

Abstract
Bestrophinopathies, one of the most common forms of inherited macular degenerations, are caused by mutations in the BEST1 gene expressed in the retinal pigment epithelium (RPE). Both human and canine BEST1-linked maculopathies are characterized by abnormal accumulation of autofluorescent material within RPE cells and bilateral macular or multifocal lesions; however, the specific mechanism leading to the formation of these lesions remains unclear. We now provide an overview of the current state of knowledge on the molecular pathology of bestrophinopathies, and explore factors promoting formation of RPE-neuroretinal separations, using the first spontaneous animal model of BEST1-associated retinopathies, canine Best (cBest). Here, we characterize the nature of the autofluorescent RPE cell inclusions and report matching spectral signatures of RPE-associated fluorophores between human and canine retinae, indicating an analogous composition of endogenous RPE deposits in Best Vitelliform Macular Dystrophy (BVMD) patients and its canine disease model. This study also exposes a range of biochemical and structural abnormalities at the RPE-photoreceptor interface related to the impaired cone-associated microvillar ensheathment and compromised insoluble interphotoreceptor matrix (IPM), the major pathological culprits responsible for weakening of the RPE-neuroretina interactions, and consequently, formation of vitelliform lesions. These salient alterations detected at the RPE apical domain in cBest as well as in BVMD- and ARB-hiPSC-RPE model systems provide novel insights into the pathological mechanism of BEST1-linked disorders that will allow for development of critical outcome measures guiding therapeutic strategies for bestrophinopathies.
Keywords: Bestrophinopathies, canine model, macula, lipofuscin, hiPSC-RPE, RPE apical microvilli, RPE-photoreceptor interface
1. Introduction
The retinal pigment epithelium (RPE) is an organized monolayer of highly specialized cells responsible for a sustained interaction with photoreceptors, a mission pivotal for normal visual function and retinal preservation (Hageman and Johnson, 1991; Bok, 1993; Strauss, 2005). These diverse functions are orchestrated by a broad array of molecules designed to access specific cascades of intra- and intercellular interactions in the retina (reviewed in: Steinberg, 1985; Strauss, 2005; Kevany and Palczewski, 2010; Bonilha, 2014). One of such RPE-specific molecules is BESTROPHIN 1 (OMIM #607854), a transmembrane channel protein encoded by BEST1 gene and associated with the RPE basolateral membrane (Marmorstein et al., 2000; Sun et al., 2002; Gomez et al., 2013). Although BESTROPHIN 1 has been extensively studied and described as a multifunctional protein implicated in mediating anion transport, regulation of calcium signaling and cell volume (Rosenthal et al., 2005; Hartzell et al., 2008; Strauss et al., 2014; Kane Dickson et al., 2014; Yang et al., 2014; Milenkovic et al., 2015), its multifaceted nature and complex interactions with photoreceptors in health and disease still remain elusive.
Mutations in BEST1 have been causally associated with several clinically heterogeneous retinal disorders, collectively termed bestrophinopathies (Petrukhin et al., 1998; Seddon et al., 2001; Yardley et al., 2004; Schatz et al., 2006; Burgess et al., 2008; Davidson et al., 2009; Boon et al., 2009a). The BEST1 mutational spectrum underlying these retinopathies varies greatly, and involves over 200 distinct mutations (Boon et al., 2009a; Pasquay et al., 2015; Yang et al., 2015). Additionally, a considerable variability in age at onset, rate of disease progression and phenotypic expression has been reported, not only between unrelated patients harboring the same mutation, but also among affected individuals within a single family (Kramer et al., 2000; Kinnick et al., 2011; Bitner et al., 2012; MacDonald et al., 2012; Boon et al., 2013). This phenomenon of phenotypic and allelic heterogeneity highlights a significant phenotypic overlap among BEST1-linked disorders, posing a challenge in both determining the diagnostic specificity as well as predicting the outcomes of visual impairment. In recent years, the complexity of genotype-phenotype correlation in bestrophinopathies has proven difficult to explain with traditional models of disease pathogenesis (Allikmets et al., 1999; Boon et al., 2007; Yu et al., 2007; Querques et al., 2009; Booij et al., 2010; Liu et al., 2016), indicating a presence of potential genetic modifiers or still undetermined Best1 protein interactors, involvement of environmental components or a combination of both. This likely would point to a complex interplay of genetic susceptibility factors and modifiable environmental stimuli utilizing novel signaling pathways in the retina.
The broad spectrum of clinical presentations in bestrophinopathies ranges from the widespread symptoms affecting peripheral retina and vitreous in a rare condition of vitreoretinochoroidopathy (ADVIRC; OMIM#193220) to the well-defined clinical abnormalities restricted to the macula and central retina in Best Vitelliform Macular Dystrophy (BVMD) and autosomal recessive bestrophinopathy (ARB). BVMD (a.k.a. VMD2, OMIM#153700), inherited as an autosomal dominant trait with incomplete penetrance, and the recessive form (ARB; OMIM#611809) are the most common and best explored juvenile macular dystrophies among bestrophinopathies, characterized by a markedly abnormal electrooculogram (EOG) accompanied by an excessive accumulation of lipofuscin material within RPE cells, formation of focal and multifocal subretinal lesions, and consequently, loss of central vision (Pianta et al., 2003; Boon et al., 2009b; Pasquay et al., 2015; Fung et al., 2015).
An abnormal accumulation of lipofuscin is a major risk factor implicated in different forms of macular degeneration (Delori et al., 1995a; Marmorstein et al., 2002; Gerth et al., 2007; Biarnes et al., 2015), and also the most notable and consistent pathological finding in BEST1-linked maculopathies, serving as an indirect biomarker of metabolic activity between the photoreceptor outer segment (POS) turnover and RPE phagocytosis (Bakall et al., 2007; Piñeiro-Gallego et al., 2011; Lei et al., 2013; Singh et al., 2013a). Recent advances with noninvasive retinal imaging modalities have enabled detailed mapping and quantification of fundus autofluorescence (FAF) in vivo, and its correlation with increased levels of lipofuscin components in the aged and diseased retinae (Delori et al., 1995a, 1995b; Brunk and Terman, 2002; Boon et al., 2008; Duncker et al., 2014); however, the polymorphous nature of lipofuscin material and consequences of its buildup in the retina are still controversial.
To begin to address these questions, we used the spontaneous canine BEST1 disease model (cBest a.k.a. canine multifocal retinopathy, cmr) (Guziewicz et al., 2007; Zangerl et al., 2010; Guziewicz et al., 2011; Beltran et al., 2014; Singh et al., 2015) to characterize lipofuscin fluorophores in the cBest1 mutant RPE, and explore factors leading to the formation of subretinal lesions in BEST1-associated maculopathies. This study highlights matching spectral profiles of the native lipofuscin autofluorescence between human and dog bestrophinopathies, as well as robust biochemical and structural alterations at the RPE-photoreceptor interface that trigger formation of vitelliform lesions.
2. Canine models of human BEST1-related dystrophies
Over the recent decades, hundreds of spontaneous genetic conditions have been described in dogs, and most of them with clinically and genetically close counterparts to the human disorders (OMIA: http://omia.angis.org.au). The naturally occurring canine models of inherited retinal disorders in man have proved crucial in the investigation of disease mechanisms and development of new therapeutic strategies (Acland et al., 2001; Komaromy et al., 2010; Beltran et al., 2012; Downs et al., 2016).
We have recently characterized a naturally occurring autosomal recessive disorder in dogs, canine Best (cBest a.k.a. canine multifocal retinopathy, cmr), which is caused by the same genetic defects as human bestrophinopathies, and captures the full range of clinical manifestations observed in patients (Guziewicz et al., 2007; Zangerl et al., 2010; Guziewicz et al., 2011; Beltran et al., 2014; Singh et al., 2015). To date, cBest retinopathy affects thirteen dog breeds worldwide and results from any of the three distinct mutations identified in the canine BEST1 (cBEST1) gene. The premature stop mutation located in the first coding exon of cBEST1 (c.73C>T/p.Arg25* alias R25X) was recognized among Mastiff-related breeds (Guziewicz et al., 2007; Guziewicz et al., 2011); a missense change (c.482G>A/p.Gly161Asp alias G161D) affecting cBest1 protein folding and trafficking was discovered in a rare breed of Coton de Tulear (Guziewicz et al., 2007; Guziewicz et al., 2012); whereas the frameshift mutation (c.1388delC/p.Pro463fs alias P463fs), truncating BESTROPHIN 1 C-terminus, was shown to segregate in Lapponian Herders (Zangerl et al., 2010). All three mutations lead to the consistent clinical phenotype in homozygous affected dogs, and model all major aspects of the disease-associated mutations as well as their molecular consequences described in man (Guziewicz et al., 2007; Zangerl et al., 2010; Guziewicz et al., 2011; Guziewicz et al., 2012; Beltran et al., 2014; Singh et al., 2015).
The spectrum of clinical and molecular features recapitulated, including the salient predilection of lesions to the canine macular region, makes cBest an extremely attractive model system not only for addressing principles behind the molecular pathology of bestrophinopathies, but also for validating new therapeutic strategies (Guziewicz et al., 2013; Beltran et al., 2014; Singh et al., 2015). Currently, the loss of function model (cBest1-R25X), the C-terminal deletion (cBest1-P463fs), and the compound heterozygous model (cBest1-R25X/P463fs) harboring both mutations in a heterozygous state, are available for research study. We used these translational models as a platform for in-depth investigation of the pathogenic sequence in BEST1-related disorders, and addressed the central question in the disease pathogenesis, the intrinsic susceptibility of the macula to its primary detachment in bestrophinopathies.
3. Lipofuscin as a pathological agent in BEST1-associated maculopathies
3.1. Excessive buildup of lipofuscin within RPE: a hallmark of canine and human bestrophinopathies
Retinal pigment epithelium has been recognized as a primary target site for many retinal degenerative disorders, including bestrophinopathies. Although the disease-causative gene product, BESTROPHIN 1, has been associated with basolateral membrane of RPE cells (Marmorstein et al., 2000; Strauss et al., 2014), the predominant clinical manifestations in BVMD and ARB patients pertain to the abnormal apposition of RPE apical surface to the neurosensory retina, delineated by intense FAF signals in vivo and aggregation of autofluorescent material when examined ex vivo (O'Gorman et al., 1988; Spaide et al., 2006; Bakall et al., 2007; Sparrow et al., 2016). As the canine Best's disease model mirrors both the altered topographic cSLO-FAF distribution as well as the end-stage pathological findings described in patients (Guziewicz et al., 2007; Beltran et al., 2014; Singh et al., 2015), this model was used to examine the spectral nature of the RPE-associated autofluorescent constituents using Hyperspectral Autofluorescence Imaging (HAI) technology (Gao et al., 2012; Smith et al., 2014).
Baseline evaluation of the wild-type canine retinae (age range: 10 mos-6 yrs) demonstrated a gradual increase in native autofluorescence within RPE cells (Fig. 1A). This slow yet steady augmentation of granular yellow to brown-tan deposits reflects the natural aging process of the canine retina and, consistent with the studies in human, increases over the dog's lifespan in a linear manner (Fig. 1A). Furthermore, in agreement with clinical assessments in both BVMD and ARB patients (Boon et al., 2013; Sparrow et al., 2016), the cBest model exhibits an abnormal distribution of FAF intensities associated with focal and multifocal retinal detachments observed by in vivo imaging (Fig. 1B,C). Canine retinae harboring a nonsense mutation (c.73C>T/p.R25X) in cBEST1 gene do not express BESTROPHIN 1 protein, and show both, increased FAF signals associated with areas of separations of the neural retina from the underlying RPE, and an excessive accumulation of lipofuscin-like material within RPE when compared to the age-matched controls (Fig. 1B-E). Representative H&E and DIC images of cBest1-R25X-affected and control retinae are shown in Fig. 1D,E. Canine cBest1-R25X/P463fs compound heterozygous and cBest1-P463fs homozygous models (Zangerl et al., 2010; Beltran et al., 2014) display similar pathological characteristics.
Figure 1. Abnormal buildup of autofluorescent material within RPE cells is a hallmark of canine bestrophinopathies.

