

Abstract
Although protein identification by matching tandem mass spectra (MS/MS) against protein databases is a widespread tool in mass spectrometry, the question about reliability of such searches remains open. Absence of rigorous significance scores in MS/MS database search makes it difficult to discard random database hits and may lead to erroneous protein identification, particularly in the case of mutated or post-translationally modified peptides. This problem is especially important for high-throughput MS/MS projects when the possibility of expert analysis is limited. Thus, algorithms that sort out reliable database hits from unreliable ones and identify mutated and modified peptides are sought. Most MS/MS database search algorithms rely on variations of the Shared Peaks Count approach that scores pairs of spectra by the peaks (masses) they have in common. Although this approach proved to be useful, it has a high error rate in identification of mutated and modified peptides. We describe new MS/MS database search tools, MS-CONVOLUTION and MS-ALIGNMENT, which implement the spectral convolution and spectral alignment approaches to peptide identification. We further analyze these approaches to identification of modified peptides and demonstrate their advantages over the Shared Peaks Count. We also use the spectral alignment approach as a filter in a new database search algorithm that reliably identifies peptides differing by up to two mutations/modifications from a peptide in a database.
Database search in mass spectrometry has been very successful in protein identification. The experimental spectrum can be compared with typical spectra for each peptide in a database and the peptide with the best fit usually provides the sequence of the experimental peptide (Eng et al. 1994; Mann and Wilm 1994; Taylor and Johnson 1997; Fenyo et al. 1998; Clauser et al. 1999). However, these methods are not mutation tolerant and are not effective for detecting types and sites of sequence variations (Gatlin et al. 2000). Identification of modified peptides is an even more challenging problem. Almost all protein sequences are post-translationally modified, and as many as 200 types of covalent modifications of amino acid residues are known (Gooley and Packer 1997). Although the sites of some post-translational modifications can be predicted from DNA sequences (Blom et al. 1999), experimental verification of post-translational modifications will remain an open problem even after the human genome is completely sequenced. It raises a challenging computational problem for the post-genomic era: Given a very large collection of spectra representing the human proteome, which of 200 types of modifications are present in each human gene?
The computational analysis of modified peptides was pioneered by Mann and Wilm (1995) and Yates et al. (1995a).
The Peptide Sequence Tag approach (Mann and Wilm 1994) was successful in many applications (Shevchenko et al. 1997), but no information about the limitations and error rates of this approach for mutation-tolerant MS/MS search is available. Yates et al. (1995a) suggested an exhaustive search approach, that is, to (implicitly) generate a virtual database of all modified peptides for a small set of modifications and to match the spectrum against this virtual database. This method was recently applied to the identification of sequence variations in human hemoglobins using SEQUEST-SNP software (Gatlin et al. 2000). However, Yates et al. (1995a) noted that it leads to a large combinatorial problem, even for a small set of modification types, and they indicated that extending this approach to a larger set of modifications is an open problem. Pevzner et al. (2000) proposed a new approach to modification-tolerant database search that automatically reveals peptide modifications without the need to generate all possible modified peptides and compare them with the spectrum in a case-by-case fashion. In fact, this approach does not need any prior knowledge of the types of modifications under study.
Given a MS/MS database search algorithm, how could we estimate its error rates? It is clear that the error rates of existing MS/MS database search algorithms are small for good spectra but grow fast with diminishing spectrum quality. This is indirectly confirmed by the fact that up to 60% of spectra acquired in an automated regime cannot be matched against databases even in the case of completely sequenced genomes like yeast. The difficulties in interpreting these spectra may come either from the fact that a significant portion of peptides have poor fragmentation or from the fact that a large portion of peptides are modified and, thus, are missed by conventional database search algorithms.
Despite widespread use of MS/MS database search algorithms, we are unaware of any attempts to estimate their error rates depending on the quality of analyzed spectra. Moreover, it is not clear how to make such an estimate as there is no simple experimental method to confirm that an analyzed peptide corresponds to a peptide from the database. To estimate the error rate of an MS/MS database search one should have a collection of peptides and their mass spectra of a given quality, analyze the mass spectra via database search, and find the portion of incorrect predictions. This is an unrealistic experiment because the only reliable method to infer the sequences of peptides is MS/MS database search itself, a “Catch 22”. In addition, it is not feasible to generate experimental spectra of a given quality.
