

Abstract
Methanogenic activity was investigated in a petroleum hydrocarbon-contaminated aquifer by using a series of four push-pull tests with acetate, formate, H2 plus CO2, or methanol to target different groups of methanogenic Archaea. Furthermore, the community composition of methanogens in water and aquifer material was explored by molecular analyses, i.e., fluorescence in situ hybridization (FISH), denaturing gradient gel electrophoresis (DGGE) of 16S rRNA genes amplified with the Archaea-specific primer set ARCH915 and UNI-b-rev, and sequencing of DNA from dominant DGGE bands. Molecular analyses were subsequently compared with push-pull test data. Methane was produced in all tests except for a separate test where 2-bromoethanesulfonate, a specific inhibitor of methanogens, was added. Substrate consumption rates were 0.11 mM day−1 for methanol, 0.38 mM day−1 for acetate, 0.90 mM day−1 for H2, and 1.85 mM day−1 for formate. Substrate consumption and CH4 production during all tests suggested that at least three different physiologic types of methanogens were present: H2 plus CO2 or formate, acetate, and methanol utilizers. The presence of 15 to 20 bands in DGGE profiles indicated a diverse archaeal population. High H2 and formate consumption rates agreed with a high diversity of methanogenic Archaea consuming these substrates (16S rRNA gene sequences related to several members of the Methanomicrobiaceae) and the detection of Methanomicrobiaceae by using FISH (1.4% of total DAPI [4′,6-diamidino-2-phenylindole]-stained microorganisms in one water sample; probe MG1200). Considerable acetate consumption agreed with the presence of sequences related to the obligate acetate degrader Methanosaeata concilii and the detection of this species by FISH (5 to 22% of total microorganisms; probe Rotcl1). The results suggest that both aceticlastic and CO2-type substrate-consuming methanogens are likely involved in the terminal step of hydrocarbon degradation, while methanogenesis from methanol plays a minor role. DGGE profiles further indicate similar archaeal community compositions in water and aquifer material. The combination of hydrogeological and molecular methods employed in this study provide improved information on the community and the potential activity of methanogens in a petroleum hydrocarbon-contaminated aquifer.
Methanogenesis is a common process in many anaerobic environments such as digesters (41), cattle rumen (33), rice fields (28), oil wells (34), landfills (17), and a range of extreme habitats (19). This process plays an important role for the formation of biogas as an alternative source of energy (35), generation of CH4 as a greenhouse gas (58), and degradation of contaminants in polluted soils and aquifers (12, 61). In the absence of other electron acceptors such as oxygen, nitrate, and sulfate, methanogens are involved in the terminal anaerobic breakdown of organic matter (19). They catabolically rely on a restricted number of simple compounds, e.g., on CO2 as an oxidant with H2 as an electron donor or on acetate, methanol, or formate (63). Hence, they depend on other organisms such as fermenting or sulfate-reducing bacteria to supply their substrates. The physiology of cultured methanogenic Archaea is related to their phylogenetic relationships based on 16S rRNA sequences (63). For example, while most species of the Methanobacteriaceae and Methanomicrobiaceae prefer H2 and CO2 (or formate) as substrates for methanogenesis, Methanosaeta, a genus within the Methanosarcinaceae, is known to generate energy only from acetate fermentation. Most of the other Methanosarcinaceae preferentially use methanol and related methyl-substrates for the generation of CH4.
The diversity of methanogenic Archaea in the environment may be monitored by using laboratory molecular methods such as fluorescence in situ hybridization (FISH) or denaturing gradient gel electrophoresis (DGGE) with the subsequent cloning and sequencing of excised bands (16, 41, 43). Previous investigations indicated that while methyl compounds were the primary substrates for methanogens in marine sediments (e.g., Methanosarcina) (63), mainly acetate-degrading Methanosaeta and CO2- and H2-degrading Methanobacteriaceae and Methanomicrobiaceae were present in a range of freshwater environments (8, 11, 14, 52, 62). For example, using FISH, Ficker et al. (16) found that 17% of total microorganisms in a toluene-degrading culture enriched from a creosote-contaminated aquifer hybridized with a Methanosaeta-specific probe and that 2% hybridized with a Methanospirillum-specific probe. While Methanosaeta spp. were abundant in samples from a sewage sludge digester, only few Methanosarcina spp. were visible (41). By assessing bands of DGGE gels, Röling et al. (43) detected different archaeal community structures inside and outside the contaminant plume of a landfill leachate-polluted aquifer. Zengler et al. (61) demonstrated that Archaea related to Methanosaeta spp. as well as Methanospirillum and Methanoculleus, both belonging to the Methanomicrobiaceae, were present in a long-chain alkane-consuming methanogenic culture enriched from anaerobic ditch sediment.
Methanogenesis may contribute considerably to the mineralization of petroleum hydrocarbons (PHC) in contaminated aquifers (11). However, methanogenic microorganisms are not able to directly degrade PHC (61). Methanogens using H2 and CO2 contribute indirectly to PHC degradation by keeping H2 concentrations low so that fermentation of PHC becomes exergonic and fermenting organisms can grow (19). Methanogens using acetate or methanol contribute directly to PHC degradation by cleaving end products of fermentation. However, the role of different metabolic groups of methanogens with respect to overall methanogenic activity in PHC-contaminated aquifers is unknown. Aceticlastic methanogenesis was hypothesized to be the terminal step of hydrocarbon degradation in a PHC-contaminated aquifer, but this was inferred from molecular data alone and not based on activity measurements (14).
Although methanogenic activity in the subsurface is difficult to monitor due to the volatility of CH4 and its preferred degradation by methanotrophs under both aerobic (24) and anaerobic (6) conditions, an attempt to quantify methanogenesis was made by using push-pull tests (PPTs) (27). In these PPTs, a test solution that contained a nonreactive conservative tracer (Br−) and a reactant (H2) was injected (“pushed”) into the aquifer through an existing well. Thereafter, the test solution or groundwater mixture was extracted (“pulled”) from the same location, and the concentrations of Br−, H2, and CH4 were analyzed. Rates of microbial activities were then determined by comparing the breakthrough curves of tracer and reactant (21, 27, 47). However, while considerable consumption of the substrate H2 occurred in these tests, only minimal CH4 production was observed (27). Hence, the interpretation of such tests is difficult since processes other than methanogenesis may also be responsible for the observed substrate consumption.