(A) Accumulation of autofluorescent debris (arrows) is an age-dependent process in normal canine RPE cells. (B) Confocal scanning laser ophthalmoscopy fundus autofluorescence (cSLO FAF) images of a 20-month-old affected (OS) and an age-matched wild-type control eye (OD). Note the increased fundus autofluorescence signals scattered throughout the supero-temporal quadrant and adjacent to the optic disc in the mutant retina. Boxed areas identify regions analyzed ex vivo by Hyperspectral Autofluorescence Imaging (HAI) and immunohistochemistry (IHC). Note the missing autofluorescence over the central lesion (OS). (C) Corresponding SD-OCT images taken at the position of the green line in the above images (B) of the temporal area. A characteristic bullous separation of the neuroretina from the RPE is demonstrated in the mutant retina; the structurally intact age-matched wild-type control retina is shown for comparison. (D) Immunostaining against Best1 protein (red) in cryosections from canine wild-type and cBest1-R25X-affected retinae. Note the presence of native autofluorescent granules (green) and absence of Best1 labeling within affected RPE cells, which appear hypertrophic on the corresponding H&E image; there is a discreet separation of the outer segments from the apical RPE in the affected retina (arrowhead). Nuclei were stained with DAPI. (E) Confocal microscopy images of a 20-month-old mutant and an age-matched wild-type retinae showing an excessive accumulation of autofluorescent deposits (arrows) within the affected RPE. ONL: outer nuclear layer; PRs: photoreceptors; RPE: retinal pigment epithelium; CH: choroid; Scale bars: 10µm applies to all photomicrographs in (A) and (E); 40µm applies to all (D) panels.
To investigate the nature of these RPE inclusions and address the question of whether the buildup of the lipofuscin-like deposits promotes development of focal to multifocal retinal detachments, cBest samples with early pre-vitelliform and vitelliform lesions from supero-temporal quadrant were selected for HAI and IHC analyses (for details, see Supplementary Materials and Methods). The HAI-determined spectral signatures derived from canine Best1-R25X mutant samples were then compared to the HAI emission profiles of human BVMD (hBest1-T6R and -Y227N) RPE (Mullins et al., 2005; Mullins et al., 2007), as well as to the healthy controls from both species (Fig. 2). Comparative HAI analysis of endogenous RPE fluorophores confirmed the excessive accumulation of autofluorescent material in BEST1 mutant as a common feature of both human and canine bestrophinopathies (Fig. 2A-E), and further revealed not only matching spectral profiles between Best1-affected and control samples of dog (Fig. 2B,D) and human (Fig. 2E), but also analogous emission profiles between human and canine bestrophinopathies (Fig. 2F,G). Overall, the endogenous RPE deposits emitted intense fluorescent signals across the recorded spectrum, with emission intensity, Iem, significantly higher in cBest1-R25X versus age-matched wild-type (WT) control (Iem ≥3 fold; ***p<0.000002) across the interval as depicted in a box plot (Fig. 2C). The spectral signature was highly comparable among all samples examined, with a major emission peak at 536-569 nm consistent with lipofuscin (Ben Ami et al., 2016), followed by a shoulder peak at 650 nm (Fig. 2B-G).
Figure 2. Hyperspectral autofluorescence imaging (HAI) reveals analogous emission profile between human and canine bestrophinopathies.

(A) Representative confocal microscopy images of the native RPE autofluorescence in cryosections from BVMD patients (hBest1-T6R and -Y227N), control subjects (Ctr) of comparable age, and canine bestrophinopathy model (cBest1-R25X) by HAI analysis. Abundant accumulation of autofluorescent material within RPE cells is a common feature in both human and canine bestrophinopathies. PRs: photoreceptors; RPE: retinal pigment epithelium; CH: choroid. (B) HAI analysis of endogenous RPE fluorophores in canine cBest1-R25X-affected and age-matched WT RPE revealed a major emission peak between 536-569 nm consistent with lipofuscin signals, followed by a shoulder peak at 650 nm. (C) Statistical analysis confirmed accelerated accumulation of lipofuscin granules in the affected tissue with emission intensity (Iem) 3-fold higher in the mutant vs age-matched control (***p<0.000002). (D) Direct comparison of the individual HAI emission data points from cBest1- R25X and cBest1-WT RPE using linear regression across all wavelength intervals. After enhancing the amplitude of the baseline Iem signal (cBest1-WT adjusted), the overlapping emission peak profiles of cBest1 mutant and cBest1-WT fell within the SEM range of each other over most wavelength intervals. (E) HAI of the native RPE autofluorescence from BVMD patients (hBest1-T6R and -Y227N averaged) and control donor eyes demonstrating parallel RPE emission spectra profiles. Data analysis was performed on hyperspectral datasets derived from a mean of four BVMD-affected or control retinae with three cryosections per eye and six ROIs per section (nBVMD=72 and nCtr=72); the color-corresponding shaded areas represent mean ± SEM across the wavelength intervals. (F) HAI-profiled intrinsic RPE fluorescence from patients harboring either hBest1-Y227N or -T6R mutation (plotted individually) and cBest1-R25X model illustrating matching spectral profiles among the disease samples from both species, varying only in the emission intensity, consistently higher in samples from elderly BVMD donors than that of young cBest retinae. (G) HAI emission peak signatures of BVMD (hBest1-T6R and -Y227N averaged) and cBest1-R25X (cBest1-R25X adjusted) indicating similar profiles of RPE-associated autofluorescence constituents in human and canine bestrophinopathies. Scale bar: 10µm applies to all (A) panels.
Because Iem amplitude was considerably higher in samples from older human donor eyes versus young canine retinae, to directly compare the individual HAI emission data points across all wavelength intervals tested, irrespective of the emission intensity, we used a linear regression to identify a monotonous transformation to boost the amplitude of the baseline Iem signal without affecting the overall shape of the profile (for details, see Supplementary Materials and Methods). As indicated by the initial HAI analysis, the emission peak profiles of the cBest1-R25X mutant and canine WT-adjusted were nearly identical, and fell within the SEM range of each other over most wavelength intervals (Fig. 2D). An analogous linear regression equation was identified for cBest1-R25X mutant (cBest1-R25X adjusted) for its direct comparison to the HAI emission pattern derived from BVMD (hBest1-T6R and -Y227N averaged), revealing a highly overlapping emission spectral profile of the RPE-associated lipofuscin constituents between human and canine BEST1-maculopathies (Fig. 2G).
In conclusion, consistent with the findings in a healthy human retina (Kennedy et al., 1995; Delori et al., 2001), we now show that lipofuscin granules accumulate as a function of age in a normal canine RPE. Interestingly, no apparent disease-specific HAI emission peaks in either canine or human Best1-mutant RPE were detected when compared to age- and retinal location-matched normal controls (Fig. 2). Although this disease aspect should be investigated further using other advanced RPE diagnostic tools, our HAI data strongly suggest that no intrinsic disease-specific RPE fluorophores underlie pathophysiology of bestrophinopathies, and the anomalous increase of lipofuscin bisretinoids observed in BEST1-retinopathies reflects rather an inability of RPE cells to properly metabolize POS (Singh et al., 2013a), and, in essence, manifests as a premature senescence of the affected RPE cell monolayer.
Furthermore, HAI and linear regression analyses exposed matching spectral signatures between cBest1-R25X and hBest1-T6R and -Y227N affected RPE, suggesting a similar composition of endogenous RPE-associated fluorophores between humans and dogs. These data support the recent ex vivo findings from a set of HAI-profiled healthy human eyes, assigning the primary spectral emission peak to the abundant lipofuscin-melanolipofuscin signal, and suggesting a presence of a third candidate family of RPE-fluorophores that requires further biochemical characterization (Ben Ami et al., 2016). Considering the corresponding hyperspectral characteristics, the dog should serve as an excellent model system assisting in further studies on the biochemical composition of the undefined families of RPE fluorophores.
3.2. Bisretinoids-mediated cholesterol accumulation in BVMD and cBest model
Studies in polarized ARPE-19 cells demonstrated that A2E, the most prominent lipofuscin fluorophore, contributes to the accumulation of free and esterified cholesterol, perturbing its metabolism (Lakkaraju et al., 2007). The mechanism of this bisretinoid-induced cholesterol accumulation within RPE cells was further explored, signifying the role of trapped cholesterol macromolecules in the disruption of RPE autophagy (Toops et al., 2015; Tan et al., 2016). It is unclear however, whether the A2E-mediated cholesterol accumulation reported in vitro contributes to the pathogenesis and progression of other RPE-linked disorders such as BVMD. To that end, we examined if the chronic accumulation of lipofuscin bisretinoids in the Best1-mutant RPE is indeed associated with increased levels of cholesterol and/or abnormal cholesterol trafficking in human and canine bestrophinopathies (Fig. 3). We found that lipofuscin granules and free cholesterol (filipin-positive puncta) were strongly positively associated in the RPE cells of both human BVMD (Pearson's correlation coefficient r = 0.85±0.03) and canine Best1-mutant RPE (r = 0.77±0.02) (Fig. 3A,B). This positive correlation was in agreement with the emission profile analyses where the somewhat less intense blue fluorescent signals of filipin-positive aggregates were co-distributed with the bright green lipofuscin granules (Fig. 3B).
Figure 3. Bisretinoids-mediated intracellular cholesterol accumulation in BVMD- and cBest-affected RPE.
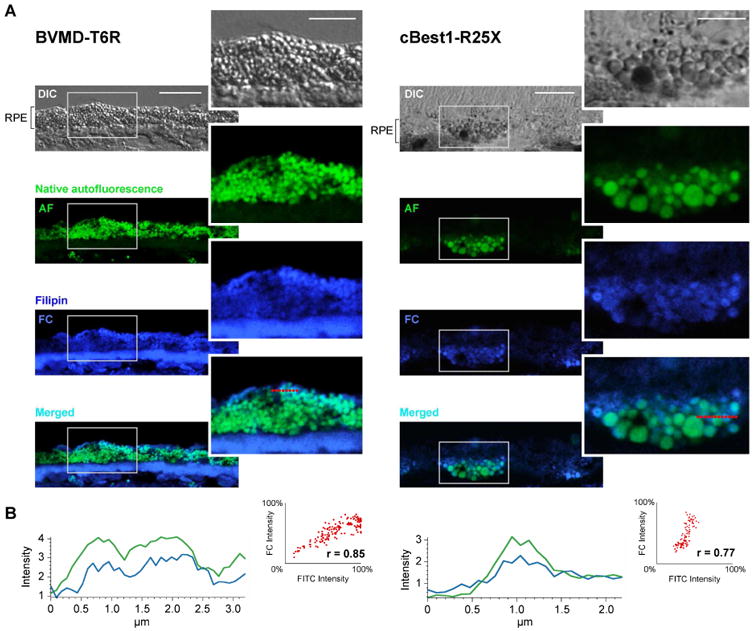
(A) Representative DIC, native autofluorescence (AF), filipin-stained free cholesterol (FC), and AF/FC-merged confocal photomicrographs of RPE cells from human BVMD (hBest1-T6R) and canine model (cBest1-R25X). Lipofuscin granules (green) and free cholesterol (filipin-positive puncta, cobalt blue) were highly associated in BVMD and cBest RPE cells. (B) Both the analysis of AF and FC emission profiles as well as Pearson correlation coefficient ‘r’ values indicated a strong positive correlation between endogenous lipofuscin fluorophores (FITC) and filipin-stained-FC in BVMD-T6R and canine cBest1-R25X RPE (r =0.85±0.03 and r =0.77±0.02, respectively). Scale bars: 10µm; magnified images: 5µm.
This strong positive correlation between lipofuscin-derived autofluorescence and cholesterol levels detected ex vivo in canine and human bestrophinopathies supports the original in vitro observations by Lakkaraju et al. (2007), and strongly suggests that the perturbed cholesterol metabolism and its extensive downstream consequences (Toops et al., 2015; Tan et al., 2016) contribute to the homeostatic imbalance in cBest- and BVMD-RPE. It has been shown that the tight control of intracellular cholesterol levels is essential for proper autophagosome-lysosome interactions, and that excess of cholesterol within RPE can interfere with intracellular trafficking, inhibiting lysosomal degradation and consequently, the autophagic clearance (Lakkaraju et al., 2007; Toops et al., 2015). Furthermore, as demonstrated in the recent study by Tan et al. (2016), overload of bisretinoids/cholesterol compromises the protective responses against complement-mediated damage, leading to mitochondrial fragmentation and oxidative stress in the RPE cells (Tan et al., 2016).
Detailed experiments focused on the gene expression controlling cholesterol metabolism and dysregulation of lipid homeostasis in Best1-mutant RPE are beyond the scope of this review. Such analyses, however, are in progress and constitute the main objective of the next phase of research. As there is no doubt that the overload of both bisretinoids and cholesterol leads to the severe perturbations of RPE cell homeostasis, a chronic accumulation of lipofuscin has been implicated in the pathology of numerous inherited retinal disorders, including AMD and Stargardt disease (Spaide, 2003; Cideciyan et al., 2004; Lakkaraju et al., 2007; Petrukhin, 2007; Tan et al., 2016), and as such constitutes a nonspecific disease biomarker that is unlikely to trigger per se the retinal separation in bestrophinopathies.