We have designed a computational protocol to estimate the error rates in MS/MS database search and to study the problem: What is the threshold for spectrum quality that leads to erroneous peptide identification by database search algorithms? We further compare the efficiency of the Shared Peaks Count, spectral convolution, and spectral alignment for MS/MS database search for both experimental and simulated spectra. In a difference from the Shared Peaks Count, spectral convolution and spectral alignment do not require generation of virtual database of all modifications while comparing a spectrum of a modified peptide against a database. This advantage of spectral alignment and spectral convolution approaches comes with a tradeoff in the accuracy of its scoring function that is somewhat lower than the accuracy of advance scoring functions like ones in SEQUEST (Eng et al. 1994), MS-Tag (Clauser et al. 1999), and SHERENGA (Dancik et al. 1999). Below, we describe how to combine the advantages of the spectral alignment with advantages of the algorithms using advanced scoring functions.
For a large peptide database, MS/MS search algorithms produce some random hits while matching spectra of modified peptides. These random hits disguise the real similarities and increase the error rate of the database search.
However, our tests revealed that even in the case when the correct solution is not the one with the highest score, it is among very few high-scoring peptides. This suggests a new two-stage approach to MS/MS database search. At the first filtration stage, the spectral alignment is used as a filter to identify t top-scoring peptides in the database, where t is chosen in such a way that it is almost guaranteed that a correct hit is present among the top t hits. These top t hits form a small database of candidate peptides subject to further analysis at the second stage. Although the spectral alignment is sometimes unable to distinguish which among t top-scoring peptides is the correct one, more accurate scoring functions (like scoring functions in SEQUEST, MS-Tag, and SHERENGA) can be used at the second verification stage to find the correct hit. At the verification stage each of these t peptides can be mutated (as suggested by spectral alignment) and compared against the experimental spectrum by an accurate scoring scheme. This approach is conceptually similar to the Yates et al. (1995a) “virtual database” approach. However, instead of exhaustive generation of all possible mutations and modifications which often makes the virtual database approach infeasible, our filtration procedure reduces the size of the database to a few hundred candidate peptides.
Estimating the Error Rates in MS/MS Database Search
Let A be an algorithm that scores a spectrum S against a peptide P from a database by assigning scores A(S; P ). How can we estimate the error rate of A while searching a database for high-scoring peptides? Given a database of peptides {P1, . . . , Pk} and their corresponding spectra {S1, . . . ,Sk} we say that the algorithm A correctly reconstructs Pi by spectrum Si if
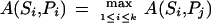 |
that is, the algorithm A assigns the highest score to peptide Pi and scores Si against the database {P1, . . . , Pk}. The error rate of A is defined as the portion of incorrect reconstructions, that is, the cases when spectra are matched against wrong peptides.
To test our algorithms, we simulate a database of peptides, induce k mutations in each peptide in this database, simulate typical tandem mass spectra for the mutated peptides, and search these spectra against the original (nonmutated) database. The percentage of correctly matched peptides in this search characterizes the efficiency of k-mutation-tolerant MS/MS database search.
This approach requires simulations of typical (theoretical) spectra. The offset frequency function, introduced in Dancik et al. (1999), allows one to simulate realistic spectra according to probabilities of different ion types. For testing purposes we restrict the number of masses in the spectra and limit our analysis to b- and y-ions only (minor ions have only a minor effect in our simulations). Some MS/MS database search applications take into account as many as 200 of the highest intensity masses in MS/MS spectra. However, Dancik et al. (1999) demonstrated that taking into account >5n (n is the peptide length) highest intensity masses hardly makes sense, because the signal in the remaining low intensity masses is indistinguishable from noise. Moreover, the signal corresponding to b- and y-ions is mainly limited to the first 2n high intensity masses (Dancik et al. 1999). Following these results, we generate theoretical spectra with the number of masses equal to twice the peptide length. Among them, 2np masses correspond to randomly chosen b- and y-ions whereas the remaining 2n(1 − p) masses are chosen randomly to simulate noise. The spectrum with quality p = 1 contains no noise (Fig. 1), whereas the spectrum with quality p = 0 is made up of noise entirely. For simplicity, we also ignore the intensities associated with these masses (although the intensities can be easily incorporated in our algorithms).
Figure 1.

Theoretical spectra of peptides PRTEIN, PRTEYN (one mutation), and PWTEYN (two mutations), representing masses of all b- and y-ions in the corresponding peptides. Shared masses between spectra of mutated peptides and the original spectrum (p = 1) are indicated by dashed lines.