Few studies so far have focused on the activity or diversity of methanogens in PHC-contaminated aquifers (8, 11, 14, 52). Furthermore, to our knowledge no attempt has been made to link the presence of metabolic or phylogenetic types of methanogens to their activity in this environment. Such information is essential for understanding the biogeochemical processes occurring in contaminated aquifers and, thus, for possibly monitoring and managing the efficiency of bioremediation.
The objective of our study was to simultaneously assess the activity and diversity of methanogens in the anoxic zone of a PHC-contaminated aquifer. In four separate PPTs, we used acetate, formate, CO2 plus H2, and methanol as substrates to examine the potential activity of different physiologic groups of methanogenic Archaea. PPT data were then compared with molecular analyses (FISH, DGGE, and sequencing of excised DGGE bands) of the methanogen community composition in water and aquifer material samples. To explore the contribution of Fe(III) reduction to H2 consumption, another PPT was performed with bromoethanesulfonate (BES) as a specific inhibitor of methanogens (36).
MATERIALS AND METHODS
Field site.
The study was conducted in a heating oil-contaminated aquifer in Studen, Switzerland, which is undergoing remediation by monitored natural attenuation (7). PPTs described in this paper were conducted in monitoring well PS5, which is located within the contaminant source zone (free-phase PHC present). Well PS5 is constructed of polyvinyl chloride casing (internal diameter, 11.5 cm) and penetrates the aquifer to a depth of ∼0.5 m below the groundwater table, which was located at ∼2.9 m below ground surface. Groundwater in PS5 exhibited reduced conditions and contained up to 1 mg of dissolved PHC (7) per liter. Previous studies have shown that PS5 is located within the methanogenic zone with O2 concentrations of <0.03 mM, NO3− and SO42− concentrations of <0.003 mM, and CH4 concentrations of 1.16 ± 0.12 mM (7, 8).
PPTs.
To assess methanogenic activity, we performed four separate PPTs by using the substrates acetate (PPTac), formate (PPTfo), CO2 plus H2 (PPTH2), or methanol (PPTme) in September 2001 (PPTac), August 2002 (PPTfo and PPTme), and September 2002 (PPTH2) in similar fashion as described by Schroth et al. (45). To assess the contribution of Fe(III) reduction to H2 consumption, another PPT in the presence of BES (PPTBES) was performed in November 2002. In all PPTs, test solutions were prepared by collecting groundwater in 500-liter plastic carboys and adding Br− (as KBr) as a nonreactive, conservative tracer along with acetate, formate, H2, or methanol as reactants to achieve final concentrations of ∼0.5 mM Br− and a ∼2.0 mM concentration of acetate, formate or methanol (Table 1). Hydrogen in PPTH2 (0.61 mM) and PPTBES (0.39 mM) was added by sparging test solutions with pure H2 gas. Carbon dioxide was not added in PPTH2 or PPTBES since CO2 was present in the groundwater of PS5 as dissolved inorganic carbon (DIC) at a concentration of 13.5 mM. In PPTBES, ∼2 mM BES was added to the test solution. Sparging test solutions in PPTH2 and PPTBES with H2 gas and in the remaining PPTs with N2 gas additionally served the purpose of keeping test solutions anoxic during preparation and injection.
TABLE 1.
Summary of experimental conditions during five PPTs performed to evaluate methanogenesis in a PHC-contaminated aquifer
| Test | Substrate injected | Substrate concentration, C0 (mM) | Br− injection concentration (mM) | Injection volume (liters) | Injection duration (h) | Initial incubation perioda (h) | Total extracted volume (liters) | Total test duration (h) |
|---|---|---|---|---|---|---|---|---|
| PPTac | Acetate | 2.11 | 0.53 | 1,000 | 1.9 | 20.4 | 1,000 | 70.8 |
| PPTfo | Formate | 2.05 | 0.59 | 500 | 0.7 | 2.3 | 1,000 | 6.9 |
| PPTH2 | H2 | 0.61 | 0.50 | 500 | 0.9 | 1.8 | 750 | 5.4 |
| PPTme | Methanol | 1.99 | 0.43 | 500 | 0.8 | 2.3 | 750 | 27.0 |
| PPTBESb | H2 | 0.39 | 0.47 | 500 | 0.6 | 2.0 | 750 | 4.2 |
Initial incubation period is defined as the time between the end of injection and the beginning of extraction.
In PPTBES, BES as a specific inhibitor of methanogenesis was added at a concentration of 2.15 mM.
For each PPT, injection of either 500 or 1,000 liters of test solution into PS5 began at time zero and was completed within 0.6 to 1.9 h (Table 1) by using gravity drainage. After an initial incubation period of 1.8 to 20.4 h, we extracted a total of 750 to 1,000 liters of test solution and groundwater mixture during a further incubation of up to 49 h. Stepwise extraction was used in PPTac and PPTme. The average total test duration ranged from 4.2 h to 70.8 h. Preliminary tests had previously shown that test durations had to be varied to accommodate different substrate degradation rates.
Sample collection procedures.
Water samples for chemical analysis were obtained during the collection of groundwater in carboys (background concentrations), injection of test solutions (injection concentrations), and at regular intervals during the extraction phases of the PPTs. Specifically, samples for the analysis of Br−, organic acids, or BES were filtered in the field by using 0.45-μm-pore-size polyvinylidene fluoride filters (Millipore, Bedford, Mass.) and stored in 12-ml plastic vials. For the analysis of methanol, unfiltered water was collected in 5-ml glass tubes with Teflon-coated screw caps. Samples for CH4 and H2 analysis were collected without headspace in 117-ml serum bottles and closed by using butyl rubber stoppers. All samples were stored at 4°C prior to analysis. Samples collected for dissolved O2, S(−II) and ferrous iron [Fe(II)] determination were analyzed immediately in the field (see below).
