

Abstract
The role of domains in defining the equilibrium and kinetic folding properties of dihydrofolate reductase (DHFR) from Escherichia coli was probed by examining the thermodynamic and kinetic properties of a set of variants in which the chain connectivity in the discontinuous loop domain (DLD) and the adenosine-binding domain (ABD) was altered by permutation. To test the concept that chain cleavage can selectively destabilize the domain in which the N- and C-termini are resident, permutations were introduced at one position within the ABD, one within the DLD and one at a boundary between the domains. The results demonstrated that a continuous ABD is required for a stable thermal intermediate and a continuous DLD is required for a stable urea intermediate. The permutation at the domain interface had both a thermal and urea intermediate. Strikingly, the observable kinetic folding responses of all three permuted proteins were very similar to the wild-type protein. These results demonstrate a crucial role for stable domains in defining the energy surface for the equilibrium folding reaction of DHFR. If domain connectivity affects the kinetic mechanism, the effects must occur in the sub-millisecond time range.
Keywords: circular permutation, equilibrium intermediate, protein folding mechanisms, thermal denaturation, urea denaturation
Introduction
Comparisons of protein sequences and structures from numerous organisms have revealed that many higher-order structural elements, or domains, are conserved across a wide-range of species (Orengo and Thornton, 2005; Todd et al., 2005). These domains can bind specific ligands, catalyze common chemical reactions or serve to stabilize protein–protein interactions. Although the conservation of function is an obvious evolutionary force driving the preservation of these domains, it is not known if the domains also play a role in determining the folding mechanism and thermodynamic stability of the proteins in which they are resident.
One common and highly conserved functional domain is the dinucleotide-binding domain, or Rossmann fold, that is found in many proteins that bind nicotinamide adenine dinucleotide [NAD(H)] or nicotinamide adenine dinucleotide phosphate [NADP(H)]. The dinucleotide-binding domain consists of an open, six-stranded, parallel β-sheet flanked on each side by α-helices (Rossmann et al., 1974). Dihydrofolate reductase (DHFR) from Escherichia coli, which features a modified form of the Rossmann fold (Carugo and Argos, 1997), is a small (159 amino acids; MW = 18.0 kDa) monomeric α/β/α sandwich protein that catalyzes the reduction of dihydrofolate by means of an NADP(H) cofactor. DHFR contains a discontinuous loop domain (DLD), residues 1–37 and 107–159, and an adenosine-binding domain (ABD), residues 38–106. The observation that the ABD moves as a unit relative to the DLD during the catalytic cycle (Sawaya and Kraut, 1997) demonstrates that the integrity of this domain is important for function. This result also suggests that domain integrity may be relevant to the thermodynamic stability and folding mechanism of the entire protein.
Analyses of the equilibrium thermal unfolding properties of wild-type (WT-DHFR) (Ohmae et al., 1996) and cysteine-free variants (Luo et al., 1995; Ionescu et al., 2000) have revealed the presence of a stable intermediate that is maximally populated at ∼50°C at pH 7.8. Although the folding status of the DLD in the thermal intermediate was not determined, spectroscopic and kinetic evidence (Kuwajima et al., 1991; Ionescu et al., 2000; O'Neill and Matthews, 2000) suggest that the ABD is largely folded. These results support the conjecture that the ABD can play a crucial role in the equilibrium folding mechanism of DHFR, but they leave unanswered the role of the DLD in the intermediate or the roles of both domains in the kinetic folding mechanism.
The most direct way to test the thermodynamic properties of a domain is to express it as a fragment and study it in isolation. Application of this approach to DHFR from E.coli (Gegg et al., 1997) revealed that neither fragment 37–107, which corresponds to the ABD, nor fragments 1–36 and 108–159, which correspond to the DLD, display the spectroscopic features of folded proteins. An alternative approach to study the role of an individual domain in stability and folding is circular permutation.
Circular permutation, in which the natural termini are connected by a linker and new termini are introduced at desired positions within the protein, makes it possible to maintain the structure while altering the connectivity of the chain. For most proteins, circular permutation results in only minor structural changes and minimal loss of stability (Zhang et al., 1993; Hahn et al., 1994; Ritco-Vonsovici et al., 1995; Vignais et al., 1995; Viguera et al., 1995; Aÿ et al., 1998; Otzen and Fersht, 1998; Feng et al., 1999; Ni and Schachman, 2001; Fishburn et al., 2002). Relevant to the present study, an exhaustive permutation analysis of DHFR, based on host survival in the presence of trimethoprim (TMP), an inhibitor of DHFR, revealed that permutation is tolerated at more than half of the 158 possible sites (Iwakura et al., 2000). If domains can act as semi-autonomous elements of structure and stability, then it is conceivable that appropriate permutations could serve to stabilize one domain by linking the natural N- and C-termini, while simultaneously disrupting another by cleaving the chain and introducing new termini.
To test the relationships between domain connectivity, domain stability and folding mechanism, three permuted variants were chosen from the DHFR set generated by Iwakura et al. (2000) to examine the effects of cleaving the polypeptide chain within each of the domains and at one of the boundaries between the two domains. Analysis of their spectroscopic features, ability to bind ligand, enzymatic activity, stability to thermal and urea denaturation, and kinetics of refolding supports the hypothesis that chain connectivity and domain stability are closely linked in DHFR.
Materials and methods
Reagents
Ultrapure urea (ICN Biomedical, Aurora, OH) was treated with AG®-501X (8D) mixed-bed resin (Bio-Rad Laboratories, Hercules City, CA) before addition of buffers to remove free cyanate and ammonia. The concentration of urea was determined by refractive index measurements according to Pace et al. (1990). All other chemicals were of reagent grade and were used without further purification. The standard buffer used in all experiments was 10 mM potassium phosphate, 0.2 mM K2EDTA and 1 mM β-mercaptoethanol, pH 7.8.
Protein purification
The permutants were purified according to methods described by Iwakura and Nakamura (1998). The concentration of each protein was determined by absorbance at 280 nm using the molar extinction coefficient of 31 100/M/cm (Touchette et al., 1986). Purity was judged to be >95% by SDS–gel electrophoresis. MALDI and LC-ESI mass spectrometry data confirmed that for all three permutants, the N-terminal formyl-methionine was not proteolytically cleaved after protein synthesis.