Although mass spectrometrists routinely use the term “spectrum quality,” there is no standard agreement on how to define this notion. Define pb = qb/n and py = qy/n as the frequencies of b-ions and y-ions correspondingly (qb and qy are the numbers of b- and y-ions of peptide P in spectrum S, and n is the length of the peptide P ). We choose p = (pb + py) / 2 to represent the spectrum quality in our simulations, and we often assume pb = py. This parameter does not reflect the presence of minor ions (like b − H2O). To account for minor ions one can use the correlation between experimental spectrum S and theoretical spectrum S(P) of peptide P and define the spectrum quality as 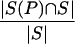 . The Backbone Cleavage Score (BCS) is another spectrum quality parameter defined as
. The Backbone Cleavage Score (BCS) is another spectrum quality parameter defined as
 |
(qb∪y is the number of positions i in a peptide for which either bi or yi ion is present). Although BCS sometimes is used to represent spectrum quality, we are not satisfied with BCS score as it does not discriminate between the case when both bi and yi ions are present and the case when only one of them is present.
Algorithms for Peptide Identification Problem
Shared Peaks Count
A match between spectrum S and peptide P is the number of masses that the experimental spectrum S and the theoretical spectrum of peptide P have in common. Let D be a database of peptides and k be a parameter (number of mutations/modifications). Let Dk be a database of all peptides that are at most k mutations/modifications apart from the peptides in D. We view Dk as a “virtual” database because it is usually so large that generation of Dk is an infeasible task. We study the following “peptide identification problem”: Given a database of peptides D, spectrum S, and parameter k, find a peptide in Dk whose theoretical spectrum has a maximal match to spectrum S.
For the sake of simplicity, we represent a spectrum S as a set of integers, corresponding to masses of fragment ions and ignore the intensities of the fragment ions (see above). Of course, real spectra are not integer, but we assume that scaling (e.g., multiplying all masses by 10) and rounding has been done already. Mass spectrometrists usually refer to masses as peaks, because every mass corresponds to an intensity peak in the experimental spectrum. Following this terminology we denote the number of masses that two spectra have in common as the Shared Peaks Count or SPC. Figure 1 presents three spectra S1, S2, and S3 with SPC(S1, S2) = 5; SPC(S2, S3) = 6 and SPC(S1, S3) = 2. Most existing database search programs are based on the SPC between the experimental and theoretical spectra. SPC is, of course, an intuitive measure of spectral similarity. However, this measure diminishes very quickly as the number of mutations increases thus leading to limitations in detecting similarities in MS/MS database search.
Spectral Convolution
Let S1 and S2 be two spectra. Following Pevzner et al. (2000) we define the spectral convolution as a multiset of integers S2 ⊖ S1 = {s2 − S1 : s1 ∈ S1,s2 ∈ S2}. Figure 2, a and b, shows the elements of this multiset in the form of a difference matrix. We define (S2 ⊖ S1)(x) as a multiplicity of an integer x in the set S2 ⊖ S1, that is, the number of pairs s1 ∈ S1, s2 ∈; S2 such that s2 − S1 = x. We view spectral convolution as both a multiset S2 ⊖ S1 and a function S2 ⊖ S1(x). The elements of S2 ⊖ S1 with high multiplicity correspond to peaks in the spectral convolution (S2 ⊖ S1)(x). If M(P) is the parent mass of peptide P with the spectrum S, then SR = M(P) − S = {M(P) − s : s ∈ S} is the reverse spectrum of S.The reverse spectral convolution (S2 ⊖ S1R)(x) is the number of pairs s1 ∈ S1,s2 ∈ S2 such that s2 + s1 − M(P) = x.
Figure 2.

(a) Elements of the spectral convolution S2 ⊖ S1 represented as elements of a difference matrix. (S1 and S2 are theoretical spectra of peptides PRTEIN and PRTEYN, correspondingly, differing by a single mutation). The elements with multiplicity >2 are shown in color, and the elements with multiplicity equal to 2 are shown in circles. The high multiplicity element 0 (red) corresponds to shared masses between these spectra, while another high multiplicity element 50 (green) corresponds to the shift of masses by δ = 50 due to mutation I → Y in PRTEIN (the mass of I is 113, and the mass of Y is 163). The SPC takes into account only the red entries in this matrix while the spectral convolution (for k = 1) takes into account both red and green entries, thus providing better peptide identification. (b) Same as a for the case of two mutations in peptide PRTEIN : R → W with δ1 = 30 and I → Y with δ2 = 50 (the mass of R is 156, and the mass of W is 186). Again, SPC takes into account only red entries. (c, d) Spectral alignment. Black lines represent the paths for k = 0 with similarity score (D(0) = 5 in c, and D(0) = 2 in d); red lines represent the paths for k = 1 (D(1) = 8 in c, and D(1) = 5 in d); blue line in d represents the path for k = 2 (D(2) = 7). The Shared Peaks Count reveals only D(0) matching peaks on the main diagonal, while spectral alignment reveals more hidden similarities between spectra and detects the corresponding mutations. Mutations/modifications are detected by jumps between the diagonals, for example,. spectral alignment with k = 1 detects a mutation with amino acid mass difference δ = 50 in c and a mutation with amino acid mass difference δ1 = 30 in d. Alignment with k = 2 detects a second mutation with amino acid mass difference δ2 = 50 in d.
