T cells to PD-1 pathway blockade should be a priority of future studies.
Fig. S7.

Minimal effects of PD-1–targeted therapies on EBV-specific PD-1+ CD8 T cells. (A, Left) Dot plots show PD-1 and EBV-TET staining on CD8 T cells. (A, Right) Dot plots show proliferation of PD-1+ EBV-TET+ CD8 T cells and PD-1+ EBV-TETneg CD8 T cells at baseline (pre) and at the best CD8 T-cell response posttreatment (post). Data from NSCLC patient 2. (B) Dot plots show EBV-TET staining on CD8 T cells at baseline (pre) and at the best CD8 T-cell response posttreatment (post). (C) As in B, but dot plots show CD38 and HLA-DR expression on EBV-specific CD8 T cells (blue dots) over the gray contour plot for total CD8 T cells. Numbers inside dot plots indicate the percentage of EBV-specific CD8 T cells that coexpress HLA-DR and CD38. (D) Dot plots show PD-1 expression on EBV-specific and total CD8 T cells at baseline. Data in B–D is from whole blood of three patients as indicated.
Associations Between CD8 T-Cell Responses and Clinical Outcomes.
Clinical responses to therapy are shown in a waterfall plot in Fig. S8A. Two patients with an unusual high frequency of Ki-67+ CD8 T cells at baseline are indicated by a dotted pattern in the waterfall plot. We noticed that these two patients had received radiation treatment within 4 wk of PD-1–targeted therapy initiation (Fig. S9), and therefore were excluded from our subsequent analysis on associations between CD8 T-cell responses and clinical responses to PD-1–targeted therapy. Fig. S8B shows that disease classification according to RECIST1.1 was consistent with patient survival. All patients with partial responses to PD-1–targeted therapy survived at least 1 y, whereas only 1 of 7 patients accounted with progressive disease survived 1 y after treatment initiation. Fig. 5A shows how tumor dimensions changed over time for each patient.
Fig. S8.

Clinical activity of PD-1–targeted therapies in NSCLC patients. (A) Waterfall plot shows maximum reduction from baseline (best response) in tumor burden assessed by RECIST 1.1. Highlighted by a dotted pattern are two patients (P30 and P44) who received previous treatment that may have contributed to tumor response: radiation treatment within 4 wk of PD-1–targeted therapy initiation. The +20% and −30% tumor burden changes are indicated by dashed lines (n = 29). (B) Survival curve of NSCLC patients according to RECIST 1.1 classification: disease progression (n = 7, no 12-mo follow-up data on P8, P24, and P32); stable disease (n = 7); partial response (n = 12).
Fig. S9.

Baseline proliferation of peripheral blood T cells on NSCLC patients. (A) Graph shows frequency of Ki-67+ cells among CD8 T cells, Foxp3neg CD4 T cells, and CD4 Treg cells at baseline. Gates were determined based on naïve T cells. Error bars show mean and SD. Outlier samples are indicated with patient number. (B) Graph shows frequency of Ki-67+ cells among CD8 T cells from patients grouped according to interval and type of previous treatments. Kruskal–Wallis test followed by Dunn’s multiple comparisons test.
Fig. 5.

Early proliferation of PD-1+ CD8 T cells is correlative to clinical outcomes. (A) Spiderplot shows changes from baseline in the tumor burden as assessed by RECIST 1.1 (n = 27). (B) Graphs show fold increase in the proliferation of CD8 T cells at all time points analyzed. Patients whose responding Ki-67+ CD8 T cells were mostly PD-1neg are indicated by a gray circle. (C) Graph shows frequency of PD-1+ cells among Ki-67+ CD8 T cells induced within 4 wk of PD-1–targeted therapy initiation (patients in B Upper). (D) Graphs show frequency of different clinical outcomes on patients that displayed early PD-1+ CD8 T-cell responses and patients that had absent, delayed, or PD-1neg CD8 T-cell responses.
