

Abstract
We describe amphiphilic tri-block copolymers containing FeIII-catecholate complexes formulated as spherical- or cylindrical-shaped micellar nanoparticles (SMN and CMN respectively) as new T1-weighted agents with high relaxivity, low cytotoxicity, and long-term stability in biological fluids. Relaxivities of both SMN and CMN exceed those of established gadolinium chelates across a wide range of magnetic field strengths. Interestingly, shape-dependent behavior was observed in terms of the particles’ interactions with HeLa cells, with CMN exhibiting enhanced uptake and contrast via magnetic resonance imaging (MRI) compared with SMN. These results suggest that control over soft nanoparticle shape will provide an avenue for optimization of particle based contrast agents as biodiagnostics. We propose those polycatechol nanoparticles as suitable for pre-clinical investigations into their viability as gadolinium-free, safe and effective imaging agents for MRI contrast enhancement.
Keywords: polycatechol, micelles, melanin, shape, relaxivity
Graphical abstract
We describe amphiphilic tri-block copolymers containing FeIII-catecholate complexes formulated as spherical- or cylindrical-shaped micellar nanoparticles as new T1-weighted imaging agents with high relaxivity, low cytotoxicity, and long-term stability in biological fluids (serum and cell media). Relaxivities of both micellar nanoparticles exceed those of established gadolinium chelates across a wide range of applied magnetic field strengths.
1. Introduction
Magnetic resonance imaging (MRI) is a frequently used radiological imaging modality that has become increasingly important in the diagnosis of human disease.[1] It is noninvasive and versatile, does not use ionizing radiation, and can be acquired at high resolution for obtaining anatomical and functional information on soft tissues.[2] However, the low sensitivity inherent to MRI has led to the development of MRI contrast agents that increase sensitivity by catalytically shortening the transverse (T1) and longitudinal (T2) relaxation times of water protons. Contrast agents belong to two categories defined by those that enhance contrast in a T1- or T2-weighted MRI experiment. Whereas most T2-agents are represented by ferromagnetic inorganic nanoparticles,[3] the majority of T1-agents consist of small molecule paramagnetic complexes.[4] Among T1-agents, GdIII contrast agents are most widely used in clinical MRI due to their high relaxivities (1/T1), a manifestation of the short water residence times, long spin relaxation times, and seven unpaired electrons fostered by GdIII ions.[4] Nonetheless, concerns have arisen regarding the release and accumulation of toxic Gd ions in vivo. For example, it has been reported that the administration of Gd-based contrast agents to patients with renal dysfunction may induce the severe disease, nephrogenic systemic fibrosis.[5] An attractive non-toxic alternative to Gd-based, T1-agents are those that feature high-spin d5 FeIII metal centers which have been shown to have significantly higher relaxivity than many other ions including CuII.[6] In addition, iron-based contrast agents remain appealing as various small molecule FeIII complexes exhibit high biocompatibility and stability under physiological conditions.[7] Indeed, a number of iron chelates are proven therapeutics for the treatment of iron overload disease[8] and cancer.[9] In short, it is clear that, although effective, Gd-agents suffer from toxicity spurring a resurgence of interest in Gd-free MRI agents. Towards this goal, we have endeavored to explore nanoscopic materials incorporating multiple FeIII-based chelates as T1-weighted MRI contrast agents with high efficiency and inherently low toxicity.
The strong coordination bonds between FeIII and catecholic ligands have been utilized extensively by natural systems in a variety of functional small molecules and biomaterials.[10] For example, the high iron affinity observed for the siderophore entorbactin is provided by a tris-catecholate ligand architecture.[11] Similarly, the mussel byssal cuticle employs 3,4-dihydroxy-L-phenylalanine (DOPA)-FeIII complexes to provide strong, yet reversible crosslinking points, resulting in self-healing properties.[12] Interestingly, while small molecule ironIII tris-catecholate (FeIII(catecholate)3) complexes have been thoroughly explored as synthetic enterobactin mimics for iron sequestration, their potential to function as T1-weighted MRI contrast agents has been largely overlooked despite their modest relaxivities.[13] Whereas marked improvements in the relaxivity and targeting specificity for Gd-based contrast agents has been achieved through the incorporation of small molecule Gd-chelates into nanomaterial scaffolds,[14] similar advances with FeIII(catecholate)3-based materials is in its infancy. Although synthetic catechol-based polymers with FeIII have been widely described for a range of applications,[10, 15] very limited examples have been explored as T1-weighted MRI contrast agents. Indeed, the only system that has been studied are natural sepia melanins[16] and synthetic melanin-based materials in the form of polydopamines generated by oxidative polymerization.[17] Considering the many promising properties offered by poly(FeIII-catecholate) materials, including high stability, low toxicity, and improved relaxivity, we considered it timely to prepare nanostructures consisting of polycatechol for the development of new MRI contrast agents.
Current strategies for the construction of poly(FeIII-catecholate) as efficient T1-weighted imaging agents for MRI generally focus on the complexation of natural sepia melanin colloidal particles with FeIII salts.[16–17] However, the development of functional and robust contrast agents from melanin-type materials has been met with profound challenges. Issues include limited control over the synthetic colloidal chemistry hindering size and shape control over the resulting melanin particles.[15a] We believe that more sophisticated strategies making use of self-assembled soft materials from amphiphilic block copolymers,[18] with the integration of FeIII-catecholate blocks, may provide an avenue for preparing particles of well-defined and predictable morphologies. Certainly, these are undoubtedly critical, and highly desirable properties if one aims to prepare materials for in vivo use.[19] This would be enabled by the use of a controlled living polymerization method, giving rise to well-defined and reproducibly accessible block copolymer architectures. We propose that the resulting supramolecular nanostructures from such copolymers with rigorously controlled physical parameters (i.e. size, shape and composition) would represent a new class of macromolecular Gd-free T1-weighted MRI contrast agent. Control over these parameters is a must as it is widely known that the fundamental physical properties of nanoparticles may affect their behavior within biological systems.[20] In this work, we report our first effort towards this goal through the design and synthesis of polycatechol-based amphiphilic block copolymers and the elucidation of the relaxation properties of the resulting self-assembled micellar nanoparticles.
2. Results and Discussion
2.1. Molecular Design and Micellar Nanoparticle Formation
Our synthetic approach employed post-polymerization functionalization[18c, 18d] for the incorporation of multiple catechol groups localized in the middle block of a tri-block copolymer amphiphile (Figure 1). The macromolecular precursors, (OEG)m-(NHS)n-(C6)p, were directly synthesized via ring-opening metathesis polymerization (ROMP)[21] (Figure S1 and S2) using a modified 2nd generation Grubbs’ catalyst (Figure 1a). Excess dopamine hydrochloride was then added to the macromolecular precursors in the presence of N,N-Diisopropylethylamine (DIPEA) to afford the final products. Notably, a post-polymerization functionalization route was taken when it was discovered that catechol-modified norbornene monomers would not polymerize utilizing this class of initiator for ROMP.[15e] Regardless, this synthetic approach achieved a near quantitative incorporation of catechol groups into the middle region of the block copolymers as determined by the 1H NMR and 13C NMR (Figure S3–S6). Both of the resulting amphiphilic tri-block copolymers (Polymers 1 and 2) are solids with limited solubility in nonpolar organic solvents. By varying the segment size of each block in the amphiphilic copolymers to tune the volume fraction of hydrophobic domain, two kinds of stable micellar morphologies (i.e. sphere and cylinder) could be obtained (Figure 1b–g).