Peaks in the spectral convolution of experimental and theoretical spectra allow one to detect mutations/modifications without an exhaustive search. If peptide P2 differs from peptide P1 by the only mutation/modification (k = 1) with an amino acid difference δ, then the spectral convolution of their spectra (S2 ⊖ S1)(x) is expected to have two approximately equal peaks at x = 0 and x = δ. The other set of correlations between the spectra of mutated peptides is captured by the reverse spectral convolution S2 ⊖ S1R, reflecting the pairings of N-terminal and C-terminal ions. S2 ⊖ S1R(x) is expected to have two peaks at the same positions 0 and δ. The spectral and the reverse spectral convolutions can be combined by introducing a multiset S:
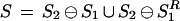 |
Pevzner et al. (2000) further introduce the shift function
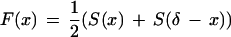 |
and define SIMk(S1,S2) as the overall height of the k highest peaks of the shift function F(x). SIMk(S1,S2) is an estimate of the similarity between spectra S1 and S2 under the assumption that the corresponding peptides are k mutations or modifications apart.
Figure 1 shows that in the case of a single mutation the Shared Peaks Count captures roughly half of the correlations between spectra, and in the case of two mutations, peptide spectra may have only few or no shared masses. The value of the shift function at x = 0 (corresponding Shared Peaks Count) decreases significantly for k = 1 and almost disappears for k = 2 (Fig. 3). On the other hand, the spectral convolution takes into account multiple peaks and captures the similarity between spectra even when the Shared Peaks Count does not (bold bars in Fig. 3). Thus, the efficiency of database search improves dramatically when the shift function is used instead of the Shared Peaks Count.
Figure 3.

Shift function F(x) for simulated spectra of pairs of peptides differing by zero, one, and two mutations. The similarity between mutated peptides is captured by multiple peaks in the shift function (indicated by bold bars).
To test the spectral convolution approach, we generate a small peptide database, induce k mutations in each database peptide, simulate typical tandem mass spectra for the mutated peptides, and search these spectra against the original (nonmutated) database. Figure 4 summarizes the statistics of errors in these computational experiments and convincingly demonstrates the advantages of the spectral convolution over the Shared Peaks Count.
Figure 4.

The matching spectra of mutated peptides with peptides in a small database (100 peptides) at different values of spectral quality p and number of mutations k. The first two pair of plots describe matching with SIM1 and SIM2 similarity scores for k = 1 and k = 2 mutations. The third pair of plots describes matching with the Shared Peaks Count (SPC). Crosses represent best matches, dots represent second-best matches. A cross at position (i, i) on the main diagonal represents the correct matching of spectra i and peptide i.
Spectral Alignment
Define a spectral product A × B of spectra A = {a1, . . . , an}and B = {b1, . . . , bm} as an an × bm two-dimensional matrix with nm 1s corresponding to all pairs of indices (ai, bj) and remaining elements being zeroes. A × B is a sparse matrix and the number of 1s at the main diagonal of this matrix describes the Shared Peaks Count between spectra A and B. The δ-shifted Peaks Count [corresponding to (A × B)(δ) in spectral convolution] is the number of 1s on the diagonal (i, + δ). The limitation of the spectral convolution is that it considers diagonals separately without combining them into feasible mutation scenarios. Following Pevzner et al. (2000) the k-similarity D(k) between spectra is defined as the maximum number of 1s on a path through the spectral matrix that uses at most k + 1 diagonals. k-optimal spectral alignment is defined as a path using these k + 1 diagonals. Because shifts between diagonals correspond to mutations or modifications in the peptide, D(k) estimates the similarity between spectra A and B under the assumption that they are k mutations/modifications apart. Figure 2 (c,d) illustrates that the spectral alignment allows one to detect more and more subtle similarities between spectra by increasing k. Below we describe a dynamic programming algorithm for spectral alignment.