Before and after the PPT series (i.e., in September 2001 and October 2002, respectively), groundwater from PS5 was sampled for biological analysis (total cell counts, FISH, and DGGE) by collecting 50 ml (each) of unfiltered water in sterile Falcon tubes. All samples for biological analysis were immediately placed on ice until further processing in the laboratory. After the PPT series, aquifer material samples were collected by using a hand-held hollow-stem auger (Humax, Lucerne, Switzerland) at a radial distance of ∼30 cm from the well casing at a depth of 3 to 3.5 m below ground surface in the anoxic zone of the aquifer. Samples were stored under N2 atmosphere on ice during transport until immediate further processing in the laboratory. Aquifer material consisted of coarse to medium size gravel with a porosity of ∼0.35.
Analytical methods.
Bromide, acetate, and formate concentrations were determined by using a DX-320 ion chromatograph (Dionex, Sunnyvale, Calif.) as described by Kleikemper et al. (29). BES, SO42−, and NO3− were quantified by using a DX-100 ion chromatograph system (Dionex). Methane and H2 concentrations were determined by gas chromatography (model GC 8000; Carlo Erba, Rodano, Italy) on a HayeSep D column with N2 as carrier gas and a Carlo Erba thermal conductivity detector according to the headspace method as described in Bolliger et al. (7). Methanol was quantified photometrically by using alcohol oxidase coupled to peroxidase and 2,2′-azino-di-(3-ethyl)-benzthiazoline-6-sulfonic acid (ABTS) as described by Herzberg and Rogerson (25). Dissolved O2, S(−II), and Fe(II) were measured colorimetrically by using a DR/890 colorimeter (Hach Co., Loveland, Colo.) following standard protocols. DIC concentrations were determined according to the method of Bolliger et al. (7).
Determination of zero-order degradation rates.
Zero-order rates for substrate degradation (in micromolar units per day) in PPTfo were determined directly from observed substrate consumption by using the method of Snodgrass and Kitanidis (47). Since the data of all other PPTs did not fit a zero-order type of reaction but, rather, a first-order type of reaction, first-order rate coefficients (per day) for substrate degradation in these tests were determined from substrate consumption by using the method of Haggerty et al. (21). To allow a comparison of substrate degradation between all tests, first-order rate coefficients were multiplied with the average substrate concentration during each test to obtain quasi-zero-order substrate consumption rates (in micromolar units per day). Methane production rates were calculated from the slope of plots of cumulatively produced CH4 versus time.
Reaction stoichiometries.
In order to relate substrate degradation to CH4 production in PPTs, the stoichiometries of the degradation reactions were taken into account. First, the total mass (in micromoles) of degraded substrate was calculated from the differences of the bromide and substrate breakthrough curves (40). Then, the theoretical stoichiometric mass of CH4 cumulatively produced from the degraded substrate was computed based on the following reaction equations (32):
 |
(1) |
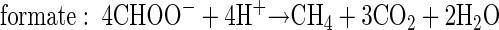 |
(2) |
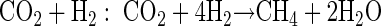 |
(3) |
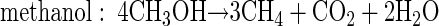 |
(4) |
The observed production of CH4 was calculated for each sampling point separately by the following equation:
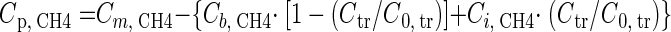 |
(5) |
where CCH4 is the CH4 concentration, the index p stands for produced, m is for measured, b is for background, and i is for injection solution. Ctr and C0,tr are the measured and the injection solution tracer concentrations, respectively. Integration of Cp,CH4 values and extraction volumes yielded the observed total cumulated mass of produced CH4 (44). Finally, the theoretical mass of produced CH4 was compared to the observed mass of produced CH4.
Cell counts and in situ hybridization.
Total cell numbers were estimated by using 4′,6-diamidino-2-phenylindole (DAPI) staining (59). For in situ hybridization, we used the Cy3-labeled 16S rRNA oligonucleotide probes (all purchased from MWG Biotech, Ebersberg, Germany) EUB338 to target Bacteria (3), Arch915 (48) for Archaea, MG1200 for Methanomicrobiaceae, MB1174 for Methanobacteriaceae, MS1414 for Methanosarcinaceae, MX825 for Methanosaetaceae (42), Rotcl1 for Methanosaeta concilii, and Rotcl2 for Methanospirillum sp. (endosymbiont of Plagiopyla nasuta) (62).
Samples for FISH and DAPI counts were processed according to Zarda et al. (59) with the following exceptions. Within a few hours after sampling, water samples were centrifuged at 2,500 × g for 10 min and the debris and cell pellet were resuspended in 1 ml of 4% paraformaldehyde in phosphate-buffered saline. Similarly, 2 g of aquifer material was fixed with 1.5 ml of 4% paraformaldehyde in phosphate-buffered saline. Formamide concentrations in the hybridization mix were 10% for probe MG1200; 20% for Arch915, MX825, and Rotcl2; 30% for probe EUB338 and Rotcl1; and 35% for MB1174 and MS1414. Sodium chloride concentrations in the wash buffer were 440 mM for probe MG1200; 308 mM for Arch915, MX825, and Rotcl2; 100 mM for EUB338 and Rotcl1; and 80 mM for MB1174 and MS1414. The slides were mounted, and visually detectable cells were counted according to the method of Zarda et al. (59).
DNA extraction, DGGE, and cloning.
To extract DNA from aquifer material, 2 g of material was stored in 1.5 ml of lysis buffer (50 mM Tris [pH 9.5], 50 mM EDTA, 50 mM NaCl, and 5% sodium dodecyl sulfate) at −80°C. Further DNA extraction from aquifer material and water samples was conducted according to the method of Kleikemper et al. (29) with the following exceptions. DNA was extracted from the aquifer material by bead beating for 30 s at 5.5 m s−1. For both water and aquifer material samples, the supernatant was transferred into a new tube after and not before digestion with lysozyme and proteinase K.