Figure 1.

General synthetic scheme for amphiphilic tri-block copolymers, and electron microscopy of the resulting micellar nanoparticles. (a) Polymer 1: m = 38, n = 34, p = 50. Polymer 2: m = 20, n = 23, p = 43; (b–d) Electron microscopy of SMN formed from Polymer 1: (b) cryo-TEM. (c) BF-STEM. (d) HAADF-STEM; (e-g) Electron microscopy of CMN formed from Polymer 2: (e) cryo-TEM. (f) BF-STEM. (g) HAADF-STEM.
The assembly of resulting catechol-based amphiphilic block copolymers was performed in a selective solvent to generate the two different micelles.[22] Specifically, to prepare a spherical micellar nanoparticle (SMN), an aqueous solution of FeCl3 (1 mg/mL) was added at a rate of 10 μL/hour to a vial containing 2 g of a stock solution of Polymer 1 in THF as a common solvent, with an initial concentration of 2.0 wt% until the final water content reached 70 wt%. The stable SMN were then obtained by dialyzing the micelle solution against deionized water for 3 days to remove the organic solvent and any unchelated FeIII ion. Cylindrical micellar nanoparticles (CMN) were generated using Polymer 2 which consists of a higher volume fraction of the hydrophobic domain, under precisely the same conditions.[22a]
Each of the two well-defined micellar morphologies were characterized by cryo-transmission electron microscopy (TEM) (Figure 1b and 1e) and dry state TEM (Figure S7), demonstrating diameters for SMN and CMN of approximately 30 nm and 25 nm, respectively. Although precise control over the micellar length of CMN is not possible,[23] it can be clearly observed that a majority of CMN possess long (> 1 μm) cylindrical lengths. The presence of high contrast metal elements (heavy nuclei) in both SMN and CMN was evident in bright field scanning transmission electron microscopy (BF-STEM) (Figure 1c and 1f) and high angle annular dark field (HAADF)-STEM (Figure 1d and 1g). Moreover, selected area BF-STEM coupled with energy dispersive X-ray spectroscopy (EDS) confirmed the presence of metal ions localized inside the micellar nanoparticles (Figures S8 and S9). Specifically, the EDS profiles suggested that the content of Fe in the testing areas of SMN and CMN were significantly higher than those on the grid surface background, which are in good agreement with the elemental mapping analysis results (Figure S8a and S9a). Furthermore, TEM was used to confirm that both SMN and CMN are stable in aqueous solution for at least 6 months (Figure S10).
2.2. Relaxometric Characterization of Micellar Nanoparticles
The basic relaxation properties of SMN and CMN were first investigated using their 1H 1/T1 nuclear magnetic relaxation dispersion (NMRD) profiles acquired under magnetic field strengths from 0.01 MHz to 70 MHz (Figure 2). Inductively coupled plasma-optical emission spectrometry (ICP-OES) was employed to calibrate the Fe concentration of SMN and CMN solutions. The NMRD profiles show a similar shape and different amplitude. In both cases there is a poorly defined plateau at low fields (ca. 0.01–0.05 MHz), followed by a wide dispersion (approx 0.05–7 MHz) and by a broad hump at higher frequencies. The ratio of the relaxivity values at low (0.01 MHz) and high fields (60 MHz) amounts to 1.6 and 1.4 for CMN and SMN, respectively. Relaxivity, r1, arises from metal-bound and/or proximate hydrogen-bonded water molecules, dipolarly interacting with the unpaired electrons of the metal ion:
| (1) |
where fM is the mole fraction of interacting water molecules, T1M their proton relaxation time due to the paramagnetic FeIII ion, μM the exchange lifetime and r1os the contribution of outer-sphere fast diffusing water protons.[24] T1M depends on r6, the distance of the interacting dipoles, on the correlation time (μC) for the proton-electron dipolar interaction and on ωI and ωS, the proton and electron Larmor frequencies, respectively. The inflection point in the profiles reflects the condition ωSμC = 1, so that we can estimate a value of approximately 0.4–0.6 ns for the correlation time. The appearance in both profiles of broad humps at high fields with relaxivity values well above 6 mM−1s−1 represents a strong indication that the relaxivity is not dominated by a simple outer-sphere mechanism. Although this high relaxivity might originate from the presence of inner-sphere water interactions afforded by dynamic FeIII(catecholate)2(H2O)2 moieties, we speculate the enhanced relaxivity may largely originate from the presence of one or more second-sphere interactions to a coordinatively saturated, six-coordinate, FeIII(catecholate)3,[25] similar to those observed for small molecule FeIII(catecholate)3 complexes.[13] These second-sphere interactions likely take the form of dynamic hydrogen bonds between H2O and polar groups on the nanoparticle proximal to FeIII(catecholate)3 sites, or to oxygen atoms of the FeIII(catecholate)3 sites themselves. Indeed, several structurally characterized FeIII(catecholate)3 complexes have been reported that feature hydrogen bonds between oxygen atoms of the FeO6 core and polar H-X units (X = O, N) (see five examples from Cambridge Structural Database listed in Figures S11–S15). Moreover, direct X-ray crystallographic evidence for hydrogen bonding between the hydrogen atoms of water and the oxygen atoms of a transition-metal catecholate has been observed in both Ni[26] and Mn[27] complexes, thereby suggesting that such second-sphere H2O interaction modes are viable in these nanoparticle systems.
Figure 2.
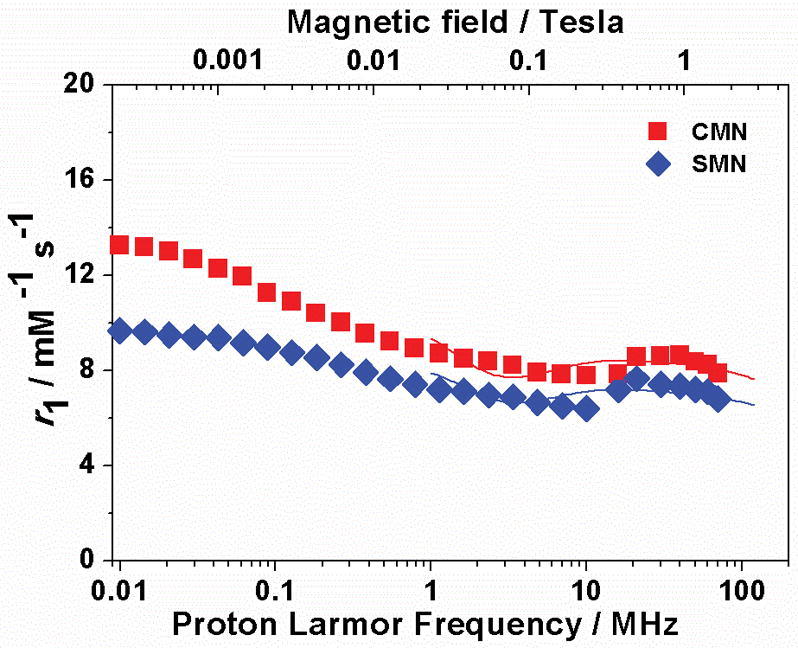
1H NMRD profiles for SMN and CMN at 298K.