Let Ai and Bj be the i-prefix of A and j-prefix of B, correspondingly. Define Dij(k) as the k-similarity between Ai and Bj such that the last elements of Ai and Bj are matched. In other words, Dij(k) is the maximum number of 1s on a path to (Ai, Bj) that uses at most k + 1 diagonals. We say that (i′, j′) and (i, j) are codiagonal if ai − ai′ = bi − bi′ and that (i′, j′) if i′ < i and j′ < j. To take care of the initial conditions, we introduce a fictitious element (0,0) with D0,0(k) = 0 and assume that (0,0) is codiagonal with any other (i,j). The dynamic programming recurrency for Dij(k) is
 |
if (i‘,j‘) and (i,j) are co-diagonal otherwise. The k-similarity between A and B is given by D(k) = maxijDij(k). The running time of the spectral alignment algorithm can be reduced to O(n2k).
Branch-and-Bound Algorithm for Mutation-Tolerant Peptide Identification
The spectral alignment approach is conceptually different from the virtual database approach because it does not rely on a prior knowledge of all possible types of modifications. If the number of mutations and modifications of interest is limited, we propose a branch-and-bound algorithm (Bushnell and Chen 1996) that is sometimes more efficient and accurate than spectral alignment for k = 2. This algorithm implements the Yates et al. (1995a,b) virtual database approach efficiently using the insights provided by the spectral alignment idea. Below we describe this algorithm for the mutations-only case and k = 2.
Let P be a peptide of mass M1 and let S be a spectrum of an (unknown) peptide of mass M2 that differs from P by two mutations. For k = 2, the alignment between P and S is given by a path that involves three diagonals: 0, δ1, and δ = M2 − M1, where δ1 is unknown. Although our algorithm exhaustively searches for δ1, preprocessing, restrictions on mass differences of amino acids, and the branch-and-bound approach significantly reduce its running time.
Consider a transformation of peptide P = p1, . . ., pn into a peptide
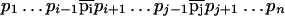 |
that differs from P by mutations at positions i and j. It corresponds to a path in the alignment graph that uses diagonal 0 for the first i − 1 steps, switches to diagonal δ1 = m( ) − m(pi) for the intermediate j − i steps and then switches to diagonal δ = δ1 + m(
) − m(pi) for the intermediate j − i steps and then switches to diagonal δ = δ1 + m(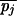 ) − m(pj) for the last n − j + 1 steps. The score for this path is given by Score = Prefix(i − 1) + Middle(i,j,δ1) + Suffix(j + 1) where Prefix(i − 1) is the precomputed score for the first (i − 1) steps on the 0-diagonal, Suffix(j + 1) is the precomputed score for the last (n − j + 1) steps on the δ-diagonal, and Middle(i,j,δ1) is the score for steps from i to j on the δ1 diagonal.
) − m(pj) for the last n − j + 1 steps. The score for this path is given by Score = Prefix(i − 1) + Middle(i,j,δ1) + Suffix(j + 1) where Prefix(i − 1) is the precomputed score for the first (i − 1) steps on the 0-diagonal, Suffix(j + 1) is the precomputed score for the last (n − j + 1) steps on the δ-diagonal, and Middle(i,j,δ1) is the score for steps from i to j on the δ1 diagonal.
Middle(i,j,δ1) depends on (unknown) δ1 and can be bounded by (precomputed) Bound(δ1) equal to the total number of 1s on the diagonal δ1. The idea of using Bound is to cut corners in the virtual database by estimating scores and not exploring variants with low estimated scores in the branch-and-bound algorithm:
Branch-and-Bound Algorithm
 |
The branch-and-bound algorithm can be adjusted for the case of modification-tolerant search for the expense of an increase in running time because of a larger alphabet of modifications. However, even in the modification-tolerant mode, the bound Bound(δ) leads to a significantly faster program than exhaustive generation of virtual database of modified peptides (for 200 types of modifications, the virtual database contains 106–107 entries per each peptide in the real database).
RESULTS
To estimate the efficiency of MS/MS database search on experimental spectra we used the sample of 36 annotated spectra of yeast tryptic peptides from Dancik et al. (1999) (Table 1). This sample contains 10 high quality spectra (p ≥ 0.4), 14 average quality spectra (0.3 < p< 0.4), and 12 poor quality spectra (p ≤ 0.3). These spectra were matched against the yeast protein database (≈120,000 peptides of 14 amino acids average length) in which every peptide was changed by k = 1 or k = 2 mutations. We also simulated a database of 10,000 peptides with typical frequencies of amino acids in human peptides, mutated every peptide in this database, and generated a typical spectrum for every peptide in the database of mutated peptides. We then searched every spectrum (of mutated peptide) against the database of (nonmutated) peptides. The length of peptides was fixed to 15 amino acids, which is close to the average length of tryptic peptides.