The PCR of partial (456 bp) archaeal 16S rRNA genes was performed by using primers ARCH915-GC (5′-GC-clamp-AGGAATTGGCGGGGGAGCAC-3′) (4) and UNI-b-rev [5′-GACGGGCGGTGTGT(A/G)CAA-3′] (9), modified from Amann et al. (4), as described in Pesaro and Widmer (38). DGGE of PCR products was performed in a denaturing gradient of 30 to 60% at 75 V for 15 h as described previously (46). DNA band patterns were digitized, photographed, and analyzed by using the GelDoc 2000 system and QuantityOne software (Bio-Rad Laboratories, Hercules, Calif.). Lane background subtraction was conducted by using the rolling disk method (disk size 2), and bands were detected with a sensitivity of 5.1. The similarity of bands was calculated by using the dice coefficient method. Dominant bands containing DNA to be sequenced were excised and incubated for 4 h in 100 μl of sterile water, followed by PCR as described above but with non-GC-clamped primers. PCR products were transformed into Escherichia coli DH5α by using the pGEM-T vector system according to manufacturer's instructions (Promega Corp., Madison, Wis.) and commercially sequenced.
Phylogenetic analyses.
By using the BLAST 2.0 algorithm, the derived sequences were compared to 16S rRNA gene sequences in the National Center for Biotechnology Information database (2). The sequences were aligned with 26 sequences of cultured organisms and environmental clones obtained from the GenBank database. The program DNApars from the PHYLIP package (version 3.5c) (15) was used to perform parsimony analyses on the original alignment and 100 bootstrap samplings with randomized species input order. A consensus tree was calculated with the program Consense from the PHYLIP package and trees were visualized by using TreeView (37). All sequences were checked for chimeric characteristics by the chimera check function of the RDP home page (13).
Nucleotide sequence accession numbers.
The partial environmental 16S rRNA gene clone sequences recovered in this study have been deposited in the GenBank nucleotide sequence database under the accession numbers AY294408 to AY294415.
RESULTS
PPTs.
In native groundwater of well PS5, acetate, formate, methanol, and H2 concentrations were below the detection limit (5 μM for acetate and formate, ∼6 μM for methanol, and ∼0.3 μM for H2). During PPTs, groundwater temperature was 16.0 ± 0.3°C (value ± standard deviation), and dissolved species concentrations, all without any obvious trend during the experiments, were as follows: O2, 4.2 ± 0.6 μmol liter−1; SO42−, 14 ± 13 μmol liter−1; NO3−, 10 ± 15 μmol liter−1; S(−II) (here defined as the sum of S2−, HS−, and H2S), 8 ± 1 μmol liter−1; Fe(II), 287 ± 24 μmol liter−1; and DIC, 14.7 ± 0.9 mmol liter−1. Methane concentrations were 0.67 ± 0.24 mM (average of all tests ± standard deviation) in background water of well PS5 and 0.04 ± 0.04 mM in injection solutions. A calculation of the total amount of Fe(II) lost or produced during the PPTs showed that minor amounts of Fe(II) evolved (at most, 18 mmol; in PPTH2) or disappeared (at most, −17 mmol; in PPTfo) (data not shown).
Breakthrough curves for Br− and each substrate showed a decline in relative concentrations (C/C0) during PPT extraction phases as the extracted test solution was increasingly diluted with native groundwater (Fig. 1a to e). Differences in the curves for relative Br− concentrations between tests show that hydrological conditions (e.g., groundwater level) were somewhat variable during the PPT series. Relative substrate concentrations were lower than relative Br− concentrations during all PPT extraction phases. This difference is significant, since the error in measurements of Br− and substrate concentrations was generally less than 5%. Of the total injected Br− mass, we recovered 44% in PPTac, 73% in PPTfo, 80% in PPTH2, 49% in PPTme, and 31% in PPTBES during the extraction phases of the PPTs (computed by integrating the solute breakthrough curves shown in Fig. 1). Furthermore, 17% of acetate, 47% of formate, 28% of H2 (8% in PPTBES), 43% of methanol, and 30% of BES were recovered.
FIG. 1.

Extraction phase breakthrough curves for Br−, acetate, formate, H2, and methanol in extraction phases during PPTs: PPTac (a), PPTfo (b), PPTH2 (c), PPTme (d), and PPTBES (e). C/C0 is relative concentration, i.e., measured concentration divided by injected concentration. Note that the time scales for the PPTs were different (Table 1) and that for PPTac the numbers on the x axis have to be divided by 2. Ac, acetate; Fo, formate; Me, methanol. The extracted/injected volume is the volume pumped during the extraction phase divided by the total injected volume.
During PPTH2 and PPTBES, formate was detected at initial concentrations of 72 and 30 μM, respectively, and concentrations declined during the tests (Fig. 1c and e). No other organic acids (except those injected and formate in PPTH2 and PPTBES) were detected during PPT extraction phases.
The computed zero-order degradation rate based on substrate disappearance was highest for formate (1.86 mM day−1) and lowest for methanol (0.11 mM day−1) (Table 2). Standard deviations ranged from 1.8 to 6.4% of zero-order degradation rates. If the rates are compared on the basis of theoretical stoichiometric CH4 production (equations 1 to 4), formate still shows the highest rate (0.47 mM CH4 day−1), followed by acetate (0.38 mM CH4 day−1), H2 in PPTH2 (0.23 mM CH4 day−1), methanol (0.083 mM CH4 day−1), and H2 in PPTBES (0.042 mM CH4 day−1) (Table 2).
TABLE 2.