Enzyme assay
Enzyme activity was determined at 15°C using the method previously described by Hillcoat et al. (1967). Enzyme assay conditions were chosen to match the buffer conditions used in fluorescence (FL) and circular dichroism (CD) spectroscopy. For all activity measurements, the reaction was started by the addition of 20 nM of enzyme. Additionally, a second set of conditions for cpN18 involved pre-incubating 200 nM of enzyme with NADPH, increasing the temperature to 30°C, and initiating the reaction by the addition of dihydrofolate (DHF). Activity was determined by monitoring the depletion of NADPH and DHF at 340 nm [ϵ340 = 11 800/M/cm, (Hillcoat et al., (1967)] for 60 or 180 s on an Aviv 14DS UV-visible spectrophotometer. The final rates were corrected for the background activity before the addition of protein, and the final calculated activity was an average of 3–5 repetitions.
Equilibrium spectroscopy
Far- and near-UV CD measurements were performed on an Aviv 62-DS spectrometer. Far-UV CD spectra of native proteins in the absence and presence of methotrexate (MTX) were collected using a 1 or 2 mm cuvette maintained at 15°C by a thermoelectric temperature control system. Near-UV CD spectra were collected using a 10 cm cylindrical cuvette maintained at 15°C by a thermostatted circulating water bath. A three-point smoothing algorithm was applied to the near-UV CD spectra because of the inherently lower signal-to-noise ratio. For both far- and near-UV measurements, the step size was 1 nm with a 2 nm bandwidth and a 4 s averaging time; the protein concentration was 5 and 10 μM for far- and near-UV CD spectroscopy, respectively.
The urea- and temperature-induced equilibrium unfolding experiments were carried out on the Aviv 62DS CD spectrometer. For the urea equilibrium experiments, the spectrometer was interfaced with a Hamilton Microlab 500 series automatic titrator (Hamilton Co., Reno, NV). Spectra were recorded from 215 to 260 nm using a 1 cm path length cell, a 2 nm bandwidth, a 1 nm step size and an integration time of 3 s. For the thermal equilibrium experiments, the CD spectra were collected from 216 to 250 nm with a 2 nm step size. An auxiliary computer controlled the addition of each denaturant concentration or each temperature increment, and the sample cell was continuously stirred to ensure proper equilibration. The denaturant titrations were collected at 15°C, and the protein concentration was 5 μM for both the urea and thermal titrations.
Fluorescence steady-state emission spectra were collected on an Aviv ATF 105 fluorometer equipped with a Peltier-style thermostatted sample compartment and interfaced to a Hamilton Microlab 500 series automatic titrator. Samples were excited at 295 nm, and emission was monitored from 310 to 450 nm at 1 nm intervals. Protein concentrations were 3 μM for both the urea and thermal titrations. Spectra were collected in a 1 cm quartz cuvette; the experimental temperature for the urea unfolding experiments was 15°C.
Equilibration times of 900 s were used after each addition of urea in both CD and fluorescence equilibrium titrations, based upon the known kinetic properties of WT-DHFR (Jennings et al., 1993). For the temperature-induced unfolding transitions, the equilibration times varied between 2 and 15 min, with the longer time at low final temperatures. In preparation for all spectroscopic measurements, the samples were dialyzed extensively against standard buffer, which had been degassed by aspiration and sparged with N2.
Thermodynamic analysis
The urea dependence of the far-UV CD and FL emission spectroscopic signals were fit to either a two-state model:
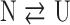 |
a three-state model:
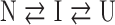 |
or a four-state model:
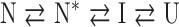 |
where N, I and U represent the native, intermediate and unfolded forms, respectively. The N* species in the four-state model represents a native-like state with altered packing of one or more of the five tryptophans. For these thermodynamic models, the free energy of unfolding in the absence of denaturant was calculated assuming a linear dependence of the apparent free-energy difference on the denaturant concentration (Schellman, 1978; Pace, 1986; Matthews, 1987),
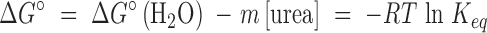 |
where ΔG° represents the free energy at a given urea concentration, ΔG°(H2O) represents the free energy of unfolding in the absence of urea and the m-value is the sensitivity of the transition to the urea concentration. A complete description of the method and equations used to obtain two-, three- and four-state fits is provided elsewhere (Tanford, 1968; Schellman, 1978; Pace, 1986; Matthews, 1987; Gualfetti et al., 1999). A discussion of the application of singular value decomposition (SVD) (Henry and Hofrichter, 1992) to discriminate between possible equilibrium mechanisms can be found in Ionescu et al. (2000). Local and global fits of the raw data and the SVD vectors were obtained using Savuka, an in-house non-linear least-squares program. Global fitting methods are also described elsewhere (Bilsel et al., 1999).
The thermal unfolding CD data were also fit to two-state or three-state equilibrium models using equations and protocols described previously (Luo et al., 1995; Ionescu et al., 2000). To obtain reliable results for the midpoint of the unfolding transitions (Tm) in the three-state model, the values for the enthalpy and heat capacity for each transition were constrained to those previously reported for the thermal unfolding of AS-DHFR, a cysteine-free version of DHFR (C85A/C152S) (Ionescu et al., 2000).
Stopped-flow FL spectroscopy
Refolding kinetics were measured using either an AVIV Model 202 stopped-flow fluorescence instrument (dead time <5 ms) or an Applied Photophysics SX-17MV (Surrey, UK) instrument (dead time <2 ms). The excitation wavelength was 295 nm, and the emission was monitored using a 320 nm low wavelength cut-off filter. In the refolding experiments, the unfolded protein was diluted 10-fold into buffer; the final concentrations were 1–3 μM protein and 0.50–0.76 M urea. All kinetic studies were performed at 15°C using standard buffer conditions. Relaxation times and associated amplitudes were determined by fitting the kinetic data to the equation
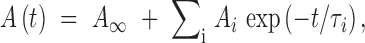 |
where A(t) is the total amplitude at time t, A∞ is the amplitude at infinite time, Ai is the amplitude of the individual phase, i, and τi is the corresponding relaxation time. Kinetic traces were best fit to a series of exponentials using Savuka, an in-house non-linear least square program (Bilsel et al., 1999).
Results
Selection of sites for permutation
Three positions within DHFR were chosen as the sites for insertion of new N- and C- termini (Figure 1). One site was within the DLD (cpN18), one was at a boundary between the DLD and ABD (cpP39), and one was within the ABD (cpD69). In this nomenclature, the circular permutants (cp) are named for the position (in the WT-DHFR primary structure) of the new N-terminal residue that follows the retained N-formyl methionine. The positions of the permutations were also selected to avoid the 10 essential folding elements that, when disrupted, prevent the folding of the protein to a stable, active form (Nakamura and Iwakura, 1999; Iwakura et al., 2000).
Fig. 1.