Thus far we analyzed the best time point with regards to degree of increase in Ki-67+ CD8 T cells following PD-1–targeted therapy. However, it is important to consider that CD8 T-cell responses exhibited different kinetics. Fig. 5B shows longitudinal CD8 T-cell responses in individual patients. In most patients, CD8 T-cell proliferation was induced early, within 4 wk after treatment initiation (16 of 20 immunologically responsive patients). CD8 T-cell responses in peripheral blood were generally not sustained, detected at only one or two time points (2- to 3-wk interval). These findings may indicate that PD-1+ CD8 T cells activated by blockade of inhibitory signals expand, and can be detected cycling in peripheral blood for a few weeks following treatment, but then migrate to tumor/inflamed sites.
We next sought to determine whether CD8 T-cell responses might be associated with clinical responses. Our data show that 80% of NSCLC patients with partial clinical responses to PD-1–targeted therapy presented early proliferative CD8 T-cell responses in the blood. In contrast, when CD8 T-cell responses were only detected after 6 wk post treatment initiation (4 of 20 immunologically responsive patients), these patients did not show clinical responses (Fig. 5B, Lower). Among the 27 patients in this study for which we investigated associations between CD8 T-cell responses in peripheral blood and clinical responses, 7 patients (∼26%) did not show CD8 T-cell responses following PD-1–targeted therapy (Fig. 5B, Lower). Among patients classified as immunological nonresponders were also two patients with partial clinical responses. However, it is important to consider that failure to detect T-cell responses in peripheral blood may be due to nonoptimal timing of blood sampling. In summary, our data highlight that CD8 T-cell responses unleashed by blockade of the PD-1 pathway usually occur within the first 4 wk after treatment initiation and may be associated with positive clinical outcomes.
We next assessed whether differences in the phenotype of CD8 T cells activated at early time points after treatment initiation might differ according to clinical outcome. Whereas all patients that developed partial clinical responses had a high frequency of PD-1+ cells among Ki-67+ CD8 T cells (Fig. 5C), the frequency of PD-1+ among Ki-67+ CD8 T cells in patients that developed progressive disease was more variable. According to this observation and our previous findings regarding the difference in phenotype between PD-1+ and PD-1neg Ki-67+ CD8 T cells, we grouped the patients according to detection of early PD-1+ CD8 T-cell responses in the blood and found that 57% of those patients achieved a partial clinical response, and 21.5% had progressive disease. In contrast, among NSCLC patients with absent, delayed, or PD-1neg peripheral blood CD8 T-cell responses following PD-1–targeted therapy, 53.8% showed progressive disease and only 15.4% demonstrated a partial clinical responses (Fig. 5D). Thus, our data with this small cohort of NSCLC patients suggest that early PD-1+ CD8 T-cell responses following PD-1–targeted therapy might be associated with positive outcomes. These preliminary findings warrant further studies analyzing peripheral blood CD8 T-cell responses in larger cohorts of cancer patients receiving PD-1–targeted therapy.
Discussion
We analyzed longitudinal peripheral blood samples from advanced NSCLC patients receiving PD-1–targeted therapy (n = 29) and found an increase in cycling CD8 T cells following therapy in most patients (∼70%). Some effects were also observed in CD4 T cells, but the magnitude of the changes in CD8 T cells was more pronounced and we focused our analysis on responding CD8 T cells. Most of proliferating CD8 T cells were PD-1+ and had an effector-like phenotype. We suggest that peripheral blood CD8 T-cell responses following PD-1–targeted therapy may indicate activation of tumor-specific CD8 T cells. Tumor-specific CD8 T cells are enriched among PD-1+ cells (20, 26), and have been identified among peripheral blood PD-1+ CD8 T cells in melanoma patients (27). In addition we did not observe significant effects on PD-1+ EBV-specific CD8 T cells, thus not all PD-1–expressing cells are activated during PD-1 blockade therapy. Herein we propose that peripheral blood can be used to study T-cell responses induced by immunotherapy, and we suggest that T-cell responses detected in the peripheral blood of cancer patients may be relevant for antitumor effects.