Therefore, based on this precedent, the data were fit to calculate rotational correlation times of 490 and 448 ps for CMN and SMN respectively, assuming the presence of two second-sphere water molecules at a long-range distance of 3.3±0.1 Å from the Fe center with a residence lifetime of ca. 2 μs[28] (Table 1, details of the best-fit parameters can be found in the SI). These results are in good agreement with similar findings reported for bovine lactoferrin[28] and methemoglobin.[29] In addition, both SMN and CMN exhibit substantially higher r1 than that of mononuclear FeIII(catecholate)3 complexes (e.g. at a field of 20 MHz, r1 values for SMN and CMN are 8.0 mM−1s−1 and 9.0 mM−1s−1, respectively, while r1 for small molecule FeIII(catecholate)3 complexes are approximately 2.0 mM−1s−1),[13c] indicating that the macromolecular scaffolds effectively increase the relaxivity of contrast agent moieties by restricting the rotational mobility of the complex (i.e. of the vector connecting FeIII and the protons of second sphere water molecules).[30] Moreover, the per FeIII r1 of SMN and CMN outperforms clinically used GdIII contrast agents across a wide range of applied magnetic field strengths (> 10 MHz) (see SI). For example, measured enhancements over Gd-DOTA are +100% and +69% for CMN and SMN, respectively, at 1.0 T and 298 K (Figure S16).
Table 1.
| Micelles | 20r1 (mM−1 s−1)a | Δ2 (1019 s–2)b | μV (ps)b | μR (ps) | r (Å) | qc | μM (μs) |
|---|---|---|---|---|---|---|---|
| SMN | 7.7 | 1.4 ± 0.1 | 54 ± 3 | 448 ± 8 | 3.3 ± 0.1 | 2 | 2.0 ± 0.1 |
| CMN | 8.6 | 1.1 ± 0.2 | 50 ± 4 | 490 ± 12 | 3.3 ± 0.1 | 2 | 1.9 ± 0.2 |
20 MHz and 298 K;
the parameters for electronic relaxation are used as empirical fitting parameters and do not have a real physical meaning for slowly tumbling nanosized systems. Low-field data, those most affected by electronic relaxation, were not included in data analysis;
fixed during the fit.
It should be noted that NMRD profiles are fitted herein only in the high field region because of the known limitations of Solomon-Bloembergen-Morgan (SBM) theory in the slowly rotating regime that render it unable to completely account for the behavior of slowly rotating systems at very low magnetic field strengths, where the Zeeman energy is smaller than the zero field splitting energy.[31] In addition, it is worth mentioning that the deviation of the profiles from the behavior of a well-defined Lorentzian might be compatible with the presence of different, non-equivalent FeIII ions in the nanoparticles.[13b] The NMRD profiles may reflect different structures and dynamics (number, distance and lifetime) of second sphere water molecules around the Fe centers, which have characteristic and different electronic relaxation times.[3b]
The longitudinal and transverse relaxation times (T1 and T2) of both nanoparticles, with various concentrations, at clinically relevant field strengths (Bo = 1.41 T) were measured using time-domain NMR to quantitatively calculate their relaxivity values (r1 and r2) (Figure 3 and Figure S17). Both SMN and CMN exhibit high relaxivity values (r1,SMN = 7.1 mM−1s−1, r1,CMN = 7.9 mM−1s−1) for potential clinical use (based on the calculated results shown in Figure 3a, 3b, Figure S17 and Table 2).[17a] In summary, the excellent contrast enhancement of the FeIII- chelated micellar nanoparticles was mainly attributed to second-sphere contributions, as discussed above. Moreover, the low r2/r1 ratios of both SMN and CMN (r2/r1 = 1.25 for SMN, and r2/r1 = 1.40 for CMN) favor positive contrast enhancement (brightening) because the interference from T2 effects (darkening) are relatively small. Overall, the high T1 relaxivity and low r2/r1 ratio of both micellar nanoparticles could make them suitable as Gd-free clinical contrast agents for T1-weighted MRI. This conclusion is supported by the bright T1-weighted MR images from SMN and CMN in aqueous solution (Figure 3c).
Figure 3.

MRI characterization of micellar nanoparticles. (a) Plots of 1/T1 vs FeIII concentration for SMN in different medium with calculated r1 (blue plot: SMN in water, red plot: SMN in freshly prepared FBS, and green plot: SMN in FBS for 3 days); (b) Plots of 1/T1 vs FeIII concentration for CMN in different medium with calculated r1 (blue plot: CMN in water, red plot: CMN in freshly prepared FBS, and green plot: CMN in FBS for 3 days); (c) T1-weighted MR images captured on a Bruker 7.0 T magnet from free FeIII, SMN and CMN in different media ([FeIII] is at 0.6 mM in each tube)
Table 2.
Relaxivity Data for Micellar Nanoparticles in Different Media.*
| Micelles | Media | r1 (mM−1s−1) | r2 (mM−1s−1) | r2/r1 |
|---|---|---|---|---|
| SMN | H2O | 7.1 | 8.9 | 1.25 |
| CMN | H2O | 7.9 | 11.1 | 1.40 |
| SMN | FBS (fresh) | 7.3 | 10.2 | 1.40 |
| CMN | FBS (fresh) | 7.8 | 10.6 | 1.36 |
| SMN | FBS (3 days) | 8.9 | 10.4 | 1.17 |
| CMN | FBS (3 days) | 8.8 | 13.3 | 1.51 |
The magnetic filed strength for T1 and T2 measurement is 1.41 T.
Variable field and temperature magnetic measurements were performed using a superconducting quantum interference device (SQUID) to confirm that the observed MRI contrast arises from multiple isolated FeIII-catecholate sites. Plots of magnetization versus applied magnetic field show significant curvature only at temperatures less than 24 K, consistent with isolated paramagnetic iron centers (Figure 4). The lack of magnetic saturation, even at 7 T and 2 K, indicates a significant magnetic anisotropy associated with the spin state and coordination environment of the FeIII; however, quantitative fitting of the data was not possible. This indicates that a range of FeIII environments may be present within SMN and CMN micelles, as suggested by the broad NMRD profiles. At temperatures exceeding 24 K, the magnetization is completely linear with the applied field, confirming that the MRI contrast ability of SMN and CMN results from many isolated FeIII-catecholate complexes inside the micellar nanoparticles.
Figure 4.

Magnetization data collected as a function of applied field for (a) SMN and (b) CMN from 2-300 K.