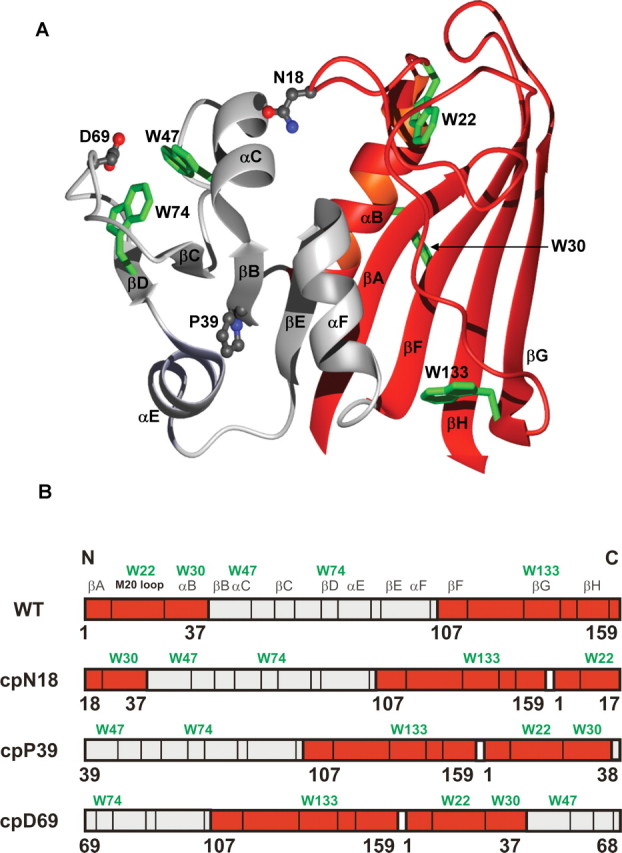
(A) Ribbon diagram and (B) schematic two-dimensional drawing of E.coli DHFR. The two domains as identified by Sawaya and Kraut (1997) are shown: red, the discontinuous loop domain (1–37; 107–159) and grey, the adenosine-binding domain (38–106). The circular permutants are named for the new N-terminal residue that follows the initial methionine. (A) The residues corresponding to the new N-termini in the circular permutants are illustrated in grey ball-and-stick representations, and the five tryptophans that are present in DHFR (W22, W30, W47, W74 and W133) are shown in green stick representations. The figure was generated using MolMol (Koradi et al., 1996) and the Protein Data Bank entry 7dfr.pdb. (B) A schematic illustrating how the connectivity of the two domains was altered in the circular permutants. The white rectangle depicts the five-glycine linker connecting the original N- and C-termini. The secondary structure assignments are identified above the WT representation and are shown by vertical black lines. To aid in the identification of the secondary structure elements in the permutant representation, the placement of the five tryptophans are indicated by green lettering.
Structural information from solution studies
Far-UV CD spectroscopy provided information on the global secondary structure of these three permutated versions of DHFR. Inspection of the far-UV CD spectra (Figure 2A–C) reveals that all of the permuted proteins have the same general spectral shape as WT-DHFR with the characteristic minimum at ∼218 nm and maximum at ∼195 nm, indicative of a mixed α/β protein. Although they are well-folded, the small decreases in ellipticity relative to WT-DHFR suggest a loosening of the secondary structure. The observed differences in the far-UV CD spectra compared with WT-DHFR are largely recapitulated when MTX, a tight-binding inhibitor, binds to the three permuted proteins (Figure 2D–F). By implication, the core structures of the permuted proteins are very similar to WT-DHFR.
Fig. 2.

Far-UV CD spectra of WT-DHFR and the circular permutants in the absence (A–C) and presence (D–F) of excess methotrexate. The spectrum for WT-DHFR is shown by a solid line in A–F. (A,D) cpN18 (dashed lines); (B,E) cpP39 (dotted lines); (C,F) cpD69 (dashed dotted lines). The final concentrations were 5 μM for protein and 25 μM for MTX (D–F only). Buffer conditions are 10 mM potassium phosphate, pH 7.8, 0.2 mM K2EDTA and 1 mM β-mercaptoethanol at 15°C.
Tryptophan FL emission spectroscopy was used to assess tertiary structure, using the five intrinsic tryptophan residues as probes. Trp22, Trp30 and Trp 133 reside in the DLD, and Trp47 and Trp74 reside in the ABD (Figure 1). The FL emission spectra (Supplementary Figure S1A is available at PEDS online) of all three permuted proteins are similar to WT DHFR, including the wavelength of maximum emission (350 ± 1 nm). Because the permutants only have 60–75% of the intensity of WT-DHFR, a change in the environment of one or more of the tryptophans must enhance their quenching.
The maxima in the ellipticity at 286 and 292 nm in the near-UV CD spectroscopy, characteristic of WT-DHFR, revealed that cpN18 and cpD69 retain chiral environments for one or more of their aromatic side chains (Supplementary Figure S1B is available at PEDS online). The substantially reduced amplitudes of these peaks for cpP39 suggest that the introduction of the charged N- and C-termini between the DLD and the ABD has disrupted the packing near one or more aromatic side chains responsible for these signals.
Enzymatic activity
The steady-state enzyme activity was determined for each permuted protein as a measure of folded, functional protein. All three permutants had reduced specific activities relative to WT-DHFR; the specific activity is neglible for cpN18 and ∼17 and ∼70% of the wild-type activity for cpP39 and cpD69, respectively. These findings are consistent with previous reports (Nakamura and Iwakura, 1999; Iwakura et al., 2000); the very low level of activity for cpN18 demonstrates that the TMP screen used to select for viable permutants is not a stringent assay for enzymatic activity. This result is not surprising considering that the cpN18 variant is disrupted in a loop region known to be involved in the catalytic cycle of DHFR (Sawaya and Kraut, 1997). The activity of cpN18 can be enhanced slightly to ∼2% of the wild-type activity by increasing the concentration of enzyme, increasing the temperature and pre-incubating with DHF, demonstrating that it is possible to access a native conformation even when the M20 loop is disrupted.
The activity assay of WT-DHFR exhibits hysteresis due to the presence of two native conformers, only one of which binds both DHF and NADPH (Penner and Frieden, 1985; Fierke et al., 1987; Falzone et al., 1991). Unlike WT-DHFR, no hysteresis was observed in the activity assays of the permutants, suggesting that the permutants have a single native conformation that represents the active enzyme conformer. The spectroscopic properties, the MTX-binding capabilities and the enzymatic activities for the three permuted proteins, when considered together, support the conclusion that cpN18, cpP39 and cpD69 largely retain the WT-DHFR fold.