4. Dysregulation of lipid metabolism and its impact on subretinal microenvironment in cBest
Besides the increase in unesterified cholesterol clearly associated with an excess of bisretinoid compounds within Best1-mutant RPE, comparable distribution and intensity of filipin staining was observed across the rest of the tissue in the wild-type and cBest retinae (Fig. 4A-C). Consistent with the studies on filipin staining patterns in human retina, free cholesterol, by far the most predominant form of sterols in the vertebrate retina (Fliesler and Bretillon, 2010), was broadly distributed throughout all layers of the canine retina with no difference detected in the cBest1-R25X-affected vs age-matched WT controls (Fig. 4A,B). However, we did observe a noticeable difference in the esterified cholesterol (EC) levels between cBest model and WT controls after subjecting the samples to ethanol-based extraction followed by cholesterol esterase pretreatment (for details, see Supplementary Materials and Methods) to visualize the EC content (Fig. 4D,E). While in a healthy human retina EC is mainly associated with Bruch's membrane (Rudolf and Curcio, 2009; Zheng et al., 2012), in a normal canine retina this form of sterol was faintly detected within choroid (Fig. 4D), whereas in cBest retina a substantial increase of filipin-stained EC fluorescence was associated with photoreceptor outer segment (Fig. 4E, inset). Because filipin-sterol fluorescence undergoes rapid photobleaching, it is not quantifiable; therefore, the EC findings were verified by alternative and more stable neutral lipid stains, a fluorescent probe BODIPY 493/503 and the classic lipid-soluble dye Oil Red O (ORO), both known to stain EC (Rudolf and Curcio, 2009).
Figure 4. Dysregulation of lipid metabolism defines canine bestrophinopathy as an RPE-photoreceptor interface disease.
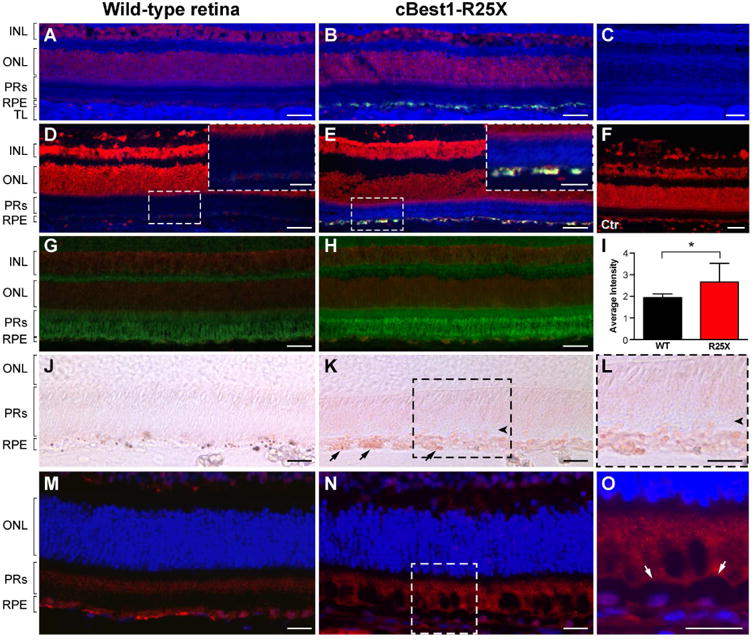
Spatial distribution of unesterified (free) cholesterol visualized by sterol-binding probe filipin (cobalt blue) in a normal (A) and cBest1-R25X-affected (B) retinae. Note the excess of autofluorescent RPE deposits in the diseased tissue (B). Filipin-bound free cholesterol was found to be widely distributed across all layers of the canine retina, and appeared to be primarily associated with plasma membrane in ONL and INL, in contrast to the remaining retinal layers where it was found in both cell membranes and somata (C); tapetum lucidum (TL), a choroidal modification characteristic to carnivores, showed the most intense staining (A-C). (D, E) Histochemical detection of esterified cholesterol (EC, cobalt blue) in a 12-month-old cBest1-affected vs age-matched control retina. Note the abnormal buildup of autofluorescent material (yellow-green) within RPE and an increase of EC-filipin associated with photoreceptor layer in the mutant (E, close-up). (F) Non-enzyme control: unesterified cholesterol was extracted with ethanol and sections stained with filipin, but omitting the cholesterol esterase treatment (no filipin labeling). (G, H) Representative retinal cryosections from cBest (H) and age-matched control (G) stained with a fluorescent neutral lipids tracer dye BODIPY 493/503 (green). (I) Quantification of EC-BODIPY 493/503 signals in photoreceptor outer segment layer between wild-type and cBest1-R25X-affected retinae. The observed difference was assessed as statistically significant using unpaired t-test (*p<0.05). (J-L) EC distribution profile in canine wild-type and cBest1-affected retinae assayed with a lysochrome Oil Red O (ORO, rose). ORO-positive inclusions within the affected RPE (arrows) and in the subretinal space are shown in (K) and (L, close-up); arrowheads point to the discreet separation of the outer segments from the RPE apical surface in cBest (K, L). (M-O) Anti-4-HNE labeling (red) in the mutant and control retinae. A scattered distribution of HNE-adducts within outer segments was observed in the affected retina (N and O, close-up) outlining the apical contour of the hypertrophic RPE cells (O, arrows). Nuclei were counterstained with propidium iodide (A, B, D-H) or DAPI (M-O). INL: inner nuclear layer; ONL: outer nuclear layer; PRs: photoreceptors; RPE: retinal pigment epithelium; TL: tapetum lucidum; Scale bars: 40µm (A-H), and 20µm (D-E close-ups, and J-O).
A series of experiments with BODIPY 493/503 as well as ORO confirmed the EC staining pattern in the Best1-mutant retina (Fig. 4G-L). The bright green fluorescent signals emitted by BODIPY 493/503 were present across all layers in both the normal control and cBest retinae (Fig. 4G,H). In the cBest retina, however, a significant increase (*p<0.05) of EC-BODIPY 493/503 was detected in the photoreceptor outer segment layer with the highest staining intensity in the region bordering the inner segment (IS) and at the OS tips (Fig. 4H,I). Similar EC distribution profile in the mutant retina was observed with ORO assay where, in addition, numerous EC-rich inclusions were found within cBest1-R25X mutant RPE and in the subretinal space as ORO-positive intracellular and extracellular deposits (Fig. 4K arrows, Fig. 4L, higher magnification). Note the subtle RPE detachment from the retinal surface in cBest retina (Fig. 4K,L, arrowheads).
To determine whether the observed dysregulation of cholesterol homeostasis in the mutant retina is associated with enhanced lipid peroxidation, we examined the levels of 4-hydoxy-2-nonenal (HNE), one of the most abundant by-products of lipid peroxidation (Esterbauer et al., 1985). As shown in Fig. 4, increased levels of HNE-adducts were present in cBest vs age-matched control retinae (Fig. 4M-O). While in the WT canine retina HNE was primarily associated with RPE and IS/OS border, in the mutant retina, the staining pattern of HNE showed in addition a patchy distribution across OS layer that was characteristic for the retinal regions located within close proximity to the areas of established subretinal lesions, and associated with hypertrophic RPE cells featuring their abnormal scalloped apical membrane (Fig. 4O, arrows).
Numerous studies have documented the essentiality of cholesterol and its tight homeostatic regulation in the maintenance of outer segment structure and function (Fliesler and Bretillon, 2010; Albert et al., 2016). Metabolic defect in any aspect of this process can lead to abnormal accumulation of cholesterol and its toxic derivatives resulting in retinal degeneration impacting both the structural integrity and function of POS (Fliesler and Bretillon, 2010). Such alterations are likely due to a combination of factors, including the requirement for cholesterol in rhodopsin activation, formation of cholesterol-rich signaling microdomains as well as the role of cholesterol in regulating pools of phosphoinositides (Albert et al., 2016). One such cholesterol-derived compound, 7-ketocholesterol (7KCh), is an oxidized form of cholesterol produced by non-enzymatic oxidation at C7 (Moreira et al., 2009; Rodriguez and Fliesler, 2009). This oxysterol is considered a potent pro-inflammatory agent playing a role in activation of NLRP3 inflammasome and associated with choroidal neovascularization, formation of AMD drusen and aging (Rodriguez and Fliesler, 2009; Moreira et al., 2009; Rodriguez et al., 2014). At the molecular level, excess 7KCh not only alters the biophysical properties of plasma membranes (Bach et al., 2008; Ruggiero et al., 2012), but also constitutes a ready substrate for esterification, producing a highly unstable esterified 7-keto molecule that re-releases 7KCh. The higher the degree of fatty acid unsaturation, the more unstable the esterified 7-KCh molecule is (Bretillon et al., 2008; Lee et al., 2015). Thus, the highly unsaturated fatty acids of the photoreceptor OS would provide a highly unstable variant of this compound, compromising the subretinal microenvironment, and consequently, the interplay of RPE-photoreceptor complex. Such changes in concentration of metabolites surrounding POS may also disturb the hydration, bonding properties and viscosity of the interphotoreceptor matrix (IPM), and misregulate the volume of subretinal space affecting retinal adhesion (Marmor, 1994; Pianta et al., 2003; Milenkovic et al., 2015).
It is possible, therefore, that the altered distribution and increased levels of cholesterol esters and HNE-adducts within photoreceptor layer of the cBest1-affected retinae may indeed indicate a compromised IPM domain, exposing thereby a new role of Best1 in modulating the homeostatic milieu of subretinal space. While the excess of EC could function as an atypical chronic inflammatory stimulus interrelated to the impaired calcium signaling and fluid flow, such as 7-KCh (Lakkaraju et al., 2007; Moreira et al., 2009; Wang et al., 2010; Singh et al., 2013a; Strauss et al., 2014), the lipoperoxidation product, HNE, may act as a stress signaling molecule (Shen et al., 2007; Perluigi et al., 2012), both predisposing retina to the loss of adhesive forces in response to these pathological stimuli.
5. Bestrophinopathy: an RPE-photoreceptor interface disease
5.1. Compromised RPE-photoreceptor interaction in cBest
The RPE-photoreceptor interface is an area of crucial importance to proper retinal function (Hageman and Johnson, 1991). Dysregulation of lipid metabolism documented in cBest1-mutant retina pointed to the presence of biochemical perturbations in the subretinal space reflecting homeostatic imbalance in the delicate IPM microenvironment. To gain additional insight into the spectrum of abnormalities at the RPE-photoreceptor interface in cBest, and identify further potential factors that may play a role in lesion formation in bestrophinopathies, a set of RPE-, photoreceptor- and IPM-specific markers were used for IHC evaluation of cBest vs control retinae (Fig. 5). A striking difference between the two was the retraction of RPE apical projections (a.k.a. microvilli, MV) in all cBest1-R25X-affected retinae as evidenced by the absence of MV labeling using a set of markers known to be expressed at the RPE apical membrane and along its extensions (Fig. 5A-F). Specific immunostaining against MCT1 (Monocarboxylate Transporter, Member 1), RLBP1 (Retinaldehyde Binding Protein 1), and EZRIN was present in both the structurally normal and mutant retinae, but in the latter the immunolabeling was limited to the apical surface of RPE monolayer, which lacked the MV extensions that interdigitated with POS (Fig. 5B, D, and F). This finding was age- and sex-independent and observed in all canine BEST1 mutant eyes examined after disease onset, in both the central retina as well as in the periphery. In contrast, the immunolabeling pattern of EZRIN, RLBP1 and MCT1 in cBest carrier retinae was comparable to that of the WT (data not shown). Confocal imaging of the mutant retinae immunolabeled by anti-EZRIN in combination with PNA lectin, known for its selective binding to the cone insoluble extracellular matrix microdomains (Johnson and Hageman, 1991; Mieziewska et al., 1993), confirmed MV retraction and revealed disordered and considerably disrupted cone-associated IPM ensheathment (Figs. 5G-J, and 6A-D). While the retraction of the longer RPE processes embracing cones was evident throughout the cBest1 mutant retinae, in both regions with bullous detachments as well as in non-detached areas, the much shorter residual MV associated with the longer rod outer segments were occasionally seen. However, the RPE apical extensions associated with rods are much shorter and finer than the cone-ensheathing processes, hence somewhat more difficult to visualize and track; yet at higher magnification, these small structures of rod-associated RPE extensions appeared disordered and interlaced (Fig. 5J). An expanded immunohistochemical study defined a bilayered extracellular compartmentalization of the cone specific escheatment responsible for a normal apposition of RPE-cone OS (Fig. 6C-D). This well-defined extracellular structure was lost in the cBest1-affected retinae, revealing a dearth of the intrinsic RPE-associated apical microvillus layer accompanied by a fragmented external cone-specific matrix domain (Fig. 6C-D, insets). As a result, cone OS stripped of their protective sheaths appeared undermined with no direct contact with the underlying RPE (Fig. 6).
Figure 5. Compromised cone-associated RPE apical domain and insoluble interphotoreceptor matrix (IPM) in canine bestrophinopathy.