CD8 T cells responding to PD-1–targeted therapy shared many features with effector cells elicited by vaccination with live attenuated viruses that cause acute infection (16, 17), such as low expression of Bcl-2, CCR7, and CD45RA, and high expression of HLA-DR, CD38, and ICOS. HLA-DR+ CD45RAneg and CCR7neg phenotype was also described for neoantigen-specific CD8 T cells that increased in frequency in the peripheral blood in one NSCLC patient with an exceptional response to PD-1–targeted therapy (21). We also found that many PD-1+ Ki-67+-responding CD8 T cells expressed granzyme B, indicative of cytotoxic potential. Similar to tumor-infiltrating CD8 T cells, peripheral blood CD8 T cells proliferating after PD-1–targeted therapies had high PD-1 expression and coexpressed CTLA-4. Higher levels of PD-1 and CTLA-4 expression are a result of T-cell activation (stronger TCR signaling due to therapeutic blockade of PD-1 inhibitory signals) and suggest that rescued CD8 T cells reduce their activation by coexpressing inhibitory receptors. Thus, therapies that block other inhibitory pathways may maximize CD8 T-cell activation when given in combination to PD-1–targeted therapy. Accordingly, in melanoma patients, combining PD-1–targeted therapies with CTLA-4 blockade has shown numerical higher response rates than PD-1–targeted monotherapy (28–30).
Most CD8 T-cell responses were detected in the peripheral blood within 4 wk of treatment initiation, and responding CD8 T cells expressed PD-1. Both PD-1 expression and early detection of expanding CD8 T cells are in accordance with our understanding of T-cell exhaustion and the PD-1 pathway. PD-1–targeted therapy operates by restoring preexisting T-cell responses that were suppressed by the PD-1 pathway. Hence, T cells rescued by blockade of the PD-1 pathway should express PD-1. However, because PD-1 expression is modulated by antigenic load, as tumor burden diminishes, tumor-specific CD8 T cells may become PD-1lo/neg. CD8 T cells cycling after blockade of the PD-1 pathway also expressed the costimulatory molecule CD28, as well as CD27 and increased levels of ICOS. We recently reported on the importance of CD28 engagement for expansion of PD-1+ CD8 T cells during PD-1–targeted therapy (18), but the role of other costimulatory molecules still needs to be addressed.
In mice, a particular subset of PD-1+ CD8 T cells possesses stem cell-like features and is responsible for CD8 T-cell expansion following PD-1 pathway blockade (31). Stem cell-like PD-1+ CD8 T cells can be identified by the transcription factor TCF-1 and have the capacity to differentiate into TCF-1neg PD-1+ CD8 T cells with effector like-features. Stem cell-like PD-1+ CD8 T cells have higher expression of costimulatory molecules and lower expression of inhibitory molecules than PD-1+ CD8 T cells that fail to expand. Preferential localization of stem cell-like PD-1+ CD8 T cells in lymphoid organs suggests that activation of PD-1+ CD8 T cells following PD-1–targeted therapy might occur in lymph nodes, by B7-expressing antigen-presenting cells. We hypothesize that following expansion in lymph nodes, CD8 T cells transition through the blood and migrate into inflamed/tumor site. CD8 T-cell expansion and migration results in higher number of tumor-specific CD8 T cells at the tumor site, consistent with observations reporting an increase in CD8 T-cell infiltration in early on-treatment tumor biopsies from melanoma patients that achieve clinical responses to PD-1–targeted therapy (12, 32, 33). Finally, therapeutic anti–PD-1 antibodies may also minimize inhibitory signals at the tumor site, allowing for CD8 T-cell effector function and cytotoxic activity against tumor cells.