Equilibrium folding mechanism
The thermodynamic features of the folding energy surface for WT-DHFR and the cpN18, cpP39, and cpD69 permuted proteins were probed by urea and thermal denaturation. The urea-induced unfolding transitions monitored by far-UV CD were highly reversible (data not shown). Unfolding profiles were inspected at individual wavelengths obtained from FL and CD, and SVD analyses were performed across the spectrum obtained by both techniques (CD: 220–260 nm; FL: 310–450 nm) to provide a comprehensive description of the unfolding reaction.
Urea denaturation studies
The urea denaturation curves monitored by both CD at 226 nm and FL at 320 nm for all three circular permutants are shown in Figure 3A–C. The unfolding profiles exhibit a sigmoidal loss of signal at moderate (2–4 M) urea concentrations, as well as small linear changes in the optical signal at low (0–2 M) and high (>4 M) urea concentrations where the native and unfolded forms are predominately populated. cpP39 exhibits an additional change in FL signal in the native baseline region (0–1 M urea), which is presumed to reflect a reorganization of the native-like packing of the tryptophans in this permutant (Ervin et al., 2002), as reflected also in the near-UV CD spectrum (Supplementary Figure S1 is available at PEDS online).
Fig. 3.

The urea-induced equilibrium unfolding transitions (A–C) and the corresponding normalized unfolding curves (D–F) for the circular permutants monitored by far-UV CD at 226 nm and FL emission at 320 nm. (A) cpN18, far-UV CD (closed circle) and FL (open circle); (B) cpP39, far-UV CD (closed square) and FL (open square); and (C) cpD69, far-UV CD (closed triangle) and FL (open triangle). The normalized curves shown in the right panels are generated directly from the corresponding raw data in the left panels and are model independent. In contrast, the continuous lines in the left panels represent combined global fits of the CD (220–260 nm) and FL (310–450 nm) datasets to a two-state (cpN18), four-state (cpP39) or three-state (cpD69) equilibrium unfolding model, respectively. The protein concentrations were 5 and 3 μM for CD and FL experiments, respectively. Buffer conditions are described in the legend to Figure 2.
The raw data shown in Figure 3A–C were converted to apparent fraction unfolded plots to allow direct visual comparison of the CD and FL data (Pace et al., 1990; Finn et al., 1992). The coincidence of the normalized CD and FL unfolding transition curves for cpN18, when coupled with the ideal sigmoidal behavior, supports a two-state folding mechanism (Figure 3D). As noted above, WT-DHFR, with a discontinuous DLD similar to cpN18, also unfolds by a two-state mechanism in response to urea denaturation (Touchette et al., 1986; Frieden, 1990; Jennings et al., 1993). In contrast, the presence of a stable intermediate in the urea-induced unfolding of cpP39 and cpD69 is implied by the non-coincidence of their CD and FL transition curves (Figure 3E and F, respectively).
The presence of a stable intermediate for cpP39 and cpD69 is further confirmed by a SVD analysis that simultaneously incorporates data from the entire CD and FL spectra. Although only two vectors are required to describe the unfolding data for cpN18, three vectors are required for both cpP39 and cpD69, indicating the presence of an intermediate (Supplementary Figure S2 is available at PEDS online). Even though the weights for the second and third vectors are small for cpD69, the single point autocorrelation analysis indicates that they are above baseline values.
The resulting significant vectors (Supplementary Figure S2 is available at PEDS online) were fit to either a two-state (cpN18; N⇄U), three-state (cpD69; N⇄I⇄U) or a four-state (cpP39; N⇄N*⇄I⇄U) model to extract the thermodynamic parameters. The N⇄N* transition in the four-state model represents a reorganization of the tryptophan packing in cpP39. The fits are in excellent agreement with the observed transition curves as indicated by the superposition of the lines and symbols in Figure 3A–C, and the resulting thermodynamic parameters are shown in Table I.
Table I.
Thermodynamic parameters obtained from urea denaturation of wild-type and circular permutants of DHFRa
|
|
WT-DHFRb |
cpN18 |
cpP39c |
cpD69 |
|---|---|---|---|---|
| ΔG°(H2O)NU (kcal/mol) | 6.4 ± 0.2 | 6.1 ± 0.2 | ||
| m NU (kcal/mol/M) | 2.1 ± 0.1 | 1.9 ± 0.1 | ||
| ΔG°(H2O)NI (kcal/mol) | 3.2 ± 0.3 | 3.5 ± 1.2 | ||
| m NI (kcal/mol/M) | 1.2 ± 0.1 | 1.3 ± 0.7 | ||
| ΔG°(H2O)IU (kcal/mol) | 5.4 ± 0.7 | 3.4 ± 2.5 | ||
| m IU (kcal/mol/M) | 1.6 ± 0.2 | 1.3 ± 0.6 | ||
| ΔG°(H2O)tot (kcal/mol) | 6.4 ± 0.2 | 6.1 ± 0.2 | 8.6 ± 0.8 | 6.9 ± 1.8 |
| m tot (kcal/mol/M) | 2.1 ± 0.1 | 1.9 ± 0.1 | 2.8 ± 0.2 | 2.5 ± 0.9 |
Obtained from global fits of svd vectors from urea-induced denaturation profiles monitored by CD and FL and calculated assuming a two-state (WT and cpN18), three-state (cpD69) or four-state (cpP39) unfolding transition and a linear dependence of the free energy on the urea concentration as outlined in Materials and methods.
Data for WT-DHFR taken from O'Neill and Matthews, 2000.
The free energy for the N to N* transition for cpP39, which is thought to represent a structural reorganization of the native-like packing of the tryptophans in the native baseline between 0 and 1 M urea observed by FL, is negligible, implying equal populations of N and N* in the absence of urea.
The thermodynamic stability for cpN18, 6.1 kcal/mol, is comparable to WT and AS-DHFR, consistent with the disruption of the DLD in these three proteins. The difference in free energy between the native and unfolded forms for cpD69, 6.9 kcal/mol is also comparable, even though disruption of the ABD yields a three-state unfolding transition. Interestingly, cpP39 is ∼2 kcal/mol more stable to unfolding than WT-DHFR, suggesting that the presence of both an intact ABD and DLD can enhance the stability of the protein.
For cpP39 and cpD69, the relative populations of the intermediate species as a function of urea were calculated from the thermodynamic parameters obtained in the global fits [see (Ionescu et al., 2000) for a description of the process]. For cpP39, the population of the intermediate is ∼50% at ∼3 M urea. The intermediate observed in cpD69 has a maximum population of ∼60% at ∼3 M urea. Apparently, the continuous DLD found in cpP39 and cpD69 is required for a stable intermediate in the urea denaturation reaction. The stable intermediate in cpD69 has about ∼40% of the native CD ellipticity at 222 nm in the absence of urea, as determined from the global fit of the svd vectors to a three-state model (Supplementary Figure S3 is available at PEDS online). A similar reconstruction of the CD spectrum for the intermediate in cpP39 is hampered by the presence of the N⇄N* transition.