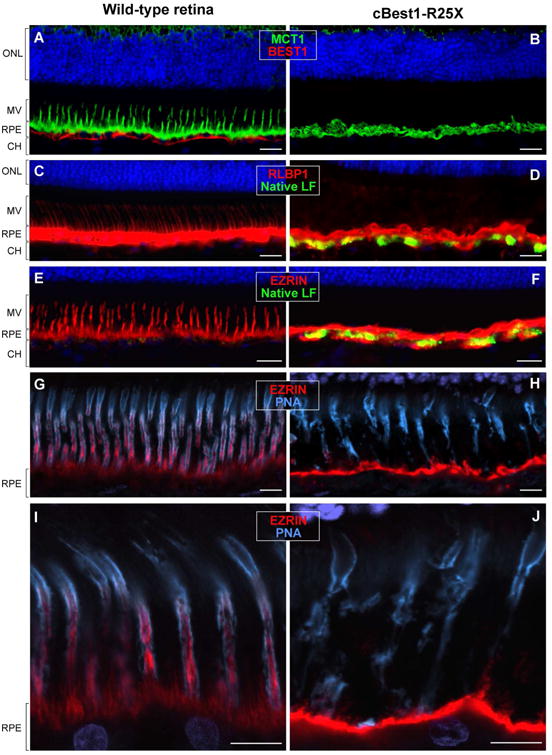
(A-J) A striking absence of cone-associated RPE apical microvilli was observed across cBest1-R25X-affected retinae as confirmed by immunolabeling against various RPE-specific markers. (A, B) Anti-MCT1 (green) and anti-BEST1 (red) double IHC labeling; Note the absence of BESTROPHIN 1 basolateral labeling in the mutant retina, accompanied by a dramatic retraction of RPE apical projections (B). (C, D) Representative images of RLBP1 (a.k.a. CRALBP) immunolabeling (red); native lipofuscin fluorescence is visualized as yellow-green RPE inclusions in the mutant RPE (D). (E, F) Direct comparison of anti-EZRIN immunostaining (red) between canine wild-type and cBest1-R25X-affected retinae. The massive inclusions (yellow-green) within the mutant RPE represent native lipofuscin (F). (G-J) Confocal photomicrographs of anti-EZRIN (red) and peanut agglutinin lectin (PNA, cyan) double-labeling in mutant (H and J) and age-matched controls (G and I). Note that in the WT retinae PNA-stained insoluble extracellular matrix closely invest cone-associated RPE apical microvilli (G-I); in the cBest mutant, however, the structure of both the intrinsic RPE-associated- and the external IPM-derived cone-specific domain is largely compromised (H, J), affecting the adhesive forces of RPE-photoreceptor complex. ONL: outer nuclear layer; MV: microvilli; RPE: retinal pigment epithelium; CH: choroid. Scale bars: 20µm (A-F) and 10µm (G-J).
Figure 6. Impaired RPE-cone interaction and photoreceptor alignment in canine bestrophinopathy.
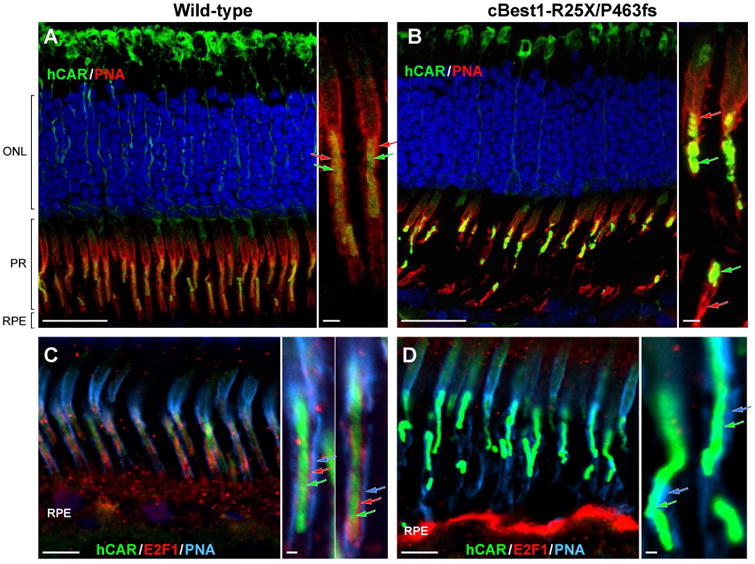
(A, B) Confocal photomicrographs of canine wild-type (A) and cBest1-R25X/P463fs-affected (B) retinae immunolabeled for human cone arrestin (hCAR, green) and PNA lectin (red). PNA-positive cone extracellular matrix sheaths tightly embrace the E2F1-labelled cone-specific RPE projections, extending along the outer- and toward the inner segment of cone photoreceptor cells in the control retina (A, close-up); in the cBest mutant, this IPM domain appears ruptured, exposing OS to the extracellular space (B, close-up). Together with the impairment in structural integrity of cone-specific IPM escheatment, cone photoreceptor OS lose their proper orientation (B), necessary for the normal interactions with RPE cells. (C, D) Multi-label IHC images of canine wild-type (C) and cBest mutant (D) retina illustrating bilayered extracellular compartmentalization of the cone-specific escheatment responsible for a normal apposition of RPE-cone OS (C, inset). This well-defined extracellular structure was lost in the affected retinae (D), revealing a dearth of the intrinsic RPE-associated apical microvillus layer, accompanied by rather fragmented external insoluble cone-specific matrix domain (D, close-up). Cone OS stripped of their protective sheaths appeared undermined with no direct contact with the underlying RPE (B, D). hCAR: human cone arrestin (shown in green A-D, and delineated by green arrows on the magnified images); PNA: peanut agglutinin lectin (shown in red A-B, and defined by red arrows on the corresponding close-ups; PNA in C-D is represented in cyan, and by cyan-colored arrows on the magnified photomicrographs); E2F1: E2F transcription factor 1 (shown in red C-D, and indicated by red arrows in C inset). ONL: outer nuclear layer; PRs: photoreceptors; RPE: retinal pigment epithelium. Scale bars: 20µm (A, B), 10µm (C, D), and 1µm (applies to all magnified images). Photomicrographs C & D: modified from Guziewicz et al. Adv. Exp. Med. Biol. (in press).
The structural abnormalities related to the impaired microvillar ensheathment of photoreceptor outer segment tips and compromised insoluble IPM constitute two major pathological culprits responsible for weakening of the RPE-neuroretina physical and electrostatic interactions (Mieziewska, 1996; Bonilha et al., 2006). Despite the fact that IPM occupies a strategic location in the retina, for many years it was considered as an amorphous substance of unknown function and significance. We now know that this intricate yet highly organized structure with interconnected domains designed to meet the specific needs of both rods and cones, is critical for maintaining the homeostatic milieu of subretinal space and formation of an adhesive bond between RPE and photoreceptors (Johnson and Hageman, 1991; Mieziewska et al., 1993; Ishikawa et al. 2015). The key location between RPE apical surface and photoreceptors allows IPM to fulfill a range of biochemical and physical tasks fundamental for normal retinal function, among which the most critical ones like regulation of oxygen, nutrients and retinoid transport or preservation of cytoskeleton and retinal adhesion by providing electrostatic support for photoreceptors (Johnson and Hageman, 1991; Mieziewska et al., 1993; Hauck et al., 2005; Ishikawa et al. 2015). It is therefore not unexpected that any genetic or metabolic defect found in the soluble and insoluble components of IPM play a role in the etiology of retinal degenerative disorders, such as IMPG1-and IMPG2-associated vitelliform macular dystrophy (Manes et al., 2013; Meunier et al., 2014; Ishikawa et al. 2015).
The dramatic retraction of the intrinsic RPE apical microvilli that underlies the loss of bilayered extracellular compartmentalization is of particular importance to the molecular pathology of bestrophinopathies. As highlighted earlier, the RPE performs a series of highly specialized tasks essential for homeostasis and structural integrity of the neural retina, including bidirectional transepithelial transport, supplying nutrients into and eliminating waste products from the photoreceptor layer, daily POS phagocytosis, light absorption, transport and regeneration of visual pigment; and all of these functions involve RPE microvilli that normally extend from its apical surface interdigitating with and tightly investing the POS (Bok, 1993; Bonilha et al., 2004; Bonilha et al., 2006). It is also important to note that these RPE projections greatly increase the apical surface area and, consequently, the number of transport and signaling molecules it contains, thereby enhancing the epithelial functional capacity (Bonilha et al., 2004; Miura et al., 2015). Thus, it is very likely that most of the major hallmarks of bestrophinopathies, e.g. disrupted ion and fluid transport (Sun et. al, 2002; Milenkovic et al., 2015), impaired phagocytosis (Singh et al., 2013a), abnormal photoreceptor equivalent thickness (PET) documented in BVMD patients (Kay et al., 2012), an excessive build-up of autofluorescent debris in the subretinal space, and finally, the RPE-neuroretinal detachment and formation of serous lesions (Boon et al., 2009a, 2009b), arise directly from the dearth of properly formed microvilli, and impaired interaction/adhesiveness with OS. We posit that these salient interface defects may also account for the abnormal EOG and Arden ratio present clinically in bestrophinopathy patients (Pianta et al., 2003; Boon et al., 2013).
5.2. Underdeveloped RPE apical domain and lesion formation in bestrophinopathies
Based on these fresh insights from the spontaneous canine model of BEST1-maculopathies, it is reasonable to hypothesize that the cone-associated microvilli as well as the insoluble cone matrix sheaths are more sensitive to the biochemical changes in the delicate subretinal microenvironment and retract first, which would explain the predilection of the highly metabolically active cone-packed macular region to its primary detachment in human and canine bestrophinopathies. Alternatively, it is possible that the BEST1 mutations impair the ability of RPE apical cone sheaths to form along with the shorter rod-associated MV processes. To test the premise, comparable IHC studies were performed in young (6-week-old) cBest retinae before disease onset, yet at the time point when the canine retina completes its development (Acland and Aguirre, 1987).
As shown in Fig. 7, both the immunolabeling against MCT1 and EZRIN revealed an underdeveloped RPE apical domain in the young cBest retinae, with only a few longer RPE microvilli and a highly disorganized distribution of EZRIN (Fig. 7A-D). In comparison to the age-matched controls, the 6-week-old cBest retina showed greatly diminished expression of EZRIN at the RPE apical surface, accompanied by an anomalous increase of EZRIN-positive signals associated with microvilli of Müller cells (Fig. 7D, close-up). While at this early time point cBest1-R25X/P463fs-affected retinae appeared histologically intact, the hCAR-labeled cone OS seemed more fragile and susceptible to destabilization following the natural course of disease.
Figure 7. Underdeveloped RPE apical domain underlies formation of subretinal lesions in canine bestrophinopathies.
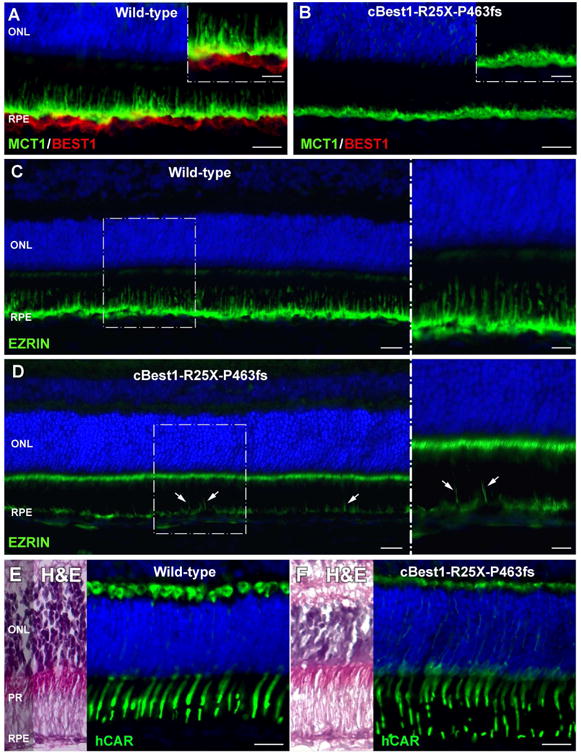
(A-D) IHC evaluation of 6-week-old cBest1-R25X/P463fs-affected and age-matched wild-type control retinae showed underdevelopment of RPE apical domain in cBest mutant, as demonstrated by comparative labeling with anti-MCT1 (A, B), and anti-EZRIN (C, D). In sharp contrast to the young, yet fully matured WT canine retina (A), the age-matched mutant one exhibited loss of the longer MCT1-positive RPE apical microvilli, even before the onset of clinical disease (B, close-up); note the absence of BESTROPHIN 1 labeling (B) compared to the robust Best1 basolateral signals detected in the control RPE (A, red). (C, D) Contrary to the specific anti-EZRIN immunostaining in the WT (C), evaluation of the mutant retina revealed largely diminished fluorescent signals at the RPE apical surface with only a few EZRIN-positive RPE processes (D, arrows). Note the dramatic increase of EZRIN labeling associated with microvilli of Müller cells (D, close-up). (E, F) Although not obvious on H&E images, the cone outer segments of cBest-affected retinae already lack the uniform regularity when immunolabeled with hCAR (F, green) and compared to the WT control (E). Scale bars: 20µm (A-F), and 10µm (magnified images in A-D). Photomicrographs D-F: modified from Guziewicz et al. Adv. Exp. Med. Biol. (in press).