2.3. Long-Term Stability of Micellar Nanoparticles
The stability of SMN and CMN in serum was also examined in two respects: 1) No collapse or change of the micellar nanoparticle morphologies were observed over time; 2) The total metal ion content and relaxivity values of the FeIII-chelated nanoparticles does not change significantly over time. With regards to morphological stability, we propose that although many previously established micelle-based therapeutic systems have suffered from an inherent instability in vivo (generally undergoing collapse or break-up in the presence of serum lipids and proteins),[32] the micellar particles described here benefit from cross-linking networks of individual polymer chains in the micellar interface via the formation of interchain, multiple FeIII-catecholate coordination bonds.[12, 33] These crosslinks lead to the particles being stable for more than 20 days in fetal bovine serum (FBS, 100%) even at a high concentration (>20 mg/mL). With regards to the strong binding capacity and affinity of catechols for FeIII,[10] we performed a stability assay of FeIII-chelated SMN and CMN in PBS solution to show that there was no iron release from micellar nanoparticles at different incubation time points (Figure S18). It can be clearly seen that the metal ion content of SMN and CMN in PBS are approximately 100% after 7 days. Accordingly, the r1 and r2 values of both SMN and CMN after 3 days incubation in FBS are almost identical to the ones corresponding to freshly-prepared micellar nanoparticles (Figure 3a, 3b, Figure S17 and Table 2), which is in good agreement with that observed in T1-weighted MR images (Figure 3c). The very slight increase in relaxivity values of SMN and CMN might be due to interactions between nanoparticles and proteins in FBS.[34] The intensity of bright MR images for several SMN and CMN samples in different media with various incubation times is also found to be very similar. However, the MRI signal from free FeIII was observed to be immediately quenched in FBS (Figure 3c), likely due to the participation of free ions in redox reactions in the biological fluid.[35] This result also suggests that metal ions were not leaking from the nanoparticles after the 3 day incubation in serum, as the MR images do not lose intensity over that time frame (Figure 3c). We conclude that the materials exhibit promising stabilities in biological media.
2.4. Shape-Dependent MR Imaging of Micellar Nanoparticles in HeLa Cells
Prior to investigating the MR imaging performance of the micellar nanoparticles in HeLa cells, their cytotoxicity at various concentrations of FeIII was assessed using a CCK-8 (cell counting kit-8) assay. Similar to FeIII-chelated melanin colloidal nanoparticles [17a] and many other kinds of polycatechol-based biomaterials,[15a, 36] both SMN and CMN also show high biocompatibility and promisingly low toxicity with respect to live cells. Cell viability was measured in HeLa cells using micellar nanoparticles with various dosages from 0.5 μM to 100 μM FeIII for 24 h and 48 h incubation. Under these conditions, cell viability was maintained at approximately 100% in all groups (Figure S19).
Next, SMN and CMN, with identical [FeIII] concentrations (67.5 μM), were incubated with HeLa cells for different periods of time. Quantitative analysis of cellular iron uptake and the corresponding T1 relaxation values were measured by ICP-OES and MRI respectively (Figure 5). HeLa cells incubated with SMN and CMN exhibited enhanced positive contrast in T1-weighted MR images compared with control cells (incubation times: 24 h or 48 h) (Figure 5a). Surprisingly, it was found that T1-weighted MR images of HeLa cell pellets incubated with CMN exhibited much stronger T1 signal enhancement (shorter T1 relaxation time) compared with those incubated with SMN after short incubation times (4 h or 12 h) (Figure 5a). Quantitative analysis of intracellular iron indicated cell uptake (Figure 5b and 5c) was indeed shape- and time-dependent (Figure 5d). For CMN, cell uptake significantly increased in the first 4 h, then slowed, reaching a plateau at 12 h, which is in good agreement with the T1 values of each corresponding cell pellet. By contrast the intracellular FeIII content of SMN gradually increased without saturation over a period of 48 h (Figure 5d), matching the decreasein T1 values of the cell pellets over the incubation time. Additionally, MR imaging of CMN-treated HeLa cells exhibits brighter positive contrast and shorter relaxation times than SMN-treated cells at identical initial [FeIII] concentrations and incubation times, particularly when short incubation times (i.e. 4 h) were employed.
Figure 5.

(a) In vitro T1-weighted MR images of HeLa cells incubated with SMN and CMN ([FeIII] is 67.5 μM) for different periods of time; TEM image of SMN (b) and CMN (c), trapped inside vesicles of HeLa cells (see Figure S20 for additional cellular TEM images); (d) Quantitative determination of intracellular FeIII content (per 106 cells) for HeLa cells incubated with SMN and CMN for different periods of time and their corresponding T1 relaxation values.
In terms of mechanism, it is possible that these differences are explained by the fact that CMN is capable of making multiple contacts with the cell surface providing an initially stronger association leading to faster and more efficient uptake. Indeed, in this context it is important to note that various observations have been made concerning shape-dependent polymeric nanoparticle cell internalization.[20b, 37] However, general trends and mechanisms are yet to be elucidated, as uptake efficiencies vary by nanoparticle composition, flexibility, surface charge, overall dimensions and aspect ratio, and other confounding factors.[20b, 37–38] In this study, poly(FeIII-catecholate)-based nanoparticles were internalized into HeLa cells to a higher extent when they were of a cylindrical morphology than when they were in the form of spherical particles (Figure 5b and 5c). A similar trend was also reported for shell-crosslinked spherical and cylindrical micelles,[39] as well as polymer brush-based spherical and long rodlike nanostructures.[38] Considering that shape not only plays an important role in cell internalization, but can also be a determining factor in overall biodistribution patterns in vivo (i.e. blood circulation and extravasation),[20a, 38] these preliminary results introduce exciting opportunities for optimization and tunability in the design of self-assembled nanoparticles. This highlights the power of the approach for the preparation of nanoparticles from well-defined polymers generated utilizing a living polymerization. Indeed, as a demonstration of this concept, the approach taken here provides direct access to two well-defined morphologies of polycatechol-based nanoparticle, not easily achievable utilizing analogous synthetic melanin colloidal nanoparticles prepared by oxidative polymerization.
3. Conclusion
In summary, we have developed a new class of efficient and biocompatible MRI contrast agents based on micellar nanoparticles formed from amphiphilic poly(FeIII-catecholate)-based copolymers. Compared with recently reported natural or synthetic melanin-based T1 agents, our approach utilizes well-defined tri-block copolymers prepared via a controlled living polymerization method. This synthetic route gives access to a tunable polymer system and hence, differently shaped self-assembled nanoparticles with controlled physical parameters. These nanoparticles have the potential to be used for various applications in diagnostic radiology and imaging, due to their enhanced relaxivity, and long-term stability in biological media. Moreover, we further demonstrate that the resulting nanoparticles provide enhanced positive contrast for MR imaging in HeLa cells. Notably, we observed shape-dependent behavior in terms of cellular uptake with cylindrical micelles exhibiting brighter contrast and shorter relaxation times than the analogous spherical micelles. We present this study with the goal that it stimulates further investigation of polycatechol nanoparticles in terms of their potential as Gd-free MRI contrast agents. Work in our own laboratory on shape-dependent behavior in vivo is underway with respect to both intraperitoneal and intravenous delivery of polycatechol nanoparticle-based contrast agents.