Thermal denaturation studies
The equilibrium thermal denaturations of cpN18, cpP39 and cpD69 were also monitored by tryptophan FL between 310 and 450 nm (data not shown) and far-UV CD between 215 and 250 nm (Figure 4). Despite the presence of two free cysteines, Cys85 and Cys152, which are also present in the wild-type sequence, the thermally induced unfolding profiles were highly reversible (>85%) for all three permuted proteins and the wild-type sequence when a slow temperature gradient was applied. AS-DHFR (C85A/C152S), which has a comparable thermodynamic stability to urea denaturation as wild-type DHFR (Iwakura et al., 1995), was used as an additional control in this set of experiments because its thermal denaturation properties have previously been extensively studied (Ionescu et al., 2000).
Fig. 4.

The temperature-induced equilibrium unfolding transitions (A–C) and the corresponding normalized unfolding curves (D–F) for the circular permutants monitored by far-UV CD at 226 and 236 nm. (A,D) cpN18, 226 nm (closed circle) and 236 nm (open circle), (B,E) cpP39, 226 nm (closed square) and 236 nm (open square), and (C,F) cpD69, 226 nm (closed triangle) and 236 nm (open triangle). The normalized unfolding curves were generated as described in the legend to Figure 3, and the continuous lines represent the global fits to a two-state (cpD69) and three-state (cpN18 and cpP39) equilibrium model. The protein concentration was 5 μM. Buffer conditions are described in the legend to Figure 2.
Although the steep sloping baselines in the fluorescence intensity unfolding profiles (Ohmae et al., 1996; Ionescu et al., 2000) make it impossible to fit the data reliably to a thermodynamic model, the far-UV CD data were tractable. The thermally induced unfolding of cpD69, which is cleaved within the ABD, was best described by a two-state model, regardless of whether single wavelengths, multiple wavelengths or SVD vectors were analyzed. The midpoint for the thermal transition, Tm, is 46°C for cpD69. In contrast, cpN18 and cpP39, with intact ABDs, were both found to have non-coincident transition curves when monitored by CD at two different wavelengths, 226 and 236 nm (Figure 4). The temperature-dependent SVD basis vectors derived from a global analysis of the CD data (Supplementary Figure S4 is available at PEDS online) were used to extract the midpoints of the thermal transitions. The Tm for the native/intermediate, TmNI, and intermediate/unfolded, TmIU, transitions for cpN18 and cpP39 are shown in Table II. Both cpN18 and cpP39 have comparable melting temperatures to WT-DHFR and slightly lower melting temperatures for both transitions as compared with AS-DHFR. The existence of a stable thermal intermediate for WT-DHFR, cpN18 and cpP39, but not for cpD69, suggests that a continuous ABD is required to stabilize this partially folded form.
Table II.
Transition temperatures for AS-DHFR and circular permutantsa
|
|
AS-DHFRb |
WTc |
cpN18c |
cpP39c |
cpD69 |
|---|---|---|---|---|---|
| T mNI (°C) | 46 ± 1 | 45 ± 1 | 44 ± 1 | 44 ± 1 | |
| T mIU (°C) | 59 ± 2 | 53 ± 2 | 55 ± 2 | 53 ± 2 | |
| T mNU (°C) | 46 ± 1 |
Obtained from global fits of svd vectors calculated from the thermally induced denaturation profiles monitored by CD.
The transition temperatures for AS-DHFR are taken from Ionescu et al., 2000.
To obtain melting temperatures in the three-state global fit, the change in heat capacity (ΔCp) and entropy (ΔS) were fixed to the values determined previously for the temperature-induced unfolding of AS-DHFR at a series of urea concentrations (Ionescu et al., 2000). Variations in the values of ΔCp and ΔS by as much as 2-fold had no significant effects on the values for Tm.
Kinetic folding mechanism
The refolding reactions of the circular permutants and WT-DHFR were examined by intrinsic Trp stopped-flow FL spectroscopy. The kinetic traces for jumps to strongly refolding conditions are shown in Figure 5. The refolding reaction of WT-DHFR, which has been documented in a series of previous studies (Jennings et al., 1993; Wallace and Matthews, 2002; Svensson et al., 2003), involves a sub-millisecond, stopped-flow burst phase reaction followed by a complex process that requires five exponentials for a satisfactory fit. The hundreds of milliseconds portion of the kinetic trace is characterized by a simple exponential increase in fluorescence intensity. This non-monotonic change in intensity between the unfolded and native forms reflects the formation of a hyperfluorescent intermediate (or a set of such intermediates) that results from the burial of Trp74 in a native-like hydrophobic core in the ABD (Garvey and Matthews, 1989). The subsequent folding reaction is well described by four phases of decreasing fluorescence intensity, the products of which are all capable of binding MTX (Jennings et al., 1993). The latter results have been interpreted in terms of four parallel folding channels leading from the hyperfluorescent intermediate(s) to native or native-like species.
Fig. 5.