These intriguing insights open a new avenue toward understanding the pathogenesis of bestrophinopathies and warrant further studies on RPE-PRs complex before disease onset. A number of research studies provided strong evidence that EZRIN is a major determinant in the maturation of surface differentiations of RPE, promoting morphogenesis of apical microvilli (Bonilha et al., 1999; Saotome et al., 2004; Miura et al., 2015). Our findings in the canine model support these results and strongly suggest that underdevelopment of cone-associated apical projections along with abnormalities of the insoluble cone extracellular matrix domain constitutes a key factor that underlies formation of the macular-selective subretinal lesions in bestrophinopathies. The fact that these prominent structural alterations associated with cone ensheathment are observed in cBest dogs as early as 6 weeks of age would also explain the early onset of ARB and BVMD along with the predisposition of the macular region to its primary detachment in affected dogs and patients.
5.3. RPE microvilli in ARB- and BVMD patient-specific hiPSC-RPE
In vitro, BVMD patient-specific human induced pluripotent stem cell-derived RPE (hiPSC-RPE) have been shown to successfully recapitulate several key cellular features of Best disease pathophysiology, including delayed POS degradation, increased accumulation of autofluorescent material after POS feeding, changes in endoplasmic reticulum-dependent Ca2+ homeostasis, and impaired fluid flux in response to physiological stimuli (Singh et al., 2013a; Singh et al., 2015). Both hiPSC-RPE and cBest models have complemented each other not only in gaining insights into BVMD pathophysiology, but also in testing potential therapeutic strategies (Singh et al., 2015). Using this hiPSC model system, we have followed up on observations from the cBest model to look for differences in the apical processes of WT, BVMD and ARB hiPSC-RPE cultures. Human iPSC-RPE from all hiPSC sources were immunolabeled for EZRIN (Singh et al., 2013b, Singh et al., 2015), which was present in a thick, vertically oriented band along the apical region of WT hiPSC-RPE, consistent with its localization within apical processes (Fig. 8A-C). BVMD (hBEST1-N296H) hiPSC-RPE also demonstrated a vertically oriented apical band of EZRIN immunolabeling (Fig. 8D-F); however, in ARB hiPSC-RPE (hBEST1-R141H/A195V) EZRIN immunolabeling was less prevalent, punctate, and localized within a much thinner apical band (Fig. 8G-I), akin to observations made in the cBest model.
Figure 8. EZRIN immunolabeling in WT, BVMD, and ARB hiPSC-RPE.
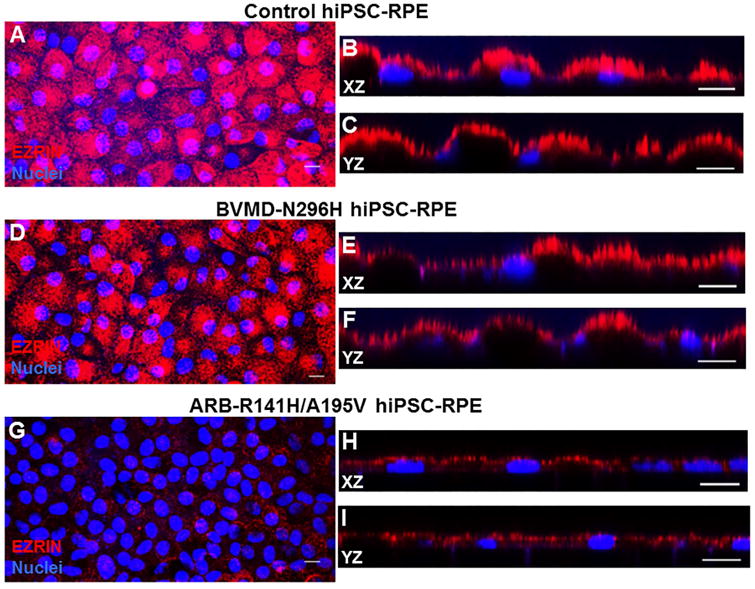
(A-C) Immunohistochemical analysis of WT hiPSC-RPE showed strong expression of EZRIN with vertically oriented, apical localization. (D-F) Similar expression and localization of EZRIN was observed in BVMD hiPSC-RPE. (G-I) In the ARB hiPSC-RPE, harboring compound heterozygous mutation in BEST1, MV are smaller in height compared to the WT hiPSC-RPE, and express EZRIN in a punctate fashion within a thin apical band. XZ and YZ labeled images are representative images for X-Z and Y-Z optical sections. Scale bar: 10µm applies to all panels.
Given that EZRIN immunostaining appeared relatively similar between WT and BVMD hiPSC-RPE, transmission electron microscopy (TEM) was performed on ultra-thin sections of RPE to further assess whether differences exist between the apical microvilli of WT and BVMD hiPSC-RPE. TEM analysis showed decreased length and density of microvilli in BEST1-N296H vs WT hiPSC-RPE (Fig. 9). Given that the hiPSC-RPE was cultured in isolation as homogenous monolayers, these differences in apical RPE microvilli between WT, BVMD, and ARB hiPSCs are independent of interactions with cone photoreceptors.
Figure 9. TEM of WT and BVMD hiPSC-RPE.
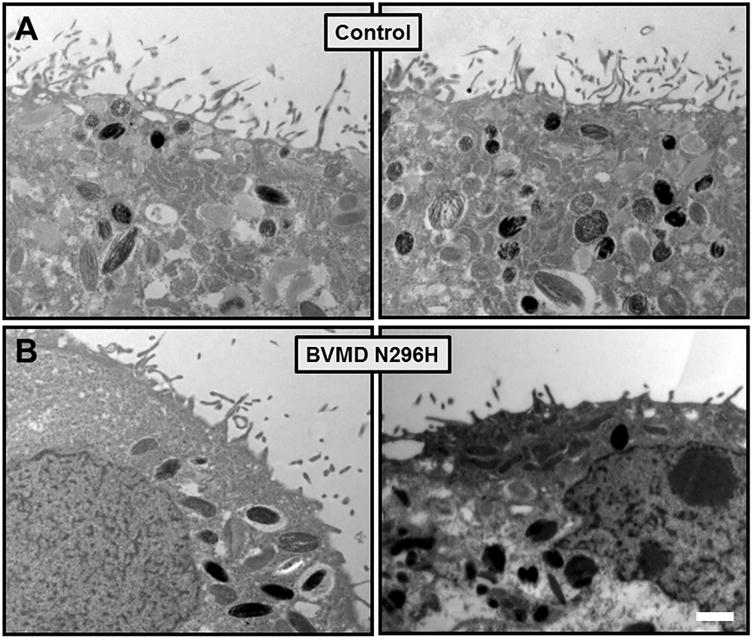
TEM analysis of WT hiPSC-RPE (A) and BVMD (BEST1-N296H) hiPSC-RPE (B) reveal shorter and more sparse microvilli in BEST1-N296H versus WT control hiPSC-RPE. Scale bar: 1µm.
6. Pathogenesis of early vitelliform lesions: insights from canine model
The structural and metabolic defects identified in the cBest model certainly contribute to, if not directly trigger, the separation of the neuroretina from the underlying RPE, the salient structural abnormality of bestrophinopathies. Further studies on molecular pathology of early focal and multifocal lesions confirmed the range of abnormalities as directly associated with subretinal detachment in cBest mutant retinae (Fig. 10). We found an abnormal accumulation of lipofuscin material within RPE cells was consistently associated with both the early and more advanced cBest lesions, remarkably aggregating not only at the detachment site, but also in its close vicinity (Fig. 10A-E). The more advanced cBest lesions represent longer standing retinal disassociations, where the autofluorescent debris starts to occupy subretinal space in transition to the pseudohypopyon stage (Fig. 10D,E). A noticeable increase in EC-BODIPY 493/503 was found to be associated with the photoreceptor layer in all early lesions examined, evident at the site of retinal separations and within still intact areas adjoining to the lesions (Fig. 10C). A series of IHC evaluations confirmed absence of microvillar elongations accompanied by a strikingly undulated RPE apical surface at the sites of detachments mapped in vivo by cSLO/SD-OCT (Fig. 10D,E). The primary lesion has a tendency to expand and secondary small satellite lesion(s) can be observed in its immediate vicinity often with a subtle retinal elevation between the individual separations (Fig. 10F,G). Eventually, after a prolonged serous detachment, the cellular debris accumulating in the subretinal space will constitute the solid phase of the gravitating pseudohypopyon material in the advanced disease. The rate of disease progression from early vitelliform to pseudohypopyon stage varies greatly between individuals and factors responsible for this mechanism are currently under investigation.
Figure 10. Canine BEST1-associated maculopathy is a disease of an RPE-photoreceptor interface.
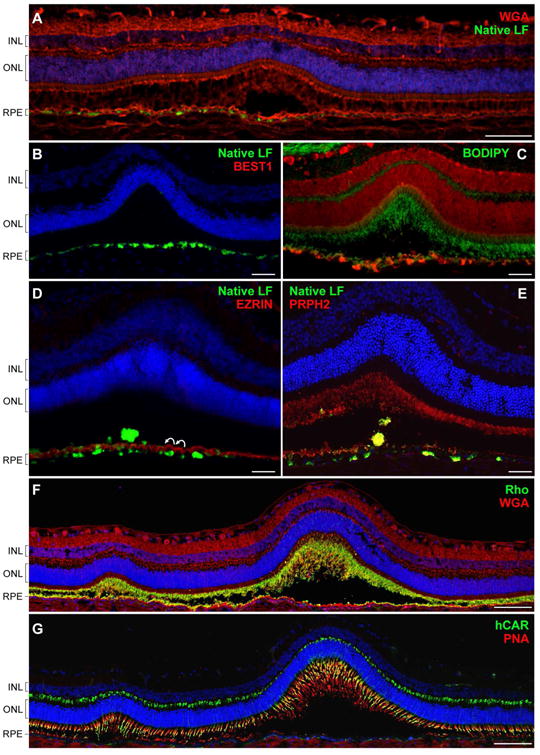
(A) Confocal photomicrograph of an early pre-vitelliform lesion. The section was stained with wheat germ agglutinin (WGA, red) and DAPI (blue). Bright green fluorescent signals of lipofuscin granules (native LF) were found to be associated with RPE cells directly underlying the primary neuroretinal disruption as well as its immediate surrounding areas. (B) Cryosection from a 30-month-old cBest1-R25X-affected dog with more advanced subretinal lesions comparable to stage II (vitelliform) in BVMD. BEST1-deficient RPE cells accumulate fluorescent debris subjacent the serous retinal detachment. (C) Retinal cross-section through an early vitelliform lesion from a 25-month-old dog harboring premature stop mutation (R25X) in the canine BEST1. The section was pretreated with ethanol before applying BODIPY 493/503 (green) and propidium iodide (red) for nuclear counterstaining. An evident increase of EC-BODIPY was recorded within the detached and more intact areas adjoining to the lesion. (D) Advanced vitelliform lesion immunolabeled with anti-EZRIN (red) and stained with DAPI (blue). Note the striking undulation of the RPE apical membrane (white curved arrows), absence of EZRIN-positive RPE apical projections, and beginning of a buildup of autofluorescent debris in the subretinal space (native LF fluorescence). (E) Similar cBest lesion in a 25-month-old cBest1-R25X-affected dog labeled with anti-PRPH2 (red). Peripherin-stained photoreceptor debris occupy RPE apical surface delimiting its scalloped nature and RPE cell hypertrophy after the serous neuroretinal detachment. Cellular debris appear as yellowish subretinal aggregates. (F, G) Montage confocal photomicrographs of a large subretinal lesion and a satellite microlesion in vitelliform stage labeled with WGA (red) and anti-Rhodopsin (Rho, green) (F) or a combination of peanut agglutinin lectin (PNA, red) and anti-cone arrestin (hCAR, green) (G). Both the Rho- and hCAR-positive cellular debris were detected in the subretinal space of the mutant retinae. Nuclei were counterstained with propidium iodide (C) or DAPI, respectively. INL: inner nuclear layer; ONL: outer nuclear layer; RPE: retinal pigment epithelium. Scale bars: 100µm (A, F, and G), and 40µm applies to (B-E) panels.