Table 1.
Matching Experimental Spectra against a Database of ≈120,000 Yeast Peptides with k = 1 and k = 2 Mutations
| No. | Peptide | Spectral quality, p | Rank in bound-and-branch algorithm | Rank in spectral alignment | ||||
|---|---|---|---|---|---|---|---|---|
| Mutations | Modifications | Modifications | ||||||
| k = 1 | k = 2 | k = 1 | k = 2 | k = 1 | k = 2 | |||
| 1 | KYNLSDQMDFVK | 0.58 | 1 | 1 | 1 | 1 | 1 | 1 |
| 2 | LSDFLHVSSGSDEK | 0.57 | 1 | 1 | 1 | 1 | 1 | 1 |
| 3 | EVTAALENAAVGLVAGGK | 0.56 | 1 | 1 | 1 | 1 | 1 | 1 |
| 4 | SPPVYSDISR | 0.55 | 1 | 3 | 1 | >500 | 3 | >500 |
| 5 | TGLSALMSK | 0.50 | 1 | 1 | 1 | 2 | 1 | 1 |
| 6 | MFHVDVAR | 0.50 | 1 | 1 | 1 | 190 | 1 | >500 |
| 7 | ATIDILHAK | 0.44 | 1 | 1 | 1 | 1 | 1 | 2 |
| 8 | HEHYLAYK | 0.44 | 1 | 1 | 1 | 230 | 1 | 16 |
| 9 | YVQNLANLATFFR | 0.42 | 1 | 1 | 1 | 1 | 1 | 1 |
| 10 | NQFDFVEGEISK | 0.42 | 1 | 1 | 1 | 1 | 1 | 2 |
| 11 | LDGIYVGIAPLVGK | 0.39 | 1 | 1 | 1 | 1 | 1 | 4 |
| 12 | LGLAPEGSK | 0.39 | 1 | 1 | 1 | 8 | 7 | 5 |
| 13 | LGWSLSFDA | 0.39 | 1 | 1 | 1 | 1 | 1 | 3 |
| 14 | AALQTYLPK | 0.39 | 1 | 3 | 1 | 3 | 1 | 1 |
| 15 | YLPDASSQVK | 0.38 | 1 | 1 | 1 | 3 | 1 | 14 |
| 16 | DTENGGEATFGGIDESK | 0.38 | 1 | 1 | 1 | 1 | 1 | 1 |
| 17 | IDSVSQLQNVAETTK | 0.37 | 1 | 1 | 1 | 1 | 1 | 69 |
| 18 | VLGAEEFPVQGEVVK | 0.37 | 1 | 1 | 1 | 1 | 1 | 23 |
| 19 | DTSHGEITLSAPYK | 0.36 | 1 | 1 | 1 | 1 | 1 | 13 |
| 20 | LEGVYSEIYK | 0.35 | 1 | 3 | 2 | 3 | 1 | 1 |
| 21 | IAYEIELGDGIPK | 0.35 | 1 | 1 | 2 | 1 | 1 | 1 |
| 22 | GAPEIDVLEGETDTK | 0.33 | 1 | 1 | 1 | 1 | 1 | 3 |
| 23 | GDLTSPDDMENAINESK | 0.32 | 1 | 1 | 1 | 4 | 1 | 3 |
| 24 | QDFAEATSEPGLTFAFGK | 0.31 | 1 | 2 | 1 | 1 | 1 | 1 |
| 25 | LFGDLNASNIDDDQR | 0.30 | 1 | 2 | 1 | 5 | 1 | 125 |
| 26 | DVDLIESMKDDIMR | 0.29 | 1 | 2 | 1 | 17 | 1 | 20 |
| 27 | LIPFLEYLATQQTK | 0.29 | 2 | 13 | 3 | 6 | 7 | 284 |
| 28 | LPNSNVNIEFATR | 0.27 | 1 | 2 | 2 | 35 | 9 | 14 |
| 29 | LFKPFLDPVTVSK | 0.27 | 1 | 14 | 2 | 18 | 8 | 10 |
| 30 | SPSALELQVHEIQGK | 0.27 | 1 | 1 | 1 | 1 | 1 | 20 |
| 31 | FYIINAPFGFSTAFR | 0.27 | 1 | 1 | 1 | 1 | 1 | 1 |
| 32 | TAPVSSTAGPQTASTSK | 0.26 | 1 | 1 | 1 | 1 | 1 | 1 |
| 33 | AHNGDLVNAIMSLSK | 0.23 | 2 | 3 | 2 | 1 | 23 | 256 |
| 34 | GSASGDLTFLASDSGEHK | 0.22 | 1 | 2 | 1 | 1 | 1 | 124 |
| 35 | DNQIYAIEKPEVFR | 0.21 | 1 | 22 | 1 | >500 | 29 | 445 |
| 36 | KPENAETPSQTSQEATQ | 0.15 | 3 | >500 | 8 | >500 | 286 | >500 |
Matching is done by three methods: Mutation-tolerant branch-and-bound algorithm; modification-tolerant branch-and-bound algorithm; and modification-tolerant spectral alignment algorithm. The table shows the ranks of the correct hits in the ranked list of top-scoring hits (rank 1 corresponds to correct peptide identification).