4. Experimental Section
Monomer and Polymer Synthesis and Characterization
All chemicals were purchased from commercial sources and used without further purification, unless otherwise indicated. Anhydrous toluene and dichloromethane were purified using a Dow-Grubbs two-column purification system (Glasscontour System, Irvine, CA).[40] (IMesH2)(C5H5N)2(Cl)2Ru=CHPh was prepared as described by Sanford et al.[41] Monomers 1, 2, 3 were synthesized as previously reported.[18a, 42] Polymerizations were performed under a dry dinitrogen atmosphere with anhydrous, degassed solvents in a glove box.
1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Varian Mercury Plus spectrometer. Chemical shifts (1H) are reported in δ (ppm) relative to the C5D5N residual proton peaks (δ 7.22, δ 7.58, and δ 8.74 ppm). Chemical shifts (13C) are reported in δ (ppm) relative to the C5D5N carbon peaks (δ 123.87, δ 135.91, and δ 150.35 ppm). All 13C NMR spectra were proton decoupled. Mass spectra were obtained at the UCSD Chemistry and Biochemistry Molecular Mass Spectrometry Facility. Polymer dispersities and molecular weights were determined by size-exclusion chromatography (Phenomenex Phenogel 5 μ 10, 1 K-75 K, 300 x 7.80 mm in series with a Phenomex Phenogel 5 μ 10, 10 K-1000 K, 300 x 7.80 mm (0.05 M LiBr in DMF) using a Shimatzu pump equipped with a multi-angle light scattering detector (DAWN-HELIOS: Wyatt Technology) and a refractive index detector (Optilab T-rEX: Wyatt Technology) normalized to a 30,000 MW polystyrene standard using dn/dc of 0.100 for all the polymers.
Micelles Preparation and Characterization
The samples were first dissolved in THF as the common solvent and stirred at room temperature overnight to ensure complete dissolution of the polymer to prepare a stock solution with an initial concentration of 2 wt%. The solution was then filtered through a filter of 0.22 μm pore size to remove any dust. FeCl3 solution (1 mg/mL) was filtered through a filter of 0.22 μm pore size and added dropwise at a rate of 10 μL/hour using a syringe pump into a vial containing 2.00 g of the stock solution. FeCl3 solution addition was continued until reaching a final water content of 70 (wt)%. Then the micelle solution was dialyzed against deionized water for three days to remove the common solvent, excess FeIII and fix the micellar morphology.
SnakeskinTM dialysis tubing was purchased from Thermoscientific, Inc. with a molecular weight cut off (MWCO) of 10,000 g/mol. Transmission Electron Microcopy (TEM) was performed on a FEI Sphera microscope operating at 200 keV. TEM grids were prepared by depositing small (3.5 μl) aliquots of sample onto grids (~ 2 min, Formvar stabilized with carbon (5–10 nm) on 400 copper mesh, Ted Pella Inc.) that had previously been glow discharged using an Emitech K350 glow discharge unit and plasma-cleaned for 90 s in an E.A. Fischione 1020 unit. Micrographs were recorded on a 2 K X 2 K Gatan CCD camera.
Cryo-TEM experiments were also performed on a FEI Sphera microscope operating at 200 keV. TEM grids were prepared by depositing small (3.5 μL) aliquots of sample onto grids (Quantifoil R2/2 holey carbon)that had previously been glow discharged using an Emitech K350 glow discharge unit and plasma-cleaned for 90 s in an E.A. Fischione 1020 unit. Sample was loaded onto the grids at 4 °C, blotted with filter paper to create a thin film on the grid, then plunged into liquid ethane and transferred into a precooled Gatan 626 cryo-transfer holder, which maintained the specimen at liquid-nitrogen temperature in a FEI Sphera microscope operated at 200 keV. Micrographs were recorded on a 2 K X 2 K Gatan CCD camera.
STEM and STEM-EDS analysis were acquired on a JEOL JEM 2100F TEM equipped with an INCA (Oxford) EDS detector at the NanoScale Fabrication and Characterization Facility (NFCF), Peterson Institute of Nanoscience and Engineering (PINSE), University of Pittsburgh, PA. Samples were prepared by drop-casting 5 μL of sample onto TEM grids (ultrathin 5 nm A-type carbon with 400 mesh copper, Ted Pella, Inc.) followed by slow drying covered on the bench top for at least 3 hours. Samples were then dried under vacuum for 24–48 hours to remove contamination that would interfere with STEM-EDS. Grids were loaded into a JEOL 31640 beryllium double tilt holder. STEM-EDS data was collected for 180 – 600 s at specific points, using the largest probe size (1.5 nm electron beam diameter) with a 200 kV accelerating voltage. Images were collected in bright field (BF) and high-angle annular dark field (HAADF) modes.
The magnetic properties of micellar nanoparticles were characterized using a Quantum Design MPMS3 superconducting quantum interference device (SQUID) with a maximum field of 7 T. Freeze-drying solid samples (~10 mg) were packed into standard Quantum Design plastic sample holders. Magnetization data were collected in DC mode and corrected for diamagnetic contributions using Pascal’s constants.
FeIII Concentration Determination in Micelles
In order to determine FeIII concentration, the metal was first stripped from the polymers using the following procedure. To an aliquot of each sample (100 μL) was added 1% HNO3 in water (1900 μL). Each mixture was then stirring for about 12 hours. Then the FeIII concentration was quantified by inductively coupled plasma-optical emission spectrometry (ICP-OES) using a Perkin Elmer Optima 3000DV spectrometer in the Scripps Institution of Oceanography, University of California, San Diego.
FeIII Stability in PBS
To determine the stability of FeIII chelated in SMN and CMN, we re-dispersed these two types of micellar nanoparticles in PBS (pH = 7.4). 300 μL of SMN and CMN solution (three replicates) were added in 500 μL dialysis tubes with Mw = 3500 respectively, and dialyzed to 500 mL PBS (pH = 7.4) under room temperature with magnetic stirring. 20 μL SMN and CMN aliquots were taken at time points as 8 h, 24 h, 48 h, 72 h, 7 days for ICP-OES analysis.
Determination of the in vitro stability of micelles in FBS by MRI analysis
Samples of both SMN and CMN were prepared in FBS at various [FeIII] concentrations (For SMN, 5 [FeIII] concentrations were used: 0.6 mM, 0.3 mM, 0.15 mM, 0.075 mM, and 0.038 mM; while for CMN, 5 [FeIII] concentrations were used: 0.67 mM, 0.34 mM, 0.17 mM, 0.08 mM, and 0.04 mM) 3 days prior to MRI analysis, and as controls, samples of identical concentration to the latter were prepared in both FBS and water immediately before MRI analysis (named, SMN/CMN in FBS for 3 days, SMN/CMN in freshly prepraed FBS, and SMN/CMN in water, respectively). Longitudinal and transverse relaxation time (T1 and T2) measurements were acquired with a Bruker Minispec mq60 (1.41 T or 60 MHz, 37 °C). Relaxivities (r1 and r2) were calculated by linearly fitting plots of 1/T1 (s−1) or 1/T2 (s−1) versus FeIII ions concentrations (μM). MR images were acquired on a Bruker 7.0 T magnet equipped with Avance II hardware and a 72 mm quadrature transmit/receive coil ([FeIII] = 0.6 mM in each tube). T1 contrast was determined by selecting regions of interest (ROI) using the ParaVision Version 5.1 software. The fitting parameters for 7 T MRI analysis are as follows: TR = 750.0 ms, TE = 12.6 ms, echo =1/1, FOV = 6.91/3.12 cm, slice thickness = 2 mm, nex = 2 mm, matrix = 256*116.