Hence we propose that blockade of PD-1 inhibitory signals leads to expansion of tumor-specific PD-1+ CD8 T cells, resulting in transient detection of cycling PD-1+ CD8 T cells in peripheral blood, followed by an increase of tumor-specific effector PD-1+ CD8 T cells at the tumor site. According to this hypothesis, in our small cohort, we found an association between early PD-1+ CD8 T-cell responses and clinical outcome. Among 10 NSCLC patients with partial responses, 8 patients showed early PD-1+ CD8 T-cell responses following PD-1–targeted therapy. It is important to note that failure to detect cycling PD-1+ CD8 T cells in peripheral blood may be related to timing of the analysis. Clinical studies with weekly analysis of peripheral blood might be necessary to ensure adequate monitoring, especially in the first month of treatment. In addition, none of the patients presenting late CD8 T-cell responses achieved partial clinical responses (≥6 wk from treatment initiation). Late CD8 T-cell responses were only detected in a few patients, and might be unrelated to blockade of the PD-1 pathway. Of note, PD-1+ Ki-67+ CD8 T cells from patient 42 differed from other patients with regards to several markers analyzed (Figs. 2–4). P42 was on corticosteroids until 9 d after the first anti–PD-1 treatment cycle. In this patient there was also an unusual threefold increase in the absolute lymphocyte count accompanied by a marked increase in CD8 T-cell frequency (4 wk posttreatment). The strategy we describe here to study CD8 T-cell responses in peripheral blood following PD-1–targeted therapy in cancer patients poses both an advantage in terms of simplicity (and thus applications) but it also has limitations because unrelated events (e.g., corticosteroids, radiation treatment) can induce or affect T-cell responses. Careful phenotypic characterization of peripheral blood CD8 T cells combined to a comprehensive analysis of clinical history may allow discerning CD8 T-cell responses to PD-1 pathway blockade from unrelated causes.
In conclusion, we show that peripheral blood analysis can provide valuable insights into dynamic changes in T-cell responses during PD-1–targeted therapy of cancer patients. Our data suggest that early proliferation of PD-1+ CD8 T cells following PD-1–targeted therapy may be associated with clinical outcome; however, this association needs to be confirmed by larger studies. Our data warrants additional studies with detailed analysis of CD8 T-cell responses in peripheral blood during the first few weeks after PD-1 blockade. If these results are confirmed and extended, peripheral blood analysis may provide a valuable strategy to monitor early responses to PD-1–targeted therapy that may assist in the management of lung cancer patients.
Materials and Methods
Study Design and Participants.
Emory University institutional review board approval was obtained before donor enrollment, and written informed consent was obtained from all donors. We approached NSCLC patients treated at Winship Cancer Institute who were initiating therapy with anti–PD-1 or anti–PD-L1 blocking antibodies on clinical trials or as standard of care therapy to participate in our study. Blood samples were collected before infusion (baseline) and at treatment cycles (up to six) and at discontinuation. PBMC samples from healthy individuals or individuals that received a single dose of s.c. 17D live-attenuated yellow fever vaccine strain were used in some analysis as comparison with cancer patients. Additional details are provided in SI Materials and Methods.
Flow Cytometry.
PBMCs were isolated and after lysis of red blood cells, samples were stained with antibodies for analysis on LSR II flow cytometer (BD). Data were analyzed using Flow Jo software (Tree Star). Fold increase in proliferation was calculated by dividing the frequency of Ki-67+ T cells in posttreatment samples to the frequency of Ki-67+ T cells at baseline before treatment initiation. Additional details are provided in SI Materials and Methods.
Statistical Analysis.
Descriptive statistics were used to summarize data. Significance levels were set at 0.05 for all tests. Additional details are provided in SI Materials and Methods and figure legends.
SI Materials and Methods
Study Design and Participants.
Study subjects, NSCLC patients receiving anti–PD-1 or anti–PD-L1 blocking antibodies, consented to the collection and storage of archived tissue and blood while on treatment, and allowed review of their medical records for past medical history, cancer tumor type, toxicity assessments, clinical response, survival, and laboratory data. Collection of at least one posttreatment sample, in addition to baseline, was necessary to be included in our analysis. In addition, one patient that was diagnosed with Herpes Zoster during treatment was excluded from the study. Patients received anti–PD-1 therapy pembrolizumab/MK-3475 (2 or 10 mg/kg) or nivolumab (3 mg/kg), or anti–PD-L1 therapy MPDL3280A (atezolizumab, 1,200 mg) as i.v. infusion every 2–3 wk (one cycle) until disease progression or unacceptable side effect. Tumor size was measured by CT or MRI and evaluated for response using Response Evaluation Criteria in Solid Tumors 1.1 (RECIST 1.1). In brief, partial response (PR): at least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters; progressive disease (PD): at least a 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study). In addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm. Appearance of one or more new lesions is also considered progression. Stable disease (SD): neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum diameters while on study.