The efficiency of our mutation-tolerant database search was tested for simulated spectra at different values of the spectral quality p and the number of mutations k. The results for spectral convolution and spectral alignment are presented in Figures 5 and 6, respectively (MS-CONVOLUTION and MS-ALIGNMENT are available by contacting Z.M.). The first plot in Figure 5 demonstrates that even for k = 0 the spectral convolution (in this case it is equivalent to the Shared Peaks Count) leads to errors for poor quality spectra (p < 0.3), and the error rate grows very fast as the spectrum quality falls below p = 0.2. Because many such spectra may be present in high-throughput MS/MS projects, the results provide an explanation as to why many spectra in such projects are hard to interpret. They also indicate that interpretations of low quality spectra should be taken with caution even for the no mutations/modifications’ case. For p ≈ 0.5, the spectral convolution approach leads to nearly perfect predictions for k = 1 and provides 70%–80% accurate peptide identification for k = 2. The efficiency of the spectral convolution approach falls significantly for k > 2 and remains below 70% even for ideal (p = 1) spectra. The spectral alignment approach further improves the accuracy of protein identification (Figure 6). For k = 1, spectral alignment leads to nearly perfect predictions as soon as the quality of spectra exceeds p > 0.4. For p ≈ 0.5, the spectral alignment provides 80%–90% accurate peptide identification for k = 2. The accuracy of spectral alignment for k = 3 improves significantly as compared to spectral convolution but remains below 70% even for high quality spectra.
Figure 5.

Database search success rate of spectral convolution approach with SIMk scores for the simulated spectra. A match is successful if one of the indicated top-scored spectra matches a correct peptide. The number shown next to a curve is the number of mutated amino acids k.
Figure 6.

Database search success rate of spectral alignment approach for the simulated spectra. A match is successful if one of indicated top-scored spectra matches a correct peptide. The number next to a curve is the number of mutated amino acids k.
Table 1 shows the results of the spectral alignment approach for the sample of experimental spectra and reveals that most errors are associated either with short peptides or with low quality spectra. It also confirms that mutation-tolerant database search is an easier problem than modification-tolerant database search. For k = 1, the mutation-tolerant branch-and-bound algorithm makes no errors for high- and average-quality spectra, whereas the modification-tolerant branch-and-bound algorithm and spectral alignment make two errors for high and average quality spectra. We emphasize that the running time of the spectral convolution and the spectral alignment is not affected by considering modifications instead of mutations. In contrast, the running time of the branch-and-bound algorithm increases in case of modifications, because the alphabet of modifications is larger than the amino acid alphabet.
Let S be a spectrum of quality p and let P be a random (unrelated) peptide from a database. The spectrum S can match the peptide P just by chance and we are interested in the probability that the score of this match is above a threshold. If S has quality above p for a random peptide P, then P causes an error in our search algorithm. These random hits disguise the real similarities and lead to an increase in the error rate of the database search. The question then arises as to how many random hits have a higher score than the correct hit? In other words, what is the rank of the real hit in the ranked list of all hits?
Figures 5 and 6 (bottom) suggest that even in the case when the correct solution is not the highest scoring one, it remains at the top of the list of high scoring peptides. Figure 7 answers the question: “What is the rank of the correct hit in the ranked list of top-scoring database hits?”
Figure 7.

Success rate of the spectral alignment approach as a function of number of top scores at qualities p = 0.3 and p = 0.5 of the simulated spectra. A match is considered correct if the correct peptide is among t top scoring peptides in the database. The database consists of 10,000 peptides.
Figure 8 studies the same question for experimental spectra and demonstrates that the spectral alignment places correct peptides among the 500 top-scoring peptides in most cases. The only exceptions are spectra of very short peptides and low quality spectra.
Figure 8.