Rates of substrate consumption and CH4 production, cumulative masses of degraded substrate or produced CH4, and a substrate-CH4 balance
| Test | Zero-order substrate degradation rate (mM day−1)a | Cumulative (total) mass of degraded substrate (mmol) | CH4 production rate (mM day−1)
|
Cumulative (total) mass of produced CH4 (mmol) | Percentage of the degraded substrate mass that was recovered as CH4 % | |
|---|---|---|---|---|---|---|
| Theoretical | Measureda | |||||
| PPTac | 0.38 ± 0.02 | 557 | 0.38 | 0.048 ± 0.004 | 158 | 28 |
| PPTfo | 1.86 ± 0.12 | 268 | 0.47 | 0.43 ± 0.043 | 44 | 65 |
| PPTH2 | 0.91 ± 0.02 | 157 | 0.23 | 0.52 ± 0.046 | 74 | 188 |
| PPTme | 0.11 ± 0.01 | 58 | 0.083 | 0.026 ± 0.0004 | 26 | 60 |
| PPTBESb | 0.17 ± 0.01 | 46 | 0.042 | −0.28 ± 0.055 | −49 | |
Value±standard deviation.
Zero-order degradation rate and cumulative substrate degraded in PPTBES refer to H2 degradation.
The CH4 concentrations in extracted groundwater increased during all PPTs from injection concentrations to background concentrations (Fig. 2a). After subtraction of the respective background CH4, the total cumulative mass of produced CH4 ranged from −45 mmol (PPTBES) to +158 mmol (PPTac) (Fig. 2b and Table 2). The negative value for PPTBES indicates that CH4 consumption instead of production occurred in this test. Methane production rates (Table 2) ranged from 0.026 (PPTme) to 0.52 (PPTH2) mM day−1, with standard deviations of 1.5 to 20.0% of CH4 production rates. BES concentrations in PPTBES remained above 0.1 mM, the suggested lower limit for inhibition (36), throughout the extraction phase of PPTBES (lowest concentration, 0.32 mM).
FIG. 2.

(a) Methane concentrations during extraction phases of five PPTs. Methane data represent the averages of two samples each. (b) Cumulative produced CH4 during extraction phases of five PPTs. Note that the time scales for the PPTs were different (Table 1). The extracted/injected volume is the volume pumped during the extraction phase divided by the total injected volume.
Cell counts and in situ hybridization.
The total cell number (DAPI-stained cells) in water samples from PS5 was (1.12 ± 0.09) × 105 cells ml−1 at the beginning of the PPT series (2001) and (0.79 ± 0.20) × 105 cells ml−1 at the end of the series (2002). Percentages of cells hybridizing with probe EUB338 and Arch915 in water samples were 7.8% ± 1.8% and 35% ± 4.3% of total (DAPI-stained) microorganisms at the beginning and 22.5% ± 3.7% and 18.2% ± 6.4% at the end of the PPT series, respectively (Fig. 3). In aquifer material samples, 13.8% ± 3.0% of total microorganisms hybridized with probe EUB338, and 9.0% ± 3.3% hybridized with probe Arch915. Hybridizations with the genera-specific probes showed that the species Methanosaeta concilii (probe Rotcl1) accounted for 22.3% of total microorganisms in water samples from 2001, 8.4% in water samples from 2002, and 5.0% in aquifer material. The more general probe MX825 for Methanosaeta spp. detected 14.3% of total microorganisms in water samples from 2001, 6.5% in water samples from 2002, and 2.5% in aquifer material samples. The family Methanomicrobiaceae (probe MG1200) accounted for 1.4% of total microorganisms in water samples after the PPT series but was not detected in water samples before the PPT series and in aquifer material (Fig. 3). Methanogenic Archaea that we probed for by using probes MB1174, MS1414, and Rotcl2 were below the detection limit of 1% (59).
FIG. 3.

Percentage of total (DAPI-stained) cells hybridizing with fluorescent probes Arch915 (Archaea), EUB338 (Bacteria), Rotcl1 (Methanosaeta concilii), MX825 (Methanosaetaceae), and MG1200 (Methanomicrobiaceae) in water samples recovered in September 2001 and October 2002 (i.e., before and after the PPT series) and aquifer material samples recovered in October 2002. Error bars indicate one standard deviation. Percentages of total cells hybridizing with MB1174, MS1414, and Rotcl2 were below the detection limit (1% of total cells).
DGGE and sequencing.
DGGE of PCR products resulted in distinct profiles, which exhibited 19 bands for each water sample and 12 bands for the aquifer material sample (Fig. 4). Profiles of water samples collected before (2001) and after (2002) the PPT series were 74.4 to 77.1% similar, while the aquifer material sample was only ∼48.8 to 52.8% similar to both water samples. The given ranges are based on a comparison of duplicate profiles for each water sample and one profile for the aquifer material sample. Especially in the lower part of the gel, some bands were present in water samples but not in aquifer material. Tree construction indicated that all sequenced clones were related to methanogenic Archaea (Fig. 5). Two sequences (5 and 8) were closely related to known methanogenic Archaea (Methanosaeta and Methanospirillum), whereas sequences 2, 4, and 6 clustered more closely with uncultured environmental archaeal clones, and sequences 3, 7, and 9 were more distantly related to methanogenic Archaea. Unfortunately, band number 1 (Fig. 4) did not reamplify after excision in repeated attempts.
FIG. 4.

DGGE profiles of DNA extracted from groundwater and aquifer material. (a) Water sample taken in 2001 before the PPT series. (b) Aquifer material from 2002. (c) Water sample taken in 2002 after the PPT series. Numbers refer to bands that were excised and sequenced (Fig. 5).
FIG. 5.

Phylogenetic relationship of the eight retrieved ∼456-bp archaeal sequences to 26 sequences of cultured organisms and environmental clones based on currently available 16S rRNA gene sequences. The percentages of 100 bootstrap samplings that supported a cluster are indicated. GenBank accession numbers of the 16S rRNA gene sequences are given in parentheses. Substrates used by members of the methanogen orders (boldface indicates close relatives of clones retrieved in this study) and FISH probes that target members of the respective order are given in parentheses after methanogen order names.
DISCUSSION
Substrate consumption and rates.