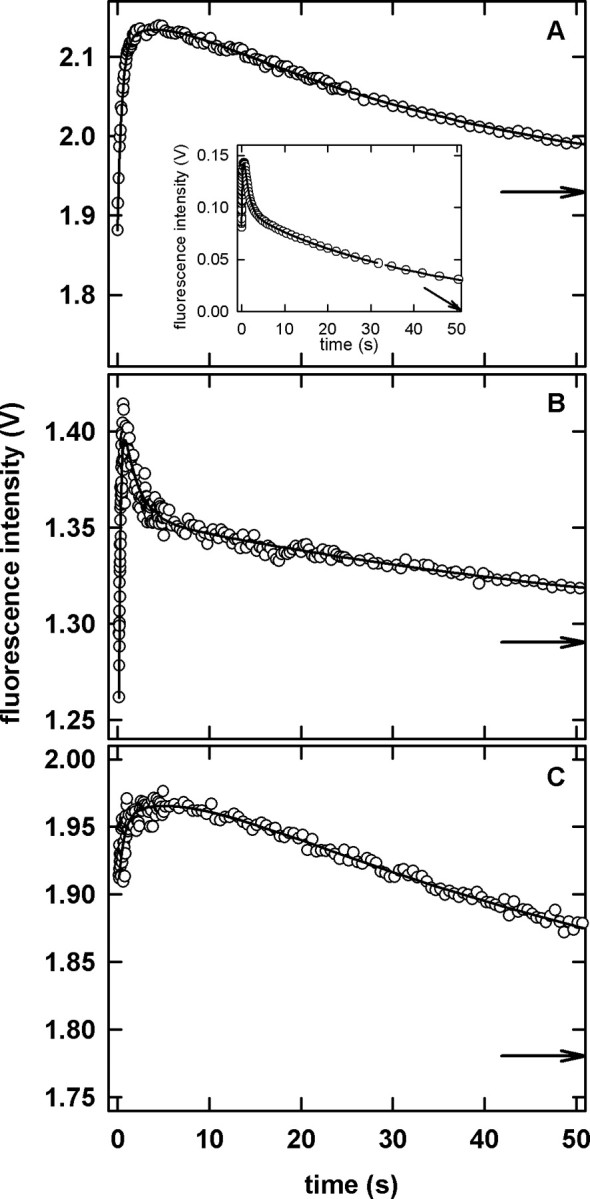
Stopped-flow fluorescence kinetic measurements for the refolding reaction of the circular permutants. (A) cpN18, (B) cpP39 and (C) cpD69. The inset in panel A illustrates the refolding reaction of WT-DHFR. The continuous lines represent the fit of the data to three (circular permutants) and five (WT-DHFR) exponentials. The arrows indicate the final fluorescence intensity. Although data were collected for 500 s, only the first 50 s are shown to better illustrate the presence of the hyperfluorescent intermediate. The final protein concentrations was 1–3 μM. Buffer conditions are described in the legend to Figure 2.
In contrast to WT-DHFR, the refolding reactions under strongly refolding conditions of the permutant proteins are best described by three exponentials. All three permutated proteins display the characteristic hyperfluorescent phase followed by a decrease in the intensity as the folding reaction proceeds (Figure 5). Because cpN18 and cpD69 require two exponentials to describe the hyperfluorescent phase, the results imply that the parallel folding channels arise prior to the formation of the corresponding intermediates and the burial of Trp74. The fewer number of phases with decreasing intensity observed for the permutants as compared with WT DHFR presumably reflects a smaller number of folding channels. It has been previously suggested that alternative folds or docking modes of the ABD and DLD may be the source of the parallel folding channels in DHFR (Svensson et al., 2003). When considered with the lack of hysteresis observed in the enzyme activity assay, the reduction in the number of folding channels is consistent with the loss of an inactive native conformer.
Discussion
Quantitative analysis of the thermodynamic and kinetic folding properties of three permuted versions of E.coli DHFR has revealed a correlation between chain connectivity and the roles of the two functional domains in folding. The rationale for these effects is based upon the expected increase in the chain entropy, and the consequent decrease in free energy, of the unfolded state when a hypothetical circular version of the chain is cleaved. If the free energy of the folded state is unperturbed by chain cleavage, the free energy difference between the folded and unfolded states, i.e. the stability, is expected to decrease when the circular chain is cleaved. For example, the decrease in stability for converting the 159 residues in DHFR from a circular to a linear form is predicted to be ∼5 kcal/mol (Arai et al., 2003). Experimentally, the reduction in free energy of 2.3 kcal/mol observed in a linear version of an intein-mediated, covalently cyclized DnaB helicase is consistent with theoretical predictions for the increased comformational entropy of the linear form (Williams et al., 2005). In contrast, linear and circular versions of an SH3 domain exhibited comparable thermodynamic stabilities (Camarero et al., 2001). Indeed, compensating enthalpic and entropic effects introduced by cyclization of the natural N- and C-terminus often mitigate the decrease in stability to be less than that predicted from theory (Zhou, 2003).
The application of the chain entropy argument to circular permutants of DHFR focuses on the connectivity of the two functional domains and the placement of the new termini within or between each of these domains. Although neither the ABD nor the DLD can fold in isolation (Gegg et al., 1997), the results of the present study suggest that both can fold if they are covalently closed and if they are in the presence of the partner domain. This phenomenon occurs whether that partner domain is folded, e.g. cpP39, or not, e.g. cpN18 and cpD69. The implication is that the decrease in stability reflecting the increase in chain entropy accompanying cleavage, or conversely the increase in stability reflecting the decrease in entropy accompanying chain closure, can be experienced at the level of an individual domain. Thus, the site of the permutation can influence the relative stabilities of various partially folded species on the folding energy surface.
The stable urea-induced folding intermediate
As is evident in Figure 3 and quantified in Table I, the stable urea-induced intermediate was observed only for the cpP39 and cpD69 variants. The absence of this intermediate in WT-DHFR and cpN18 is consistent with a requirement for a continuous DLD to stabilize this species. These results appear to contradict those of a previous study comparing the urea-induced unfolding profiles of the linear and a circularized form of DHFR created by forming a disulfide crosslink between extensions of the N- and C-termini (Arai et al., 2003). The assumption of a two-state equilibrium unfolding model in that study was based upon the near-coincidence of the normalized transition curves derived from the ellipticity at 220 nm and the absorbance difference spectra at 292 nm. However, the acknowledged small discrepancies between the two transition curves [see Figure 4 in (Arai et al., 2003)] and the relatively small absorbance changes that accompany unfolding suggest that the stable intermediate may have been present but not detected. The large changes in tryptophan FL observed for unfolding of DHFR and, perhaps a more distinctive response of the emission properties of the intermediate species, may have enhanced the detection of the partially folded species in the present study. The application of SVD methods, which incorporate information from the signal responses across a broad spectral range for both CD and FL, also enhanced the detection of the partially folded form.
The stable thermally induced intermediate
The observation of a thermal intermediate in cpN18, cpP39 and wild-type-like cysteine-free AS-DHFR (Luo et al., 1995; Ionescu et al., 2000), but not in cpD69, implies that a continuous ABD is required for its stability. Consistent with this conclusion, a Gō-model simulation by Onuchic and co-workers (Clementi et al., 2000) predicts that the ABD should be more stable than the DLD. Because the Gō-model only contains information on the structure of the backbone, it is likely that topology plays a critical role in defining the stability of the domains. In contrast, the results of a molecular dynamics (MD) simulation of the thermal unfolding reaction for DHFR (Sham et al., 2002) predicted that unfolding of the ABD should precede unfolding of the DLD. The reasons for the discrepancy between the MD result and both the present experimental results and the Gō-model results are not apparent. It is possible that increasing the number of independent simulations, to enhance the sampling, and extending the time period of the sampling, to insure equilibration, might alter the outcome of the MD simulations.