Best1 is a pentameric channel protein with defined roles in mediating anion transport and regulating calcium signaling in RPE cells (Kane Dickson et al., 2014; Yang et al., 2014; Strauss et al., 2014). Since intracellular calcium has been implicated to play important role in microvilli formation (Miura et al., 2015), the altered calcium homeostasis caused by Best1 dysfunction could underlie the defective villus architecture detected in cBest (Figs. 5-7, and 10) and hiPSC-RPE (Figs. 8-9) models, and reported in BVMD (Mullins et al., 2007) (Fig. 11). While the studies on ARB- and BVMD patient-specific hiPSC-RPE will further explore Best1 interactors as well as specific cellular pathways affected by dysregulated calcium signaling (Singh et al., 2013a; Singh et al., 2015), the studies on the natural canine model will continue to provide vital insights into the mechanism and kinetics of retinal detachment, the salient clinical structural abnormality of bestrophinopathies. Based on the studies to date, the proposed mechanism of lesion formation in canine and human bestrophinopathies is summarized in Fig. 11.
Figure 11. Proposed mechanism of lesion formation in canine and human bestrophinopathies.
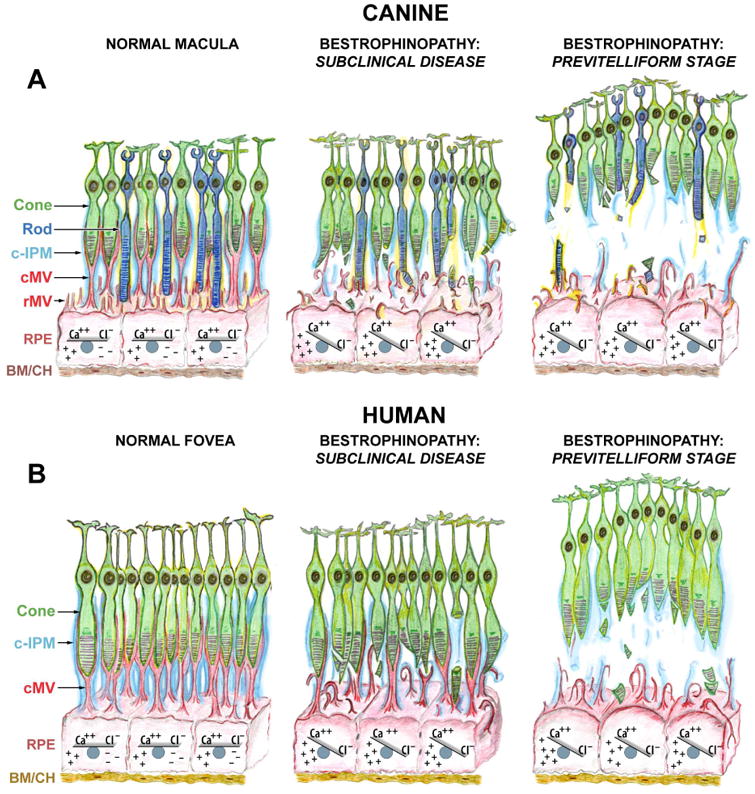
(A) Schematic representation of canine macula (a.k.a. area centralis) illustrating the normal RPE-photoreceptor apposition, sustained by intra- and extracellular ionic equilibria (Normal Macula, left panel). Ionic imbalance caused by mutation(s) in BEST1 gene initially affects RPE cell homeostasis, including expression of Ca++-dependent molecules (i. a., EZRIN). Impaired expression of EZRIN, the key determinant in the maturation of surface differentiations of RPE, leads to the underdeveloped RPE apical surface with fewer and rather immature microvilli that are not capable of investing and supporting the photoreceptor outer segments (Bestrophinopathy: Subclinical Disease, middle panel). Once the disease progresses, RPE cells gradually hypertrophy and lose their ability to mediate the ionic milieu of subretinal space. The cumulative effect of biochemical insult, accompanied by the structural abnormalities at the RPE apical surface weakens the adhesive forces of RPE-photoreceptor complex, which consequently results in separation of neuroretina from the RPE, and formation of serous lesion (Previtelliform Stage, right panel). (B) Schematic representation of human fovea. This rod-free structure is densely packed with cone photoreceptors tightly enveloped by RPE apical microvilli (cMV) and ensheathed in cone-specific interphotoreceptor matrix (c-IPM) (Normal Fovea, left panel). Due to the extremely high density of cones at this site, their outer segments in vivo will appear thinner in diameter, resembling rod photoreceptors; however, the cone outer segments are shorter than that of rods, and the RPE apical projections form long pedicles in order to reach them (cMV, red). The downstream effects of BEST1 mutation(s) manifest as both the ionic disequilibrium within RPE cells (and later within the adjacent subretinal space) and structural disintegration associated with RPE-derived cone ensheathment (Bestrophinopathy: Subclinical Disease, middle panel). As a consequence, the underdeveloped cone-associated RPE apical extensions are not able to physically support cone OS, which together with the reduced RPE functional capacity resulting directly from the decreased apical surface area, leads to the formation of macular-selective subretinal lesions at this highly metabolically active site (Bestrophinopathy: Previtelliform Stage, right panel). The compromised cone-associated insoluble IPM (c-IPM, cyan) may result from the chronic metabolic insult perturbing the delicate microenvironment of subretinal space and, secondarily, from the physical disassociation of the RPE-photoreceptor complex. The rate of disintegration of this extracellular matrix may constitute an important factor directly related to the expansion of vitelliform lesion and disease progression. Cone: cone photoreceptor cell; Rod: rod photoreceptor cell; c-IPM: cone-associated interphotoreceptor matrix; cMV: cone-associated microvilli; rMV: rod-associated microvilli; RPE: retinal pigment epithelium; BM/CH: Bruch's membrane/choroid.
7. Concluding remarks and future directions
The pathological mechanisms of pediatric retinal disorders, such as juvenile BEST1-associated maculopathies, are often challenging to explore; this can be attributed to the shortage of quality biospecimens harvested at the disease-relevant stages and, until now, to the dearth of bona fide animal models to carry out mechanistic studies. Genetic disorders caused by mutations in a single locus, yet resulting in a variable clinical expression, pose perhaps the greatest challenge, which can be extremely difficult to address if the laboratory-induced animal models fail to recapitulate the human disease (Marmorstein et al., 2004; Marmorstein and Marmorstein, 2007; Zhang et al., 2010; Xiao et al., 2010). It is now possible, however, to undertake such studies with the canine BEST1 disease model as it shares all the fundamental aspects with human bestrophinopathy, including the salient predilection of subretinal lesions to the canine macular region (Guziewicz et al., 2007; Guziewicz et al., 2011; Guziewicz et al., 2012; Beltran et al., 2014; Singh et al., 2015).
Here, we provide key insights into the complex molecular pathology of BEST1-related disorders, revealing factors predisposing macula to its primary detachment in BEST1-linked maculopathies, which may also account for the abnormal EOG and Arden ratio in BVMD and ARB patients. In view of the fact that RPE monolayer exhibits a considerable mosaicism (Burke and Hjelmeland, 2005), including not only topographic variations and regional specializations, but also cell-cell heterogeneity in gene expression (Mullins et al., 2007) and individual ability to support adjacent photoreceptors, single cell profiling may be required for elucidating the multifocal form of bestrophinopathy, in order to gain a full understanding of the spectrum of changes that each RPE cell must withstand before losing its direct interacting apposition.
Considering the rapid progress in hiPSC research, the key insights from a reliable animal model and its immediate verification in patient-specific hiPSC-RPE generate an exceptional momentum that will accelerate progress to the clinical applications (Guziewicz et al., 2013; Sinha et al., 2016). As a complementary system, hiPSC-RPE-based in vitro modeling of BVMD provides a platform for assessing pharmacological and gene therapy based interventions (e.g., lengthening of apical microvilli as a therapeutic outcome measure). As such, homeostatic restoration of RPE-photoreceptor interface should be considered an important outcome measure in the assessment of experimental therapies for bestrophinopathies that will warrant the sustained disease reversal.
Supplementary Material
Highlights.
Fresh perspective on the development of macular lesions in BEST1-linked disorders.
Novel insights into BEST1 pathogenesis derived from canine model and hiPSC-RPE.
Underdeveloped RPE apical domain underlies lesion formation in bestrophinopathies.
New disease aspects for guidance of therapeutic strategies for bestrophinopathies.
Advances in understanding of the molecular pathology of bestrophinopathies.
Acknowledgments
The authors thank Simone Iwabe and Valerie L. Dufour for assistance in cSLO/SD-OCT imaging, Emily McTish and Gordon Ruthel (University of Pennsylvania) for excellent technical assistance, Nancy J. Philp (Thomas Jefferson University) for anti-MCT1 antibody, Robert S. Molday (University of British Columbia) for anti-PRPH2, and Cheryl M. Craft (University of Southern California) for human cone arrestin antibody; Mary Leonard (Biomedical Art & Design, University of Pennsylvania) for assistance in graphic design, Theresa Jordan and the Retinal Disease Studies Facility staff for excellent animal care, and Lydia Melnyk for editorial help and research coordination.
Funding: This work was supported by the Foundation Fighting Blindness (FFB), FFB - Translational Research Acceleration Program (FFB-TRAP) and FFB-Facility grants, Macula Vision Research Foundation (MVRF), National Eye Institute/National Institutes of Health (NEI/NIH: EY06855, EY17549, EY10420, P30-EY001583, R01-EY024588), Research to Prevent Blindness, Retina Research Foundation Emmett A. Humble Distinguished Directorship of the McPherson Eye Research Institute, Sandra Lemke Trout Chair in Eye Research, Muskingum County Community Foundation, Van Sloun Fund for Canine Genetic Research, and Hope for Vision.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- RPE
retinal pigment epithelium
- BVMD
Best Vitelliform Macular Dystrophy
- VMD2
Vitelliform Macular Dystrophy, 2
- ARB
Autosomal Recessive Bestrophinopathy
- cBest
canine bestrophinopathy
- cSLO/SD-OCT
confocal scanning laser ophthalmoscopy spectral domain optical coherence tomography
- FAF
fundus autofluorescence
- EOG
electrooculogram
- hiPSC-RPE
human induced pluripotent stem cells - derived RPE
- hBest1
human BESTROPHIN 1 protein
- cBest1
canine BESTROPHIN 1 protein
- POS
photoreceptor outer segment
- IPM
interphotoreceptor matrix
- c-IPM
cone-associated interphotoreceptor matrix
- MV
microvilli
- cMV
cone-associated RPE apical microvilli
- IHC
immunohistochemistry
- HAI
Hyperspectral Autofluorescence Imaging
- Iem
emission intensity
- FC
free cholesterol
- EC
esterified cholesterol
- 7KCh
7-ketocholesterol
- HNE
4-hydoxy-2-nonenal
- ORO
Oil Red O
- MCT1
Monocarboxylate Transporter, Member 1
- RLBP1
Retinaldehyde Binding Protein 1
- hCAR
human cone arrestin
- PNA
peanut agglutinin lectin
- WGA
wheat germ agglutinin
- IMPG1
Interphotoreceptor Matrix Proteoglycan 1
Footnotes
Competing Interests Statement: The authors declare no competing or financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acland GM, Aguirre GD. Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res. 1987;44:491–521. doi: 10.1016/s0014-4835(87)80160-4. [DOI] [PubMed] [Google Scholar]
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- Albert A, Alexander D, Boesze-Battaglia K. Cholesterol in the rod outer segment: A complex role in a “simple” system. Chem Phys Lipid. 2016;199:94–105. doi: 10.1016/j.chemphyslip.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Allikmets R, Seddon JM, Bernstein PS, Hutchinson A, Atkinson A, Sharma S, Gerrard B, Li W, Metzker ML, Wadelius C, Caskey CT, Dean M, Petrukhin K. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Hum Genet. 1999;104:449–453. doi: 10.1007/s004390050986. [DOI] [PubMed] [Google Scholar]
- Bach D, Epand RF, Epand RM, Wachtel E. Interaction of 7-ketocholesterol with two major components of the inner leaflet of the plasma membrane: phosphatidylethanolamine and phosphatidylserine. Biochem. 2008;47:3004–3012. doi: 10.1021/bi702070b. [DOI] [PubMed] [Google Scholar]
- Bakall B, Radu RA, Stanton JB, Burke JM, McKay BS, Wadelius C, Mullins RF, Stone EM, Travis GH, Marmorstein AD. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2) Exp Eye Res. 2007;1:34–43. doi: 10.1016/j.exer.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Beltran WA, Cideciyan AV, Lewin AS, Iwabe S, Khanna H, Sumaroka A, Chiodo VA, Fajardo DS, Román AJ, Deng WT, Swider M, Alemán TS, Boye SL, Genini S, Swaroop A, Hauswirth WW, Jacobson SG, Aguirre GD. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012;109:2132–2137. doi: 10.1073/pnas.1118847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Cideciyan AV, Guziewicz KE, Iwabe S, Swider M, Scott EM, Savina SV, Ruthel G, Stefano F, Zhang L, Zorger R, Sumaroka A, Jacobson SG, Aguirre GD. Canine retina has a primate fovea-like bouquet of cone photoreceptors which is affected by inherited macular degenerations. PLoS One. 2014;3:e90390. doi: 10.1371/journal.pone.0090390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Ami T, Tong Y, Bhuiyan A, Huisingh C, Ablonczy Z, Ach T, Curcio CA, Smith RT. Spatial and Spectral Characterization of Human Retinal Pigment Epithelium Fluorophore Families by Ex Vivo Hyperspectral Autofluorescence Imaging. Transl Vis Sci Technol. 2016;5:e5. doi: 10.1167/tvst.5.3.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biarnes M, Arias L, Alonso J, Garcia M, Hijano M, Rodríguez A, Serrano A, Badal J, Muhtaseb H, Verdaguer P, Monés J. Increased Fundus Autofluorescence and Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration: The GAIN Study. Am J Ophthalmol. 2015;2:345–353. doi: 10.1016/j.ajo.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Bitner H, Schatz P, Mizrahi-Meissonnier L, Sharon D, Rosenberg T. Frequency, genotype, and clinical spectrum of best vitelliform macular dystrophy: data from a national center in Denmark. Am J Ophthalmol. 2012;154:403–412. doi: 10.1016/j.ajo.2012.02.036. [DOI] [PubMed] [Google Scholar]