Lower relative substrate concentrations compared to relative Br− concentrations throughout all PPTs (Fig. 1a to e) indicated that substrates were consumed during those tests, presumably due to microbial activity. Differences between recovered cumulative relative Br− and substrate masses showed that 27% of injected acetate, 26% of formate, 52% of H2 (PPTH2) or 23% of H2 (PPTBES), and 6% of methanol were degraded in the respective tests. This illustrates that total test durations (Table 1) were sufficiently long to allow detectable substrate consumption during the tests.
The evolution of formate upon injection of H2 in PPTH2 and PPTBES may be explained by the presence of hydrogen lyases both within methanogenic Archaea (57) and other bacteria (55). In both PPTs, 24% of total degraded H2 was transformed into formate. The activity of hydrogen lyases is not inhibited by BES (57).
Degradation rates determined in our study (Table 2) were similar to those determined by Istok et al. (27) also using PPTs, who found rates of 1.92 to 4.80 mM day−1 for H2 consumption and 0.048 mM day−1 for CH4 production in another PHC-contaminated aquifer. However, our rates were 1 to 2 orders of magnitude higher than those published by Hansen et al. (22), who found maximal rates of 0.011 mM day−1. Nevertheless, rates in the latter study were determined for a noncontaminated site where methanogenesis may be expected to be slower. Furthermore, since we added the substrates in concentrations higher than the indigenous levels, the rates we measured do not represent indigenous conditions. Rather than determining indigenous degradation rates, our goal was to use the substrates to test for the activity of different groups of methanogens.
Differences in substrate consumption rates between tests may be evoked by variations of groundwater temperature, geochemical conditions, contributions of processes other than methanogenesis to substrate degradation, and distinct activities of different physiological groups of methanogens. Temperature and geochemical conditions remained fairly stable among PPTs (see above). Hence, these factors probably do not explain much of the variation in rates.
Contribution of other processes to substrate consumption.
The fact that in PPTac, PPTfo, and PPTme less than 100% of degraded substrate was accounted for by CH4 production (Table 2) indicated that processes other than methanogenesis contributed to substrate degradation. We have currently no explanation why more than 100% of H2 was recovered in produced CH4 during PPTH2. Other processes possibly contributing to substrate consumption during our PPTs are O2, SO42−, NO3−, and Fe(III) reduction and acetogenesis (23, 60). For example, in PPTac, the measured average in situ concentrations of O2 (4.2 μmol liter−1), SO42− (14 μmol liter−1), and NO3− (10 μmol liter−1) together would allow for the mineralization of a total amount of 14.7 mmol of acetate in 600 liters of extracted volume (after that the acetate concentration was zero in PPTac). This corresponds to merely 2.6% of total degraded acetate in PPTac (557 mmol) (Table 2). Hence, the contribution of the electron acceptors O2, SO42−, and NO3− to substrate degradation was negligible.
Fe(III)-reducing bacteria are likely able to consume all of the added substrates (10, 49), and methanogenesis and Fe(III) reduction are known to occur simultaneously (5). However, only minor amounts of Fe(II) evolved or disappeared during our PPTs. For example, the observed production of 18 mmol of Fe(II) in PPTH2 may have been linked to the consumption of 9 mmol of H2, which is only a fraction of the total degraded H2 in PPTH2 (157 mmol) (Table 2). However, a large uncertainty is associated with the Fe(II) balances due to high concentrations of Fe(II) in the background water of PS5, possible precipitation reactions of Fe(II), e.g., with S(−II) or CO32−, and sorption of Fe(II) to the solid phase. If we consider an Fe(II) concentration of 3 × 10−4 M and an S2− concentration of 1.14 × 10−13 M (calculated for pH 6.7, and the sum of S2−, HS−, and H2S is a concentration of 8 μM), the ion activity product of [Fe2+][S2−] is equal to 3.4 × 10−17 M2, which is 43 times higher than the solubility product of FeS (7.94 × 10−19 M2) (50). Similarly, if we consider a CO32− concentration of 2.64 × 10−6 M (calculated for pH 6.7, and the sum of H2CO3, HCO3−, CO32− is a concentration of 1.47 × 10−2 M), the ion activity product of [Fe(II)][CO32−] is equal to 7.9 × 10−10 M2, which is 20 times higher than the solubility product of FeCO3 (3.98 × 10−11 M2) (50). Hence, the groundwater was supersaturated with respect to FeS and FeCO3 and precipitation was likely. Therefore, observed Fe(II) production does not allow us to quantify the contribution of Fe(III) reduction to substrate consumption during the PPTs (26).
Distinct group activities of methanogenic Archaea.
The observed CH4 production in the first four tests (Table 2) indicates that methanogenic Archaea contributed to a significant extent to substrate consumption. This was further corroborated by the inhibition of methanogenesis during PPTBES, for which a lower H2 degradation rate was determined than for PPTH2, in which no inhibitor was added (Table 2). Substantial 13C-CH4 production in recently conducted PPTs with 13C-labeled acetate or CO2 confirms these results (39). Hence, much of the variability in substrate consumption rates in our experiments may have been due to different activities of different physiologic groups of methanogens.
Microbial population analyses.
Archaea were abundant in water (18 to 38% of total microorganisms) and aquifer material (9%) (Fig. 3). The presence of 19 DGGE bands in water samples and 12 bands in the aquifer material sample indicated a diverse archaeal population in the vicinity of well PS5 (Fig. 4). Diverse and abundant archaeal populations have been found in PHC-contaminated environments before (11, 14, 52). All of the retrieved sequences were related to methanogenic Archaea (Fig. 5). Two sequences (5 and 8) were closely related to known methanogenic Archaea (Methanosaeta and Methanospirillum), whereas sequences 2, 4, and 6 clustered more closely with uncultured environmental archaeal clones. These clones were mostly derived from methanogenic habitats such as wetland soils (51) (clones OS-8, AM-10, and OS-16), a PHC-contaminated aquifer (14) (clone WCHD3-07) or a dichloropropane-dechlorinating culture (clone SHB-200; GenBank accession no. AJ312014). In contrast, clones 3, 7, and 9 were more distantly related to methanogenic Archaea. Therefore, clones 2, 4, 5, 6, and 8 were likely methanogenic Archaea, whereas the function of clones 3, 7, and 9 is less certain.