1H NMRD Profiles
Proton 1/T1 NMRD profiles were measured on a fast field-cycling Stelar SMARTracer Relaxometer (Stelar, Mede (PV), Italy) at magnetic field strengths from 0.00024 to 0.25 T (corresponding to 0.01–10 MHz proton Larmor frequencies) at room temperature. The relaxometer operates under computer control with an absolute uncertainty in 1/T1 of ± 1%. Additional data points in the range 15–70 MHz were obtained on a Bruker WP80 NMR electromagnet adapted to variable-field measurements (15–80 MHz proton Larmor frequency) Stelar Relaxometer. The 1H T1 relaxation times were acquired by the standard inversion recovery method with typical 90° pulse width of 3.5 μs, 16 experiments of 4 scans. The temperature was controlled with a Stelar VTC-91 airflow heater equipped with a calibrated copper–constantan thermocouple (uncertainty of ± 0.1 °C).
Cell Viability
In vitro cytotoxicity of micellar nanoparticles was determined in HeLa cells by the CCK-8 (cell counting kit-8) assay. HeLa cells were incubated on 96-well plates with 1*104 cells per well in High-glucose DMEM medium containing 10% fetal bovine serum and 1% antibiotics at 37 °C in 5% CO2 humidified atmosphere for 24 h and 48 h respectively. Addition of 10 μL of CCK-8 solution to each well and incubation for another 4 hours at 37 °C resulted in the formation of formazan crystals. Then the absorbance value at 460 nm was recorded using a microplate reader. The absorbance value of the untreated cells was used as the reference value of 100% cellular viability.
Shape- and Time-Dependent MR Imaging in HeLa cells
HeLa cells were seeded in 15 cm round tissue culture dishes and allowed to attach overnight. After washing twice with sterile PBS, the cells were incubated with micellar nanoparticles (the concentration of FeIII ions was approximately 67.5 μM) for different times including 4 hours, 12 hours, 24 hours, and 48 hours respectively at normal cell culture condition. The cells were washed with PBS three times in order to remove excess nanoparticles, and then treated with 0.05% trypsin to remove them from the dishes. The cells were gathered by centrifuge at 300 g for 3 min and washed with PBS buffer twice. The numberof cells in each sample was counted for further use. The cell MR images were acquired on a Bruker 7.0 T magnet with Avance II hardware equipped with a 72 mm quadrature transmit/receive coil. T1 contrast was determined by selecting regions of interest (ROI) using Software ParaVision Version 5.1. The parameters for 7 T MRI are: TR = 1000.0 ms, TE = 12.6 ms, echo length = 1, FOV = 7.91/3.22 cm, slice thickness = 1 mm, nex = 1 mm, matrix = 256*104. Then, the cells were digested by 70% HNO3 solution under bath sonication for overnight, in order to test the iron ions content by ICP-MS. The FeIII quantities of each samples were normalized to 106 cells.
Cell TEM Observation
HeLa cells were seeded in 35 mm round tissue culture dishes and allowed to proliferate till 80% fluent. After washing twice with sterile PBS buffer, the cells were incubated with micellar nanoparticles (the concentration of FeIII ions were approximately 67.5 μM) for 24 hours at 37 °C. The cells were washed with PBS three times in order to remove excess nanoparticles, and then were fixed by 2% glutaraldehyde in 0.1 M sodium cacodylate buffer with pH = 7.4 (SC buffer) on ice for more than 2 hours. After washing three times with 0.1 M SC buffer for 5 min each, the cells were postfixed with 1% osmium tetroxide in 0.1 M SC buffer for 1 hour on ice. Then cell pellets were washed with 0.1 M SC buffer 3 time for 5 min, followed by a quick rinse with H2O. The cell pellets were stained with 2% uranyl acetate (UA) for 1 hour on ice. After staining, they were dehydrated in a graded series of ethanol (50%, 70%, 90% and 100%) for 5–8 min each, and dried in acetone at room temperature. Then the cell pellets were infiltrated by 50:50 dry acetone/durcupan for 1–2 hours on a rotator, followed by 100% durcupan overnight and 2X 100% durcupan next day. Finally the cell pellets were embedded in durcupan and incubated in an oven at 60 °C for 36–48 hours. Ultrathin sections were cut and were examined via electron microscopy.
NMRD Profiles Analysis
A simplified model was utilized to analyze the data and obtain an estimation for some of the relevant molecular parameters affecting the relaxivity of the system. In this model, only the high field data were considered and the fit was performed according to the Solomon-Bloembergen-Morgan (SBM) set of equations, as discussed in the main text. The determination of electron spin relaxation parameters is almost entirely dependent upon fitting the low field data. As we stated in the manuscript, low field data was not included in the fitting. The reason for this is simply that SBM theory does not function very well across this frequency range for slowly tumbling systems – a fact that has been commonly noted for many years by several different groups.[31] For this reason it would be unwise to try to attribute any genuine physical meaning to the values of Δ2 and τV. We then used as adjustable parameters Δ2, τV, τR, τM and q/r6. Satisfactory fit was obtained with the parameters reported in Table 1. The choice of q = 2 is arbitrary and was made since associated with the reasonable value of r of 3.3 Å. Of course, setting q = 3 we would obtain values for r equal to 3.72 Å. The same value was reported in the case of FeIIIheme-HSA.[43] A distance of 3.2 ± 0.1 Å was also reported for bovine lactoferrin.[28] The value of the rotational correlation times is much shorter than that associated with a macromolecular system and in good agreement with that expected for loosely bound second-sphere water molecules. The exchange lifetime τM is also very similar to that estimated for bovine lactoferrin.[28]
Supplementary Material
Acknowledgments
Y. Li and Y. Huang contributed equally to this work. We acknowledge support from the NIH (NIBIB R01EB011633), a Director’s New Innovator Award (DP2OD008724), and Alfred P. Sloan Foundation to N.C.G. M.B. thanks support of the “Compagnia di San Paolo” (CSP-2012 NANOPROGLY Project).
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Yiwen Li, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Yuran Huang, Department of Materials Science and Engineering, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA.
Zhao Wang, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA.
Prof. Fabio Carniato, Dipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale “A. Avogadro”, Alessandria, Italy
Yijun Xie, Department of Materials Science and Engineering, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA.