In a previous fragmentation study of DHFR (Smith and Matthews, 2001), cleavage of the backbone between residues 86 and 87 within the ABD yields a pair of complementing fragments that have a micromolar dissociation constant but cannot acquire the native fold in the absence of MTX. The implication of these results, along with those in the present communication, is that at least one continuously connected domain is required to access the native, functional conformation of DHFR. In support of this conclusion are the folding properties of a circular permutation at this site, cpG86 (Smith and Matthews, 2001). Similar to cpD69, cpG86 is capable of adopting a well-folded native structure, and it displays a urea-induced, but not a thermally induced, intermediate.
It is important to recognize that the creation of new chain termini also introduces formal positive and negative charges at the N- and C-termini. Although a favorable electrostatic interaction between the juxtaposed charges might enhance stability, unfavorable electrostatic interactions could also play a role in destabilizing the domain in which the new termini are resident. This potentially destabilizing effect could play a role in the loss of the thermal intermediate for cpD69. If one assumes that the electrostatic fields around the natural N- and C-termini in DHFR (whose Cα atoms are 15 Å apart in the native structure) have been optimized for stability, the potential destabilization from their neutralization with a peptide crosslink is not sufficient to override the enhancement from covalently closing the DLD in the cpP39 and cpD69 permutants. These results imply that chain connectivity is likely to be the principal factor influencing the stabilities of the ABD and the DLD.
Because the intermediate with a continuous ABD is only revealed by thermal denaturation and the intermediate with a continuous DLD is only revealed by urea denaturation, it is likely that there are variations in the relative contributions of hydrophobic, hydrogen-bonding and ionic interactions for stabilizing these two domains. Although the complexity of fully understanding the mechanisms of denaturation makes it unrealistic to quantify the magnitude of these various stabilizing interactions for each domain, the results do highlight the utility of using multiple denaturation methods for obtaining differential views of the folding energy landscape.
Implications for the kinetic folding mechanism of DHFR
The similar observed kinetic folding responses, i.e. those that occur after 5 ms, for all of the permutants and WT-DHFR (Figure 5) suggest that if either a folded DLD or ABD plays a role in folding, it must occur in the sub-millisecond time range. This conjecture is supported by a previous quench-flow hydrogen-exchange NMR study of the product of the sub-millisecond folding reaction in DHFR (Jones and Matthews, 1995). Thirteen milliseconds after folding is initiated by dilution from 6.0 M urea, protection against exchange is observed for ∼60% of the population of folding molecules at 13 amide hydrogens located in six of the eight β strands. Three of these strands are contained in the DLD and three in the ABD, implying that both domains are folded in the sub-millisecond time range. The biphasic increase in FL intensity in the hundreds of milliseconds time range for cpN18 and cpD69 (Figure 5) provides the first opportunity to discriminate the subsequent folding reactions of two sub-millisecond intermediate populations detected in the pulse-quench hydrogen-exchange NMR experiment (Jones and Matthews, 1995).
Conclusions
The results on cpN18, cpP39 and cpD69 DHFR demonstrate a crucial role for stable subdomains in defining the energy surface for the equilibrium folding reaction. Specifically, the reduction of chain entropy as a consequence of covalently connecting either the ABD or the DLD or both is sufficient to alter the balance of forces stabilizing the protein and enhance the population of partially folded species containing a continuously connected domain. Although the specific placement of the termini within DHFR has a readily discernible effect on its thermodynamic properties, these same permutations can have a minimal effect on the global fold, the post-millisecond folding mechanism or, with one obvious exception, the enzymatic activity. One might conclude that protein folding mechanisms are sufficiently robust that variations in the connectivity of the polypeptide do not significantly perturb either the rate-limiting steps or the final product of these complex reactions.
Supplementary Material
Acknowledgments
The authors would like to thank Dr Masahiro Iwakura, National Institute of Advanced Industrial Science and Technology, Tsukuba, Ibaraki, Japan, for providing the expression plasmids for the circular permutants; Mark Signs, at The Pennsylvania State University Fermentation Facility for assistance with the growth of the permutants, and Dr A.Daniel Jones at The P.S.U. Mass Spectrometry Center for providing MALDI and ESI-MS analysis. Finally, the authors are very grateful to Jen Smith for assistance with data collection of cpP39 and Dr Osman Bilsel for assistance and helpful advice with data fitting. This work was supported by NSF grants MCB 0296053 and 0327504 to C.R.M. Lynne Regan served as editor for this manuscript.
Edited by Lynne Regan
References
- Arai,M., Kataoka,M., Kuwajima,K., Matthews,C.R. and Iwakura,M. (2003) J. Mol. Biol., 329, 779–791. [DOI] [PubMed] [Google Scholar]
- Aÿ,J., Hahn,M., Decanniere,K., Piotukh,K., Borriss,R. and Heinemann,U. (1998) Proteins, 30, 155–167. [DOI] [PubMed] [Google Scholar]
- Bilsel,O., Zitzewitz,J.A., Bowers,K.E. and Matthews,C.R. (1999) Biochemistry, 38, 1018–1029. [DOI] [PubMed] [Google Scholar]
- Camarero,J.A., Fushman,D., Sato,S., Giriat,I., Cowburn,D., Raleigh,D.P. and Muir,T.W. (2001) J. Mol. Biol., 308, 1045–1062. [DOI] [PubMed] [Google Scholar]
- Carugo,O. and Argos,P. (1997) Proteins, 28, 29–40. [DOI] [PubMed] [Google Scholar]
- Clementi,C., Jennings,P. and Onuchic,J. (2000) Proc. Natl Acad. Sci. USA, 97, 5871–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervin,J., Larios,E., Osvath,S., Shulten,K. and Gruebele,M. (2002) Biophys. J., 83, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone,C., Wright,P. and Benkovic,S. (1991) Biochemistry, 30, 2184–2191. [DOI] [PubMed] [Google Scholar]
- Feng,Y. et al (1999) Biochemistry, 38, 4553–4563. [DOI] [PubMed] [Google Scholar]
- Fierke,C.A., Johnson,K.A. and Benkovic,S.J. (1987) Biochemistry, 26, 4085–4092.3307916 [Google Scholar]
- Finn,B.E., Chen,X., Jennings,P.A., Saalau-Bethell,S.M. and Matthews,C.R. (1992) Principles of protein stability. Part 1—reversible unfolding of proteins: kinetic and thermodynamic analysis In Rees,A.R., Sternberg,M.J.E. and Wetzel,R.), Protein Engineering: A Practical Approach. IRL Press, Oxford, pp. 167–189.