Higher detection rates with the Methanosaeta concilii-specific probe Rotcl1 than with the more general Methanosaeta-specific probe MX825 possibly reflect different accessibilities of the probe binding sites on the 16S rRNA or different binding properties of the two probes (18). Hence, we refrain from comparing the results of two different hybridization probes. Nevertheless, FISH and DGGE (Fig. 5, band 5) suggested that Methanosaeta concilii was a dominant member of the microbial community both in water and aquifer material samples (Fig. 3), agreeing with earlier molecular analyses conducted at the same site (8). Except for Methanosaeta, only members of the Methanomicrobiaceae were detected by FISH (Fig. 3). However, much lower amounts of Methanomicrobiaceae were determined by FISH than suggested by the abundance of Methanomicrobiaceae-related sequences forming dominant bands in DGGE profiles (Fig. 4 and 5), by the presence of the target sequence of probe MG1200 in clones 6 and 8, and by one mismatch in clones 2 and 4. Our limited success in detecting methanogenic Archaea other than Methanosaeta by using FISH may have been associated with a low ribosome content in the target cells (53) and/or the lack of exact probe matches (e.g., targets for probe MB1174 were not present in our clones [four mismatches each with clones 3, 7, and 9]).
The higher percentages of Archaea in water compared to aquifer material samples (Fig. 3) agrees well with other studies, in which the percentage of free-living methanogens was frequently higher than that of attached methanogens (5, 20, 30). The reason for this remains unknown and deserves further research. However, archaeal community composition was similar but not identical in water and aquifer material (Fig. 4). Both similarities and differences between attached and suspended microorganisms were shown previously for other aquifers, including PHC-contaminated ones (1, 5, 31, 56).
The higher relative numbers of Archaea in water samples before (35%) than after the PPT series (18%) were accompanied by only subtle changes in DGGE profiles (Fig. 4a and c, bands 7 and 9) and the detection of Methanomicrobiaceae in the latter water samples (Fig. 3). The temporal stability of the archaeal population agrees with the results of Bolliger et al. (8), who showed that the archaeal community composition in the same well (PS5) was stable over another 1-year observation period (1998 to 1999). Furthermore, these data agree with our chemical data (see above), the study by Bolliger et al. (8), and continued monitoring of the site (unpublished data), which revealed that chemical parameters in well PS5 remained fairly invariable over long time periods. Similar DGGE profiles before and after the study period also suggested that the archaeal community likely did not diverge dramatically during that period.
Comparison of PPT data with molecular analyses.
Since Methanosaeta are known to generate energy only through aceticlastic methanogenesis (54), the abundance of Methanosaeta concilii in groundwater and aquifer material near well PS5 (Fig. 3 to 5) (8) and substantial acetate consumption and CH4 production during PPTac suggested that acetate was the main substrate for methanogenesis in this aquifer. However, CH4 was produced more slowly from acetate than from H2 and formate in PPTH2 and PPTfo (Table 2), which agrees with the sequencing data indicating that many DGGE bands represented Archaea that consumed formate and H2 plus CO2 (Fig. 5). Even though some methanol was consumed during PPTme, none of our sequences were related to methanol-degrading Archaea, which may have been represented by one or more of the nonsequenced, minor DGGE bands. Hence, the measurements of potential activities of methanogens in this study largely agreed with a molecular analysis of methanogenic Archaea populations.
Conclusions.
For the first time (to our knowledge), both the activity and diversity of methanogens were investigated in a PHC-contaminated aquifer by using PPTs and molecular analyses. The potential rates of methanogenesis from several methanogen substrates were determined and showed a large potential for methanogenesis in the examined aquifer with rates of up to 1.86 ± 0.12 mM day−1 when formate was added. In addition, this study allows us to answer the question of which methanogens are active in a petroleum-contaminated aquifer. As observed previously (8, 14), aceticlastic methanogenesis played a major role, but methanogenesis with formate or CO2 and H2 showed higher potential rates, indicating the presence of a large population of CO2-type substrate-utilizing methanogens. These findings agreed with our data from FISH and DGGE or cloning. Hence, both types of methanogenesis are likely involved in the terminal step of hydrocarbon degradation, while methanogenesis from methanol plays a minor role. However, at present the exact contribution of each process to total in situ methanogenesis cannot be determined since only potential rates of methanogenesis were measured. Furthermore, the feasibility of PPTs in the methanogenic zone was demonstrated beyond the study of Istok et al. (27), since in contrast to their experiments, we used a range of substrates for methanogenesis and were able to measure CH4 production as a result of longer incubation periods. The combination of hydrogeological and molecular methods in this study provided valuable information on the community structure and the activity of methanogens in a PHC-contaminated aquifer. One method by itself may not have provided the full picture. Future studies will focus on the role of Fe(III) reduction and the direct linkage between the activity and identity of PHC-degrading microorganisms.
Acknowledgments
Support for this study was provided by the Swiss National Science Foundation (Priority Program Environment) and the Swiss Agency for the Environment, Forests and Landscape (BUWAL).