Dr. Joseph P. Patterson, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Dr. Matthew P. Thompson, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Dr. Christopher M. Andolina, Department of Chemistry, University of Pittsburgh, 4200 Fifth Ave., Pittsburgh, PA 15260, USA
Dr. Treffly B. Ditri, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Prof. Jill E. Millstone, Department of Chemistry, University of Pittsburgh, 4200 Fifth Ave., Pittsburgh, PA 15260, USA
Prof. Joshua S. Figueroa, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Prof. Jeffrey D. Rinehart, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Prof. Miriam Scadeng, Department of Radiology, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
Prof. Mauro Botta, Dipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale “A. Avogadro”, Alessandria, Italy
Prof. Nathan C. Gianneschi, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
References
- 1.Fox MD, Raichle ME. Nat Rev Neurosci. 2007;8:700. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- 2.Terreno E, Castelli DD, Viale A, Aime S. Chem Rev. 2010;110:3019. doi: 10.1021/cr100025t. [DOI] [PubMed] [Google Scholar]
- 3.a) Na HB, Song IC, Hyeon T. Adv Mater. 2009;21:2133. [Google Scholar]; b) Laurent S, Forge D, Port M, Roch A, Robic C, Vander Elst L, Muller RN. Chem Rev. 2008;108:2064. doi: 10.1021/cr068445e. [DOI] [PubMed] [Google Scholar]
- 4.Lauffer RB. Chem Rev. 1987;87:901. [Google Scholar]
- 5.a) Aime S, Caravan P. J Magn Reson Imaging. 2009;30:1259. doi: 10.1002/jmri.21969. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kribben A, Witzke O, Hillen U, Barkhausen J, Daul AE, Erbel R. J Am Coll Cardiol. 2009;53:1621. doi: 10.1016/j.jacc.2008.12.061. [DOI] [PubMed] [Google Scholar]
- 6.a) Bertini I, Capozzi F, Luchinat C, Xia Z. J Phys Chem. 1993;97:1134. [Google Scholar]; b) Bertini I, Luchinat C, Parigi G. Adv Inorg Chem. 2005;57:105. doi: 10.1021/ic010161j. [DOI] [PubMed] [Google Scholar]
- 7.a) Hider RC, Kong X. Nat Prod Rep. 2010;27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]; b) Auerbach M, Coyne D, Ballard H. Am J Hematol. 2008;83:580. doi: 10.1002/ajh.21154. [DOI] [PubMed] [Google Scholar]; c) Yi P, Chen G, Zhang H, Tian F, Tan B, Dai J, Wang Q, Deng Z. Biomaterials. 2013;34:3010. doi: 10.1016/j.biomaterials.2013.01.022. [DOI] [PubMed] [Google Scholar]; d) Shapiro EM, Skrtic S, Sharer K, Hill JM, Dunbar CE, Koretsky AP. Proc Natl Acad Sci USA. 2004;101:10901. doi: 10.1073/pnas.0403918101. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mahmoudi M, Hosseinkhani H, Hosseinkhani M, Boutry S, Simchi A, Shane Journeay W, Subramani K, Laurent S. Chem Rev. 2011;111:253. doi: 10.1021/cr1001832. [DOI] [PubMed] [Google Scholar]
- 8.Kalinowski DS, Richardson DR. Pharmacol Rev. 2005;57:547. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 9.Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, Richardson DR. Curr Med Chem. 2012;19:2689. doi: 10.2174/092986712800609706. [DOI] [PubMed] [Google Scholar]
- 10.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Annu Rev Mater Res. 2011;41:99. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris WR, Carrano CJ, Raymond KN. J Am Chem Soc. 1979;101:2213. [Google Scholar]
- 12.Harrington MJ, Masic A, Holten-Andersen N, Waite JH, Fratzl P. Science. 2010;328:216. doi: 10.1126/science.1181044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Rodríguez E, Simoes RV, Roig A, Molins E, Nedelko N, Œlawska-Waniewska A, Aime S, Arús C, Cabañas ME, Cerdán S, García-Martín ML. Magn Reson Mater Phy. 2007;20:27. doi: 10.1007/s10334-006-0066-7. [DOI] [PubMed] [Google Scholar]; b) Davies JA, Dutremez SG, Hockensmith CM, Keck R, Richardson N, Selman S, Smith DA, Ulmer CW, Wheatley LS, Zeiss J. Acad Radio. 1996;3:936. doi: 10.1016/s1076-6332(96)80305-9. [DOI] [PubMed] [Google Scholar]; c) Schwert DD, Richardson N, Ji G, Radüchel B, Ebert W, Heffner PE, Keck R, Davies JA. J Med Chem. 2005;48:7482. doi: 10.1021/jm0501984. [DOI] [PubMed] [Google Scholar]
- 14.a) Ratzinger G, Agrawal P, Körner W, Lonkai J, Sanders HM, Terreno E, Wirth M, Strijkers GJ, Nicolay K, Gabor F. Biomaterials. 2010;31:8716. doi: 10.1016/j.biomaterials.2010.07.095. [DOI] [PubMed] [Google Scholar]; b) Turner JL, Pan D, Plummer R, Chen Z, Whittaker AK, Wooley KL. Adv Funct Mater. 2005;15:1248. [Google Scholar]
- 15.a) Liu Y, Ai K, Lu L. Chem Rev. 2014;114:5057. doi: 10.1021/cr400407a. [DOI] [PubMed] [Google Scholar]; b) Lynge ME, Van Der Westen R, Postma A, Städler B. Nanoscale. 2011;3:4916. doi: 10.1039/c1nr10969c. [DOI] [PubMed] [Google Scholar]; c) Ye Q, Zhou F, Liu W. Chem Soc Rev. 2011;40:4244. doi: 10.1039/c1cs15026j. [DOI] [PubMed] [Google Scholar]; d) d’Ischia M, Napolitano A, Ball V, Chen C-T, Buehler MJ. Acc Chem Res. 2014;47:3541. doi: 10.1021/ar500273y. [DOI] [PubMed] [Google Scholar]; e) Faure E, Falentin-Daudré C, Jérôme C, Lyskawa J, Fournier D, Woisel P, Detrembleur C. Prog Polym Sci. 2013;38:236. [Google Scholar]; f) Lee H, Dellatore SM, Miller WM, Messersmith PB. Science. 2007;318:426. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Caulder DL, Raymond KN. Acc Chem Res. 1999;32:975. [Google Scholar]
- 16.a) Enochs WS, Petherick P, Bogdanova A, Mohr U, Weissleder R. Radiology. 1997;204:417. doi: 10.1148/radiology.204.2.9240529. [DOI] [PubMed] [Google Scholar]; b) Fan Q, Cheng K, Hu X, Ma X, Zhang R, Yang M, Lu X, Xing L, Huang W, Gambhir SS, Cheng Z. J Am Chem Soc. 2014;136:15185. doi: 10.1021/ja505412p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Ju KY, Lee JW, Im GH, Lee S, Pyo J, Park SB, Lee JH, Lee JK. Biomacromolecules. 2013;14:3491. doi: 10.1021/bm4008138. [DOI] [PubMed] [Google Scholar]; b) Xiao M, Li Y, Allen MC, Deheyn DD, Yue X, Zhao J, Gianneschi NC, Shawkey MD, Dhinojwala A. ACS Nano. 2015;9:5454. doi: 10.1021/acsnano.5b01298. [DOI] [PubMed] [Google Scholar]