The success rate of database search versus the number considered top- scoring while matching experimental sample against yeast tryptic peptides database with (a) mutation-tolerant branch-and-bound algorithm, (b) modification-tolerant branch-and-bound algorithm, and (c) spectral alignment algorithm.
Considering the mutations-only case reduces the number of random hits and significantly improves the accuracy of the mutation-tolerant algorithm as compared with the modification-tolerant algorithm (Fig. 8). It justifies the two-stage filtration-and-verification approach to mutation-tolerant protein identification. At the first stage, the spectral alignment is used as a filter to identify t top-scoring peptides in the database. At the second stage each of these top-scoring peptides is verified by a comparison against the spectrum using a more accurate scoring function.
Conclusion
We described mutation-tolerant and modification-tolerant database search approaches that are based on spectral convolution, spectral alignment, and branch-and-bound algorithms. The algorithms have been tested on both experimental and simulated data and proved to be efficient for identification of mutated and modified peptides with up to two mutations/modifications. An alternative to this method is de novo interpretation followed by a BLAST-like database similarity search as proposed by Taylor and Johnson (1997) and Clauser et al. (1999). This approach gives hope for mutation-tolerant searches but is unlikely to succeed for modification-tolerant searches since de novo reconstruction of modified peptides remains an open problem.
A number of questions related to modification-tolerant MS/MS database searches remain open. One of them is the choice of parameter k (number of mutations) that is not known in advance. We propose to run MS-CONVOLUTION and MS-ALIGNMENT with a range of k and analyze top-scoring peptides for each k. Our tests indicate that the difference between the score of the top-scoring and the second-scoring peptides may provide an insight for the choice of k. The correct k often corresponds to the case when the gap in the scores of two top-scoring peptides is relatively large (compare with hikes in energy landscapes; Tiana et al. 2000). However, this is an empirical rule and further statistical analysis of MS/MS database search is needed.
Acknowledgments
The authors thank Karl Clauser for many critical comments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ppevzner@hto.usc.edu; FAX (213) 740-2424.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.154101.
REFERENCES
- Blom N, Gammeltoft S, Brunak S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Bushnell ML, Chen X. Efficient Branch and Bound Search With Application to Computer-Aided Design. Kluwer Academic Publishers; 1996. [Google Scholar]
- Clauser KR, Baker PR, Burlingame AL. The role of accurate mass measurement ( + /– 10ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- Dancik V, Addona T, Clauser K, Vath J, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. J Comp Biol. 1999;6:327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- Eng J, McCormack A, Yates J. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Amer Soc Mass Spect. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Fenyo D, Qin J, Chait BT. Protein identification using mass spectrometric information. Electrophoresis. 1998;19:998–1005. doi: 10.1002/elps.1150190615. [DOI] [PubMed] [Google Scholar]
- Gatlin C, Eng J, Cross S, Detter J, Yates J. Automated identification of amino acid sequence variations in proteins by hplc/microspray tandem mass spectrometry. Anal Chem. 2000;72:757–763. doi: 10.1021/ac991025n. [DOI] [PubMed] [Google Scholar]
- Gooley A, Packer N. The importance of co- and post-translational modifications in proteome projects. In: Wilkins W, Williams K, Appel R, Hochstrasser D, editors. Proteome Research: New Frontiers in Functional Genomics. Springer-Verlag; 1997. pp. 65–91. [Google Scholar]
- Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- ————— Electrospray mass-spectrometry for protein characterization. Trends Biochem Sci. 1995;20:219–224. doi: 10.1016/s0968-0004(00)89019-2. [DOI] [PubMed] [Google Scholar]
- Pevzner PA, Dancik V, Tang C. Mutation-tolerant protein identification by mass-spectrometry. J Comput Biol. 2000;7:761–770. doi: 10.1089/10665270050514927. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Mann M. Peptide mass spectrometry for homology searches and cloning of genes. J Protein Chem. 1997;5:481–490. doi: 10.1023/a:1026361427575. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Johnson RS. Sequence database searches via de novo peptide sequencing by tandem mass spectrometry. Rapid Comm Mass Spect. 1997;11:1067–1075. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1067::AID-RCM953>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Tiana G, Broglia RA, Shakhnovich EI. Hiking in the energy landscape in sequence space: A bumpy road to good folders. Proteins. 2000;39:244–251. [PubMed] [Google Scholar]
- Yates J, Eng J, McCormack A. Mining genomes: Correlating tandem mass-spectra of modified and unmodified peptides to sequences in nucleotide databases. Anal Chem. 1995b;67:3202–3210. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- Yates J, Eng J, McCormack A, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995a;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]