- Bok D. The retinal pigment epithelium: a versatile partner in vision. J Cell Sci Suppl. 1993;17:189–195. doi: 10.1242/jcs.1993.supplement_17.27. [DOI] [PubMed] [Google Scholar]
- Bonilha VL, Finnemann SC, Rodriguez-Boulan E. Ezrin promotes morphogenesis of apical microvilli and basal infoldings in retinal pigment epithelium. J Cell Biol. 1999;147:1533–1548. doi: 10.1083/jcb.147.7.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilha VL, Bhattacharya SK, West KA, Crabb JS, Sun J, Rayborn ME, Nawrot M, Saari JC, Crabb JW. Support for a proposed retinoid-processing protein complex in apical retinal pigment epithelium. Exp Eye Res. 2004;79:419–422. doi: 10.1016/j.exer.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Bonilha VL, Rayborn ME, Bhattacharya SK, Gu X, Crabb JS, Crabb JW, Hollyfield JG. The retinal pigment epithelium apical microvilli and retinal function. Adv Exp Med Biol. 2006;572:519–524. doi: 10.1007/0-387-32442-9_72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilha VL. Retinal pigment epithelium (RPE) cytoskeleton in vivo and in vitro. Exp Eye Res. 2014;126:38–45. doi: 10.1016/j.exer.2013.09.015. [DOI] [PubMed] [Google Scholar]
- Booij JC, Boon CJ, van Schooneveld MJ, Ten Brink JB, Bakker A, de Jong PT, Hoyng CB, Bergen AA, Klaver CC. Course of visual decline in relation to the Best1 genotype in vitelliform macular dystrophy. Ophthalmol. 2010;7:1415–1422. doi: 10.1016/j.ophtha.2009.11.044. [DOI] [PubMed] [Google Scholar]
- Boon CJF, Klevering BJ, den Hollander AI, Zonneveld MN, Theelen T, Cremers FP, Hoyng CB. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol. 2007;125:1100–1106. doi: 10.1001/archopht.125.8.1100. [DOI] [PubMed] [Google Scholar]
- Boon CJ, Jeroen Klevering B, Keunen JE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vis Res. 2008;48:2569–2577. doi: 10.1016/j.visres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009a;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Boon CJ, Theelen T, Hoefsloot EH, van Schooneveld MJ, Keunen JE, Cremers FP, Klevering BJ, Hoyng CB. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina. 2009b;6:835–847. doi: 10.1097/IAE.0b013e31819d4fda. [DOI] [PubMed] [Google Scholar]
- Boon CJ, van den Born LI, Visser L, Keunen JE, Bergen AA, Booij JC, Riemslag FC, Florijn RJ, van Schooneveld MJ. Autosomal recessive bestrophinopathy: differential diagnosis and treatment options. Ophthalmol. 2013;120:809–820. doi: 10.1016/j.ophtha.2012.09.057. [DOI] [PubMed] [Google Scholar]
- Bretillon L, Thuret G, Gregoire S, Acar N, Joffre C, Bron AM, Gain P, Creuzot-Garcher CP. Lipid and fatty acid profile of the retina, retinal pigment epithelium/choroid, and the lacrimal gland, and associations with adipose tissue fatty acids in human subjects. Exp Eye Res. 2008;87:521–528. doi: 10.1016/j.exer.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33:611–619. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E, Brown PD, Robson AG, Wright GA, Kestelyn P, Holder GE, Webster AR, Manson FD, Black GC. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82:19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JM, Hjelmeland LM. Mosaicism of the retinal pigment epithelium: seeing the small picture. Mol Interv. 2005;5:241–249. doi: 10.1124/mi.5.4.7. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Swider M, Schwartz SB, Steinberg JD, Brucker AJ, Maguire AM, Bennett J, Stone EM, Jacobson SG. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004;13:525–534. doi: 10.1093/hmg/ddh048. [DOI] [PubMed] [Google Scholar]
- Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O'Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85:581–592. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt's disease - Fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1995a;11:2327–2331. [PubMed] [Google Scholar]
- Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995b;3:718–729. [PubMed] [Google Scholar]
- Delori FC, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 2001;42:1855–1866. [PubMed] [Google Scholar]
- Downs LM, Scott EM, Cideciyan AV, Iwabe S, Dufour V, Gardiner KL, Genini S, Marinho LF, Sumaroka A, Kosyk MS, Swider M, Aguirre GK, Jacobson SG, Beltran WA, Aguirre GD. Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw254. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Greenberg JP, Ramachandran R, Hood DC, Smith RT, Hirose T, Woods RL, Tsang SH, Delori FC, Sparrow JR. Quantitative fundus autofluorescence and optical coherence tomography in best vitelliform macular dystrophy. Invest Ophthalmol Vis Sci. 2014;55:1471–1482. doi: 10.1167/iovs.13-13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H, Zollner H, Lang J. Metabolism of the lipid peroxidation product 4-hydroxynonenal by isolated hepatocytes and by liver cytosolic fractions. Biochem J. 1985;228:363–373. doi: 10.1042/bj2280363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliesler SJ, Bretillon L. The ins and outs of cholesterol in the vertebrate retina. J Lipid Res. 2010;12:3399–3413. doi: 10.1194/jlr.R010538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung AT, Yzer S, Goldberg N, Wang H, Nissen M, Giovannini A, Merriam JE, Bukanova EN, Cai C, Yannuzzi LA, Tsang SH, Allikmets R. New best1 mutations in autosomal recessive bestrophinopathy. Retina. 2015;4:773–782. doi: 10.1097/IAE.0000000000000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Smith RT, Tkaczyk TS. Snapshot hyperspectral retinal camera with the Image Mapping Spectrometer (IMS) Biomed Opt Exp. 2012;3:48–54. doi: 10.1364/BOE.3.000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth C, Zawadzki RJ, Choi SS, Keltner JL, Park SS, Werner JS. Visualization of lipofuscin accumulation in Stargardt macular dystrophy by high-resolution Fourier-domain optical coherence tomography. Arch Ophthalmol. 2007;125:575. doi: 10.1001/archopht.125.4.575. [DOI] [PubMed] [Google Scholar]
- Gomez NM, Tamm ER, Strauβ O. Role of bestrophin-1 in store-operated calcium entry in retinal pigment epithelium. Pflugers Arch. 2013;465:481–495. doi: 10.1007/s00424-012-1181-0. [DOI] [PubMed] [Google Scholar]
- Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest Ophthalmol Vis Sci. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Slavik J, Lindauer SJ, Aguirre GD, Zangerl B. Molecular consequences of BEST1 gene mutations in canine multifocal retinopathy predict functional implications for human bestrophinopathies. Invest Ophthalmol Vis Sci. 2011;52:4497–4505. doi: 10.1167/iovs.10-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Aguirre GD, Zangerl B. Modeling the structural consequences of BEST1 missense mutations. Adv Exp Med Biol. 2012;723:611–618. doi: 10.1007/978-1-4614-0631-0_78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Zangerl B, Komáromy AM, Iwabe S, Chiodo VA, Boye SL, Hauswirth WW, Beltran WA, Aguirre GD. Recombinant AAV-mediated BEST1 transfer to the retinal pigment epithelium: analysis of serotype-dependent retinal effects. PLoS One. 2013;8:e75666. doi: 10.1371/journal.pone.0075666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Johnson LV. The Photoreceptor - Retinal Pigment Epithelium Interface. In: Heckenlively JR, Arden GB, editors. Principles and Practise of Clinical Electrophysiology of Vision. Mosby-Year Book Inc; St Louise: 1991. pp. 53–68. [Google Scholar]
- Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- Hauck SM, Schoeffmann S, Deeg CA, Gloeckner CJ, Swiatek-de Lange M, Ueffing M. Proteomic analysis of the porcine interphotoreceptor matrix. Prot. 2005;5:3623–3636. doi: 10.1002/pmic.200401223. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Sawada Y, Yoshitomi T. Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp Eye Res. 2015;133:3–18. doi: 10.1016/j.exer.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Hageman GS. Structural and compositional analyses of isolated cone matrix sheaths. Invest Ophthalmol Vis Sci. 1991;32:1951–1957. [PubMed] [Google Scholar]
- Kane Dickson V, Pedi L, Long SB. Structure and insights into the function of a Ca(2+)-activated Cl(-) channel. Nature. 2014;516:213–218. doi: 10.1038/nature13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay CN, Abramoff MD, Mullins RF, Kinnick TR, Lee K, Eyestone ME, Chung MM, Sohn EH, Stone EM. Three-dimensional distribution of the vitelliform lesion, photoreceptors, and retinal pigment epithelium in the macula of patients with best vitelliform macular dystrophy. Arch Ophthalmol. 2012;130:357–364. doi: 10.1001/archophthalmol.2011.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy CJ, Rakoczy PE, Constable IJ. Lipofuscin of the retinal pigment epithelium: a review. Eye (Lond) 1995;9:763–771. doi: 10.1038/eye.1995.192. [DOI] [PubMed] [Google Scholar]
- Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010;25:8–15. doi: 10.1152/physiol.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnick TR, Mullins RF, Dev S, Leys M, Mackey DA, Kay CN, Lam BL, Fishman GA, Traboulsi E, Iezzi R, Stone EM. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina. 2011;31:581–595. doi: 10.1097/IAE.0b013e318203ee60. [DOI] [PubMed] [Google Scholar]
- Komáromy AM, Alexander JJ, Rowlan JS, Garcia MM, Chiodo VA, Kaya A, Tanaka JC, Acland GM, Hauswirth WW, Aguirre GD. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19:2581–2593. doi: 10.1093/hmg/ddq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer F, White K, Pauleikhoff D, Gehrig A, Passmore L, Rivera A, Rudolph G, Kellner U, Andrassi M, Lorenz B, Rohrschneider K, Blankenagel A, Jurklies B, Schilling H, Schütt F, Holz FG, Weber BH. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- Lakkaraju A, Finnemann SC, Rodriguez-Boulan E. The lipofuscin fluorophore A2E perturbs cholesterol metabolism in retinal pigment epithelial cells. Proc Natl Acad Sci U S A. 2007;26:11026–11031. doi: 10.1073/pnas.0702504104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Huang JD, Rodriguez IR. Extra-hepatic metabolism of 7-ketocholesterol occurs by esterification to fatty acids via cPLA2alpha and SOAT1 followed by selective efflux to HDL. Biochim Biophys Acta. 2015;1851:605–619. doi: 10.1016/j.bbalip.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Tzekov R, McDowell JH, Smith WC, Tang S, Kaushal S. Formation of lipofuscin-like material in the RPE Cell by different components of rod outer segments. Exp Eye Res. 2013;112:57–67. doi: 10.1016/j.exer.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang Y, Xuan Y, Liu W, Wang M. Novel BEST1 Mutations and Special Clinical Features of Best Vitelliform Macular Dystrophy. Ophthalmic Res. 2016;56:178–185. doi: 10.1159/000444681. [DOI] [PubMed] [Google Scholar]
- MacDonald IM, Gudiseva HV, Villanueva A, Greve M, Caruso R, Ayyagari R. Phenotype and genotype of patients with autosomal recessive bestrophinopathy. Ophthalmic Genet. 2012;3:123–129. doi: 10.3109/13816810.2011.592172. [DOI] [PubMed] [Google Scholar]
- Manes G, Meunier I, Avila-Fernández A, Banfi S, Le Meur G, Zanlonghi X, Corton M, Simonelli F, Brabet P, Labesse G, Audo I, Mohand-Said S, Zeitz C, Sahel JA, Weber M, Dollfus H, Dhaenens CM, Allorge D, De Baere E, Koenekoop RK, Kohl S, Cremers FP, Hollyfield JG, Sénéchal A, Hebrard M, Bocquet B, Ayuso Garcia C, Hamel CP. Mutations in IMPG1 cause vitelliform macular dystrophies. Am J Hum Genet. 2013;93:571–578. doi: 10.1016/j.ajhg.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF. Mechanisms of normal retina adhesion. In: Ryan SJ, editor. Retina. Mosby-Year Book Inc; St Louise: 1994. pp. 1931–1953. [Google Scholar]
- Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Marmorstein LY, Sakaguchi H, Hollyfield JG. Spectral profiling of autofluorescence associated with lipofuscin, Bruch's Membrane, and sub-RPE deposits in normal and AMD eyes. Invest Ophthalmol Vis Sci. 2002;7:2435–2441. [PubMed] [Google Scholar]
- Marmorstein AD, Stanton JB, Yocom J, Bakall B, Schiavone MT, Wadelius C, Marmorstein LY, Peachey NS. A model of best vitelliform macular dystrophy in rats. Invest Ophthalmol Vis Sci. 2004;45:3733–3739. doi: 10.1167/iovs.04-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Marmorstein LY. The challenge of modeling macular degeneration in mice. Trends Genet. 2007;23:225–231. doi: 10.1016/j.tig.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Meunier I, Manes G, Bocquet B, Marquette V, Baudoin C, Puech B, Defoort-Dhellemmes S, Audo I, Verdet R, Arndt C, Zanlonghi X, Le Meur G, Dhaenens CM, Hamel CP. Frequency and clinical pattern of vitelliform macular dystrophy caused by mutations of interphotoreceptor matrix IMPG1 and IMPG2 genes. Ophthalmol. 2014;121:2406–2414. doi: 10.1016/j.ophtha.2014.06.028. [DOI] [PubMed] [Google Scholar]
- Mieziewska K, Van Veen T, Aguirre GD. Development and fate of interphotoreceptor matrix components during dysplastic photoreceptor differentiation: a lectin cytochemical study of rod-cone dysplasia 1. Exp Eye Res. 1993;56:429–441. doi: 10.1006/exer.1993.1056. [DOI] [PubMed] [Google Scholar]
- Mieziewska K. The interphotoreceptor matrix, a space in sight. Microsc Res Tech. 1996;35:463–471. doi: 10.1002/(SICI)1097-0029(19961215)35:6<463::AID-JEMT5>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Milenkovic A, Brandl C, Milenkovic VM, Jendryke T, Sirianant L, Wanitchakool P, Zimmermann S, Reiff CM, Horling F, Schrewe H, Schreiber R, Kunzelmann K, Wetzel CH, Weber BH. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc Natl Acad Sci U S A. 2015;112:E2630–9. doi: 10.1073/pnas.1418840112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S, Sato K, Kato-Negishi M, Teshima T, Takeuchi S. Fluid shear triggers microvilli formation via mechanosensitive activation of TRPV6. Nat Commun. 2015;6:8871. doi: 10.1038/ncomms9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira EF, Larrayoz IM, Lee JW, Rodriguez IR. 7-Ketocholesterol is present in lipid deposits in the primate retina: potential implication in the induction of VEGF and CNV formation. Invest Ophthalmol Vis Sci. 2009;50:523–532. doi: 10.1167/iovs.08-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Oh KT, Heffron E, Hageman GS, Stone EM. Late development of vitelliform lesions and flecks in a patient with best disease: clinicopathologic correlation. Arch Ophthalmol. 2005;123:1588–1594. doi: 10.1001/archopht.123.11.1588. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM. Differential macular and peripheral expression of bestrophin in human eyes and its implication for best disease. Invest Ophthalmol Vis Sci. 2007;48:3372–3380. doi: 10.1167/iovs.06-0868. [DOI] [PubMed] [Google Scholar]
- O'Gorman S, Flaherty WA, Fishman GA, Berson EL. Histopathologic findings in Best's vitelliform macular dystrophy. Arch Ophthalmol. 1988;106:1261–1268. doi: 10.1001/archopht.1988.01060140421045. [DOI] [PubMed] [Google Scholar]
- Pasquay C, Wang LF, Lorenz B, Preising MN. Bestrophin 1-Phenotypes and Functional Aspects in Bestrophinopathies. Ophthalmic Genet. 2015;36:193–212. doi: 10.3109/13816810.2013.863945. [DOI] [PubMed] [Google Scholar]
- Pianta MJ, Aleman TS, Cideciyan AV, Sunness JS, Li Y, Campochiaro BA, Campochiaro PA, Zack DJ, Stone EM, Jacobson SG. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp Eye Res. 2003;76:203–211. doi: 10.1016/s0014-4835(02)00280-4. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxid Redox Signal. 2012;17:1590–1609. doi: 10.1089/ars.2011.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, Vujic M, Bergen AA, McGarty-Dugan V, Figueroa D, Austin CP, Metzker ML, Caskey CT, Wadelius C. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- Petrukhin K. New therapeutic targets in atrophic age-related macular degeneration. Expert Opin Ther Targets. 2007;11:625–639. doi: 10.1517/14728222.11.5.625. [DOI] [PubMed] [Google Scholar]
- Piñeiro-Gallego T, Álvarez M, Pereiro I, Campos S, Sharon D, Schatz P, Valverde D. Clinical evaluation of two consanguineous families with homozygous mutations in BEST1. Mol Vis. 2011;17:1607–1617. [PMC free article] [PubMed] [Google Scholar]
- Querques G, Zerbib J, Santacroce R, Margaglione M, Delphin N, Rozet JM, Kaplan J, Martinelli D, Delle Noci N, Soubrane G, Souied EH. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol Vis. 2009;15:2960–2972. [PMC free article] [PubMed] [Google Scholar]
- Rodriguez IR, Fliesler SJ. Photodamage generates 7-keto- and 7-hydroxycholesterol in the rat retina via a free radical-mediated mechanism. Photochem Photobiol. 2009;85:1116–1125. doi: 10.1111/j.1751-1097.2009.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez IR, Clark ME, Lee JW, Curcio CA. 7-ketocholesterol accumulates in ocular tissues as a consequence of aging and is present in high levels in drusen. Exp Eye Res. 2014;128:151–155. doi: 10.1016/j.exer.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Bakall B, Kinnick T, Peachey N, Wimmers S, Wadelius C, Marmorstein A, Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2005;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- Rudolf M, Curcio CA. Esterified cholesterol is highly localized to Bruch's membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J Histochem Cytochem. 2009;57:731–739. doi: 10.1369/jhc.2009.953448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero L, Connor MP, Chen J, Langen R, Finnemann SC. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5-/- or Mfge8-/- mouse retina. Proc Natl Acad Sci U S A. 2012;109:8145–8148. doi: 10.1073/pnas.1121101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome I, Curto M, McClatchey AI. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell. 2004;6:855–864. doi: 10.1016/j.devcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Schatz P, Klar J, Andreasson S, Ponjavic V, Dahl N. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006;27:51–56. doi: 10.1080/13816810600677990. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Afshari MA, Sharma S, Bernstein PS, Chong S, Hutchinson A, Petrukhin K, Allikmets R. Assessment of mutations in the Best macular dystrophy (VMD2) gene in patients with adult onset foveomacular vitelliform dystrophy, age-related maculopathy, and bull's-eye maculopathy. Ophthalmol. 2001;108:2060–2067. doi: 10.1016/s0161-6420(01)00777-1. [DOI] [PubMed] [Google Scholar]
- Shen JK, Dong A, Hackett SF, Bell WR, Green WR, Campochiaro PA. Oxidative damage in age-related macular degeneration. Histol Histopathol. 2007;12:1301–1308. doi: 10.14670/HH-22.1301. [DOI] [PubMed] [Google Scholar]
- Singh R, Shen W, Kuai D, Martin JM, Guo X, Smith MA, Perez ET, Phillips MJ, Simonett JM, Wallace KA, Verhoeven AD, Capowski EE, Zhang X, Yin Y, Halbach PJ, Fishman GA, Wright LS, Pattnaik BR, Gamm DM. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2013a;23:593–607. doi: 10.1093/hmg/dds469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Phillips MJ, Kuai D, Meyer J, Martin JM, Smith MA, Perez ET, Shen W, Wallace KA, Capowski EE, Wright LS, Gamm DM. Functional analysis of serially expanded human iPS cell-derived RPE cultures. Invest Ophthalmol Vis Sci. 2013b;54:6767–6778. doi: 10.1167/iovs.13-11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kuai D, Guziewicz KE, Meyer J, Wilson M, Lu J, Smith M, Clark E, Verhoeven A, Aguirre G, Gamm DM. Pharmacological Modulation of Photoreceptor Outer Segment Degradation in a Human iPS Cell Model of Inherited Macular Degeneration. Mol Ther. 2015;11:1700–1711. doi: 10.1038/mt.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha D, Phillips J, Joseph Phillips M, Gamm DM. Mimicking Retinal Development and Disease With Human Pluripotent Stem Cells. Invest Ophthalmol Vis Sci. 2016;57:ORSFf1–9. doi: 10.1167/iovs.15-18160. in press. [DOI] [PubMed] [Google Scholar]
- Smith RT, Post R, Johri A, Lee MD, Ablonczy Z, Curcio CA, Ach T, Sajda P. Simultaneous decomposition of multiple hyperspectral data sets: signal recovery of unknown fluorophores in the retinal pigment epithelium. Biomed Opt Exp. 2014;5:4171–4185. doi: 10.1364/BOE.5.004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaide RF. Fundus autofluorescence and age-related macular degeneration. Ophthalmol. 2003;110:392–399. doi: 10.1016/S0161-6420(02)01756-6. [DOI] [PubMed] [Google Scholar]
- Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform macular dystrophy. Ophthalmol. 2006;113:1392–1400. doi: 10.1016/j.ophtha.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Duncker T, Woods R, Delori FC. Quantitative Fundus Autofluorescence in Best Vitelliform Macular Dystrophy: RPE Lipofuscin is not Increased in Non-Lesion Areas of Retina. Adv Exp Med Biol. 2016;854:285–290. doi: 10.1007/978-3-319-17121-0_38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg RH. Interactions between the retinal pigment epithelium and the neural retina. Doc Ophthalmol. 1985;60:327–346. doi: 10.1007/BF00158922. [DOI] [PubMed] [Google Scholar]
- Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- Strauss O, Müller C, Reichhart N, Tamm ER, Gomez NM. The role of bestrophin-1 in intracellular Ca(2+) signaling. Adv Exp Med Biol. 2014;801:113–119. doi: 10.1007/978-1-4614-3209-8_15. [DOI] [PubMed] [Google Scholar]
- Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci U S A. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan LX, Toops KA, Lakkaraju A. Protective responses to sublytic complement in the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2016;113:8789–8794. doi: 10.1073/pnas.1523061113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toops KA, Tan LX, Jiang Z, Radu RA, Lakkaraju A. Cholesterol-mediated activation of acid sphingomyelinase disrupts autophagy in the retinal pigment epithelium. Mol Biol Cell. 2015;26:1–14. doi: 10.1091/mbc.E14-05-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Clark ME, Crossman DK, Kojima K, Messinger JD, Mobley JA, Curcio CA. Abundant lipid and protein components of drusen. PLoS One. 2010;5:e10329. doi: 10.1371/journal.pone.0010329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Hartzell HC, Yu K. Bestrophins and retinopathies. Pflugers Arch. 2010;460:559–569. doi: 10.1007/s00424-010-0821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Liu Q, Kloss B, Bruni R, Kalathur RC, Guo Y, Kloppmann E, Rost B, Colecraft HM, Hendrickson WA. Structure and selectivity in bestrophin ion channels. Science. 2014;346:355–359. doi: 10.1126/science.1259723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Justus S, Li Y, Tsang SH. BEST1: the Best Target for Gene and Cell Therapies. Mol Ther. 2015;12:1805–1809. doi: 10.1038/mt.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM, Perveen R, Reddy MA, Bhattacharya SS, Traboulsi E, Baralle D, De Laey JJ, Puech B, Kestelyn P, Moore AT, Manson FD, Black GC. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest Ophthalmol Vis Sci. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- Yu K, Qu Z, Cui Y, Hartzell HC. Chloride channel activity of bestrophin mutants associated with mild or late-onset macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:4694–4705. doi: 10.1167/iovs.07-0301. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Stanton JB, Wu J, Yu K, Hartzell HC, Peachey NS, Marmorstein LY, Marmorstein AD. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum Mol Genet. 2010;19:1108–1118. doi: 10.1093/hmg/ddp583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Reem RE, Omarova S, Huang S, DiPatre PL, Charvet CD, Curcio CA, Pikuleva IA. Spatial distribution of the pathways of cholesterol homeostasis in human retina. PLoS One. 2012;5:e37926. doi: 10.1371/journal.pone.0037926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangerl B, Wickström K, Slavik J, Lindauer SJ, Ahonen S, Schelling C, Lohi H, Guziewicz KE, Aguirre GD. Assessment of canine BEST1 variations identifies new mutations and establishes an independent bestrophinopathy model (cmr3) Mol Vis. 2010;16:2791–2804. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.