- 18.a) Ku TH, Chien MP, Thompson MP, Sinkovits RS, Olson NH, Baker TS, Gianneschi NC. J Am Chem Soc. 2011;133:8392. doi: 10.1021/ja2004736. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rush AM, Nelles DA, Blum AP, Barnhill SA, Tatro ET, Yeo GW, Gianneschi NC. J Am Chem Soc. 2014;136:7615. doi: 10.1021/ja503598z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chien MP, Rush AM, Thompson MP, Gianneschi NC. Angew Chem Int Ed. 2010;49:5076. doi: 10.1002/anie.201000265. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chien MP, Thompson MP, Barback CV, Ku TH, Hall DJ, Gianneschi NC. Adv Mater. 2013;25:3599. doi: 10.1002/adma.201300823. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nguyen MM, Carlini AS, Chien MP, Sonnenberg S, Luo C, Braden RL, Osborn KG, Li Y, Gianneschi NC, Christman KL. Adv Mater. 2015;27:5547. doi: 10.1002/adma.201502003. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang Z, Li Y, Huang Y, Thompson MP, LeGuyader CLM, Sahu S, Gianneschi NC. Chem Commun. 2015 doi: 10.1039/C5CC05653E. [DOI] [PubMed] [Google Scholar]; g) Callmann CE, Barback CV, Thompson MP, Hall DJ, Mattrey RF, Gianneschi NC. Adv Mater. 2015;27:4611. doi: 10.1002/adma.201501803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duncan R. Nat Rev Drug Discov. 2003;2:347. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 20.a) Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nature Nanotech. 2007;2:249. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Champion JA, Katare YK, Mitragotri S. J Control Release. 2007;121:3. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1. [Google Scholar]
- 22.a) Zhang L, Eisenberg A. J Am Chem Soc. 1996;118:3168–3181. [Google Scholar]; b) Wang Z, Li Y, Dong XH, Yu X, Guo K, Su H, Yue K, Wesdemiotis C, Cheng SZD, Zhang WB. Chem Sci. 2013;4:1345. [Google Scholar]
- 23.Mai Y, Eisenberg A. Chem Soc Rev. 2012;41:5969. doi: 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- 24.a) Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]; b) Caravan P. Chem Soc Rev. 2006;35:512. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]; c) Aime S, Botta M, Terreno E. Adv Inorg Chem. 2006;57:173. [Google Scholar]
- 25.a) Lauffer RB, Greif WL, Stark DD, Vincent AC, Saini S, Wedeen VJ, Brady TJ. Int J Rad Appl Instrum B. 1988;15:47. doi: 10.1016/0883-2897(88)90160-2. [DOI] [PubMed] [Google Scholar]; b) Botta M. Eur J Inorg Chem. 2000:399. [Google Scholar]; c) Stack TDP, Karpishin TB, Raymond KN. J Am Chem Soc. 1992;114:1512. [Google Scholar]
- 26.Brückner C, Caulder DL, Raymond KN. Inorg Chem. 1998;37:6759. doi: 10.1021/ic980620p. [DOI] [PubMed] [Google Scholar]
- 27.Kabir MK, Kawahara M, Kumagai H, Adachi K, Kawata S, Ishii T, Kitagawa S. Polyhedron. 2001;20:1417. [Google Scholar]
- 28.Fasano M, Fanali G, Polticelli F, Ascenzi P, Antonini G. J Inorg Biochem. 2004;98:1421. doi: 10.1016/j.jinorgbio.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Koenig SH, Brown RD, III, Lindstrom TR. Biophys J. 1981;34:397. doi: 10.1016/S0006-3495(81)84858-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Xu H, Regino CAS, Bernardo M, Koyama Y, Kobayashi H, Choyke PL, Brechbiel MW. J Med Chem. 2007;50:3185. doi: 10.1021/jm061324m. [DOI] [PubMed] [Google Scholar]; b) Ananta JS, Godin B, Sethi R, Moriggi L, Liu X, Serda RE, Krishnamurthy R, Muthupillai R, Bolskar RD, Helm L, Ferrari M, Wilson LJ, Decuzzi P. Nature Nanotech. 2010;5:815. doi: 10.1038/nnano.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fries PH, Belorizky E. J Chem Phys. 2005;123:124510. doi: 10.1063/1.2011389. [DOI] [PubMed] [Google Scholar]
- 32.Barkey NM, Preihs C, Cornnell HH, Martinez G, Carie A, Vagner J, Xu L, Lloyd MC, Lynch VM, Hruby VJ, Sessler JL, Sill KN, Gillies RJ, Morse DL. J Med Chem. 2013;56:6330. doi: 10.1021/jm4005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.a) Yang J, Stuart MAC, Kamperman M. Chem Soc Rev. 2014;43:8271. doi: 10.1039/c4cs00185k. [DOI] [PubMed] [Google Scholar]; b) Holten-Andersen N, Jaishankar A, Harrington MJ, Fullenkamp DE, Dimarco G, He Ld, McKinley GH, Messersmith PB, Lee KYC. J Mater Chem B. 2014;2:2467. doi: 10.1039/C3TB21374A. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH. Proc Natl Acad Sci U S A. 2011;108:2651. doi: 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ejima H, Richardson JJ, Liang K, Best JP, Van Koeverden MP, Such GK, Cui J, Caruso F. Science. 2013;341:154. doi: 10.1126/science.1237265. [DOI] [PubMed] [Google Scholar]
- 34.Caravan P. Acc Chem Res. 2009;42:851. doi: 10.1021/ar800220p. [DOI] [PubMed] [Google Scholar]
- 35.Sadler NP, Chuang C-C, Milburn RM. Inorg Chem. 1995;34:402. [Google Scholar]
- 36.a) Liu Y, Ai K, Liu J, Deng M, He Y, Lu L. Adv Mater. 2013;25:1353. doi: 10.1002/adma.201204683. [DOI] [PubMed] [Google Scholar]; b) Fisher OZ, Larson BL, Hill PS, Graupner D, Nguyen-Kim MT, Kehr NS, De Cola L, Langer R, Anderson DG. Adv Mater. 2012;24:3032. doi: 10.1002/adma.201104437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao L, Hu W, Liu Y, Huang G, Sumer BD, Gao J. Exp Bio Med. 2011;236:20. doi: 10.1258/ebm.2010.010243. [DOI] [PubMed] [Google Scholar]
- 38.Müllner M, Dodds SJ, Nguyen T-H, Senyschyn D, Porter CJH, Boyd BJ, Caruso F. ACS Nano. 2015;9:1294. doi: 10.1021/nn505125f. [DOI] [PubMed] [Google Scholar]
- 39.Zhang K, Rossin R, Hagooly A, Chen Z, Welch MJ, Wooley KL. J Polym Sci, Part A: Polym Chem. 2008;46:7578. doi: 10.1002/pola.23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 41.Sanford MS, Love JA, Grubbs RH. Organometallics. 2001;20:5314. [Google Scholar]
- 42.a) Hahn ME, Randolph LM, Adamiak L, Thompson MP, Gianneschi NC. Chem Commun. 2013;49:2873. doi: 10.1039/c3cc40472b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Culberson CF, Wilder P., Jr J Org Chem. 1960;25:1358. [Google Scholar]
- 43.Fanali G, Pariani G, Ascenzi P, Fasano M. FEBS J. 2009;276:2241. doi: 10.1111/j.1742-4658.2009.06952.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.