- Fishburn,A.L., Keeffe,J.R., Lissounov,A.V., Peyton,D.H. and Anthony-Cahill,S.J. (2002) Biochemistry, 41, 13318–13327. [DOI] [PubMed] [Google Scholar]
- Frieden,C. (1990) Proc. Natl Acad. Sci. USA, 87, 4413–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey,E. and Matthews,C. (1989) Biochemistry, 28, 2083–2093. [DOI] [PubMed] [Google Scholar]
- Gegg,C.V., Bowers,K.E. and Matthews,C.R. (1997) Protein Sci., 6, 1885–1892.9300488 [Google Scholar]
- Gualfetti,P.J., Bilsel,O. and Matthews,C.R. (1999) Protein Sci., 1623–1635.10452606
- Hahn,M., Piotukh,K., Borriss,R. and Heinemann,U. (1994) Proc. Natl Acad. Sci. USA, 91, 10417–10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry,E.R. and Hofrichter,J. (1992) Methods Enzymol., 210, 129–192. [Google Scholar]
- Hillcoat,B.L., Nixon,P.F. and Blakley,R.L. (1967) Anal. Biochem., 21, 178–189.4384177 [Google Scholar]
- Ionescu,R.M., Smith,V.F., O'Neill,J.C.Jr and Matthews,C.R. (2000) Biochemistry, 39, 9540–9550. [DOI] [PubMed] [Google Scholar]
- Iwakura,M. and Nakamura,T. (1998) Protein Eng., 11, 707–713. [DOI] [PubMed] [Google Scholar]
- Iwakura,M., Jones,B.E., Luo,J. and Matthews,C.R. (1995) J. Biochem., 117, 480–488. [DOI] [PubMed] [Google Scholar]
- Iwakura,M., Nakamura,T., Yamane,C. and Maki,K. (2000) Nat. Struct. Biol., 7, 580–585. [DOI] [PubMed] [Google Scholar]
- Jennings,P.A., Finn,B.E., Jones,B.E. and Matthews,C.R. (1993) Biochemistry, 32, 3783–3789. [DOI] [PubMed] [Google Scholar]
- Jones,B.E. and Matthews,C.R. (1995) Protein Sci., 4, 167–177.7757007 [Google Scholar]
- Koradi,R., Billeter,M. and Wuthruch,K. (1996) J. Mol. Graph., 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Kuwajima,K., Garvey,E.P., Finn,B.E., Matthews,C.R. and Sugai,S. (1991) Biochemistry, 30, 7693–7703. [DOI] [PubMed] [Google Scholar]
- Luo,J., Iwakuro,M. and Matthews,C.R. (1995) Biochemistry, 34, 10669–10675. [DOI] [PubMed] [Google Scholar]
- Matthews,C.R. (1987) Methods Enzymol., 154, 498–511. [DOI] [PubMed] [Google Scholar]
- Nakamura,T. and Iwakura,M. (1999) J. Biol. Chem., 274, 19041–19047. [DOI] [PubMed] [Google Scholar]
- Ni,X. and Schachman,H.K. (2001) Protein Sci., 10, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill,J.C. and Matthews,C.R. (2000) J. Mol. Biol., 295, 737–744.10656786 [Google Scholar]
- Ohmae,E., Kurumiya,T., Makino,S. and Gekko,K. (1996) J. Biochem., 120, 946–953. [DOI] [PubMed] [Google Scholar]
- Orengo,C.A. and Thornton,J.M. (2005) Annu. Rev. Biochem., 74, 867–900. [DOI] [PubMed] [Google Scholar]
- Otzen,D. and Fersht,A. (1998) Biochemistry, 37, 8139–8146. [DOI] [PubMed] [Google Scholar]
- Pace,C.N. (1986). Methods Enzymol. 131, 266–280. [DOI] [PubMed] [Google Scholar]
- Pace,C.N., Shirley,B.A. and Thomson,J.A. (1990) Measuring the conformation stability of a protein In Creighton,T.E. (ed.) Protein Structure: A Practical Approach. IRL Press, Oxford, pp. 311–330.
- Penner,M.H. and Frieden,C. (1985) J. Biol. Chem., 260, 5366–5369. [PubMed] [Google Scholar]
- Ritco-Vonsovici,M., Minard,P., Desmadril,M. and Yon,J.M. (1995) Biochemistry, 34, 16543–16551. [DOI] [PubMed] [Google Scholar]
- Rossmann,M.G., Moras,D. and Olsen,K.W. (1974) Nature, 250, 194–199.4368490 [Google Scholar]
- Sawaya,M.R. and Kraut,J. (1997) Biochemistry, 36, 586–603. [DOI] [PubMed] [Google Scholar]
- Schellman,J.A. (1978) Biopolymers, 17, 1305–1322. [Google Scholar]
- Sham,Y.Y., Ma,B., Tsai,C.J. and Nussinov,R. (2002) Proteins, 46, 308–320. [DOI] [PubMed] [Google Scholar]
- Smith,V.F. and Matthews,C.R. (2001) Protein Sci., 10, 116–128.11266600 [Google Scholar]
- Svensson,A.K., O'Neill,J.C.Jr and Matthews,C.R. (2003) J. Mol. Biol., 326, 569–583.12559923 [Google Scholar]
- Tanford,C. (1968) Adv. Protein Chem., 23, 121–282. [DOI] [PubMed] [Google Scholar]
- Todd,A.E., Marsden,R.L., Thornton,J.M. and Orengo,C.A. (2005) J. Mol. Biol., 348, 1235–1260. [DOI] [PubMed] [Google Scholar]
- Touchette,N.A., Perry,K.M. and Matthews,C.R. (1986) Biochemistry, 25, 5445–5452.3535877 [Google Scholar]
- Vignais,M., Corbier,C., Mulliert,G., Branlant,C. and Branlant,G. (1995) Protein Sci., 4, 994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera,A., Blanco,F. and Serrano,L. (1995) J. Mol. Biol., 247, 670–681. [DOI] [PubMed] [Google Scholar]
- Wallace,L.A. and Matthews,C.R. (2002) J. Mol. Biol., 315, 193–211.11779239 [Google Scholar]
- Williams,N.K., Liepinsh,E., Watt,S.J., Prosselkov,P., Matthews,J.M., Attard,P., Beck,J.L., Dixon,N.E. and Otting,G. (2005) J. Mol. Biol., 346, 1095–1106. [DOI] [PubMed] [Google Scholar]
- Zhang,T., Bertelsen,E., Benvegnu,D. and Alber,T. (1993) Biochemistry, 32, 12311–12318. [DOI] [PubMed] [Google Scholar]
- Zhou,H.X. (2003) J. Mol. Biol., 332, 257–264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.