Flow Cytometry.
Peripheral blood samples were collected into cell preparation tubes (BD Biosciences) and PBMCs were isolated. Single-cell suspensions were obtained, and after lysis of red blood cells using ammonium-chloride-potassium buffer, PBMCs were immediately stained or frozen for posterior analysis. Samples were stained with anti-CD8 (RPA-T8), -CD4 (RPA-T4), -CD3 (UCHT1), -CD45RA (2H4LDH11LDB9), -CCR7 (150503), -PD-1 (EH12.2H7), -HLA-DR (l243), -CD38 (HIT2), -CD28 (CD28.2), -CD27 (323), -ICOS (ISA-3) -Bcl-2 (Bcl-2/100), -Ki-67 (B56), -CTLA-4 (BNI3), -granzymeB (GB11), Foxp3 (236A/E7). Antibodies were purchased from BD Bioscience, BioLegend, eBioscience, or Beckman Coulter. After anti–PD-1 treatment, PD-1 expression was detected by combining anti–human IgG4 biotin (HP-6025; Sigma) with anti–PD-1 antibody (conjugated to PE or BV421), followed by a secondary staining with streptavidin conjugated to PE or BV421. In patients treated with anti–PD-L1, PD-1 expression was detected with anti–PD-1 antibody. Live/dead cell discrimination was performed using fixable Yellow Dead Cell Stain Kit (Life Technology). Intracellular staining for Ki-67, CTLA-4, granzyme B, Bcl-2, and Foxp3 were performed using Foxp3 Fixation Kit (eBioscience). EBV-specific MHC Class I tetramers were made in house. EBV-specific tetramers staining was performed by using a mix of EBV tetramers HLA-A*0201: GLCTLVAML (from EBV-lytic protein BMLF1); HLA-B*0702:RPPIFIRRL (from EBV-latent protein EBNA3A), and HLA-B*0801:FLRGRAYGL (from EBV-latent protein EBNA3A). Some analysis where performed in whole blood as indicated in figures. After staining whole-blood samples with antibodies, lysis of RBC was performed with FACS lysing solution (BD). All samples were acquired with LSR II flow cytometer (BD) and analyzed using Flow Jo software (Tree Star). Gates for expression of different markers were determined based on naïve T cells (CD8+ CCR7+ CD45RA+ or CD4+ CCR7+CD45RA+).
Statistical Analysis.
Continuous variables were presented with mean with SD or median with range. Category variables were summarized as frequency and percentage. Paired t test and repeated-measures ANOVA were used in the longitudinal data analyses to compare different T-cell subsets within the same sample. Kruskal–Wallis test followed by Dunn’s test for multiple comparisons were conducted to compare the difference across independent groups. The survival estimates of different groups of patients were calculated with Kaplan–Meier method and presented graphically as survival curve. The SAS statistical package V9.4 (SAS Institute, Inc.) and Prism software (GraphPad Software) were used for data managements and statistical analyses. Significance levels were set at 0.05 for all tests.
Acknowledgments
This work was funded in part by the National Institutes of Health and the T. J. Martell Foundation.
Footnotes
Conflict of interest statement: R.A. is an inventor on patents held by Emory University that cover the topic of PD-1–directed immunotherapy. S.S.R. served on the scientific advisory board and received an honorarium from BMS, Merck, Genentech, and Astra Zeneca.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1705327114/-/DCSupplemental.
References
- 1.Zajac AJ, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallimore A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 4.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 5.Yokosuka T, et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hui E, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taube JM, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerdes J, et al. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 14.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol. 2015;194:4073–4080. doi: 10.4049/jimmunol.1500046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duraiswamy J, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol. 2011;186:4200–4212. doi: 10.4049/jimmunol.1001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JD, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Akondy RS, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183:7919–7930. doi: 10.4049/jimmunol.0803903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamphorst AO, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daud AI, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmadzadeh M, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gubin MM, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Rooij N, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31:e439–e442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. 2017;18:255–262. doi: 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gros A, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gros A, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22:433–438. doi: 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Postow MA, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Im SJ, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen PL, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ribas A, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4:194–203. doi: 10.1158/2326-6066.CIR-15-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]