REFERENCES
- 1.Alfreider, A., C. Vogt, and W. Babel. 2002. Microbial diversity in an in situ reactor system treating monochlorobenzene contaminated groundwater as revealed by 16S ribosomal DNA analysis. Syst. Appl. Microbiol. 25:232-240. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amann, R. I., L. Krumholz, and D. A. Stahl. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 172:762-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amann, R. I., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekins, B. A., E. M. Godsy, and E. Warren. 1999. Distribution of microbial physiologic types in an aquifer contaminated by crude oil. Microb. Ecol. 37:263-275. [DOI] [PubMed] [Google Scholar]
- 6.Boetius, A., K. Ravenschlag, C. J. Schubert, D. Rickert, F. Widdel, A. Gieseke, R. Amann, B. B. Jørgensen, U. Witte, and O. Pfannkuche. 2000. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407:623-626. [DOI] [PubMed] [Google Scholar]
- 7.Bolliger, C., P. Höhener, D. Hunkeler, K. Häberli, and J. Zeyer. 1999. Intrinsic bioremediation of a petroleum hydrocarbon-contaminated aquifer and assessment of mineralization based on stable carbon isotopes. Biodegradation 10:201-217. [DOI] [PubMed] [Google Scholar]
- 8.Bolliger, C., F. Schönholzer, M. H. Schroth, D. Hahn, S. Bernasconi, and J. Zeyer. 2000. Characterizing intrinsic bioremediation in a petroleum hydrocarbon-contaminated aquifer by combined chemical, isotopic and biological analyses. Bioremediat. J. 4:359-371. [Google Scholar]
- 9.Bundt, M., F. Widmer, M. Pesaro, J. Zeyer, and P. Blaser. 2001. Preferential flow paths: biological “hot spots” in soils. Soil Biol. Biochem. 33:729-738. [Google Scholar]
- 10.Caccavo, F., R. P. Blakemore, and D. R. Lovley. 1992. A hydrogen-oxidizing, Fe(III)-reducing microorganism from the Great Bay estuary, New Hampshire. Appl. Environ. Microbiol. 58:3211-3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapelle, F. H., P. M. Bradley, D. R. Lovley, K. O'Neill, and J. E. Landmeyer. 2002. Rapid evolution of redox processes in a petroleum hydrocarbon-contaminated aquifer. Ground Water 40:353-360. [DOI] [PubMed] [Google Scholar]
- 12.Christensen, T. H., P. Kjeldsen, H. J. Albrechtsen, G. Heron, P. H. Nielsen, P. L. Bjerg, and P. E. Holm. 1994. Attenuation of landfill leachate pollutants in aquifers. Crit. Rev. Environ. Sci. Technol. 24:119-202. [Google Scholar]
- 13.Cole, J. R., B. Chai, T. L. Marsh, R. J. Farris, Q. Wang, S. A. Kulam, S. Chandra, D. M. McGarrell, T. M. Schmidt, G. M. Garrity, and J. M. Tiedje. 2003. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Research 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dojka, M. A., P. Hugenholtz, S. K. Haack, and N. R. Pace. 1998. Microbial diversity in a hydrocarbon- and chlorinated-solvent-contaminated aquifer undergoing intrinsic bioremediation. Appl. Environ. Microbiol. 64:3869-3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felsenstein, J. 1989. PHYLIP: phylogeny inference package (version 3.2). Cladistics 5:164-166. [Google Scholar]
- 16.Ficker, M., K. Krastel, S. Orlicky, and E. Edwards. 1999. Molecular characterization of a toluene-degrading methanogenic consortium. Appl. Environ. Microbiol. 65:5576-5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fielding, E. R., D. B. Archer, E. C. Demacario, and A. J. L. Macario. 1988. Isolation and characterization of methanogenic bacteria from landfills. Appl. Environ. Microbiol. 54:835-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs, B. M., G. Wallner, W. Beisker, I. Schwippl, W. Ludwig, and R. Amann. 1998. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 64:4973-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia, J. L., B. K. C. Patel, and B. Ollivier. 2000. Taxonomic phylogenetic and ecological diversity of methanogenic Archaea. Anaerobe 6:205-226. [DOI] [PubMed] [Google Scholar]
- 20.Godsy, E. M., D. F. Goerlitz, and D. Grbicgalic. 1992. Methanogenic biodegradation of creosote contaminants in natural and simulated groundwater ecosystems. Ground Water 30:232-242. [Google Scholar]
- 21.Haggerty, R., M. H. Schroth, and J. D. Istok. 1998. Simplified method of “push-pull” test data analysis for determining in situ reaction rate coefficients. Ground Water 36:314-324. [Google Scholar]
- 22.Hansen, L. K., R. Jakobsen, and D. Postma. 2001. Methanogenesis in a shallow sandy aquifer, Romo, Denmark. Geochim. Cosmochim. Acta 65:2925-2935. [Google Scholar]
- 23.Hansen, T. A. 1994. Metabolism of sulfate-reducing prokaryotes. Antonie Leeuwenhoek 66:165-185. [DOI] [PubMed] [Google Scholar]
- 24.Hanson, R. S., and T. E. Hanson. 1996. Methanotrophic bacteria. Microbiol. Mol. Biol. Rev. 60:439-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herzberg, G. R., and M. Rogerson. 1985. Use of alcohol oxidase to measure the methanol produced during the hydrolysis of d-methyl-3-hydroxybutyric and l-methyl-3-hydroxybutyric acid. Anal. Biochem. 149:354-357. [DOI] [PubMed] [Google Scholar]
- 26.Hunkeler, D., P. Höhener, S. Bernasconi, and J. Zeyer. 1999. Engineered in situ bioremediation of a petroleum hydrocarbon-contaminated aquifer: assessment of mineralization based on alkalinity, inorganic carbon and stable carbon isotope balances. J. Contam. Hydrol. 37:201-223. [Google Scholar]
- 27.Istok, J. D., M. D. Humphrey, M. H. Schroth, M. R. Hyman, and K. T. O'Reilly. 1997. Single-well, “push-pull” test for in situ determination of microbial activities. Ground Water 35:619-631. [Google Scholar]
- 28.Joulian, C., B. Ollivier, B. K. C. Patel, and P. A. Roger. 1998. Phenotypic and phylogenetic characterization of dominant culturable methanogens isolated from rice field soils. FEMS Microbiol. Ecol. 25:135-145. [Google Scholar]
- 29.Kleikemper, J., M. H. Schroth, W. V. Sigler, M. Schmucki, S. M. Bernasconi, and J. Zeyer. 2002. Activity and diversity of sulfate-reducing bacteria in a petroleum hydrocarbon-contaminated aquifer. Appl. Environ. Microbiol. 68:1516-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehman, R. M., and S. P. O'Connell. 2002. Comparison of extracellular enzyme activities and community composition of attached and free-living bacteria in porous medium columns. Appl. Environ. Microbiol. 68:1569-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehman, R. M., F. F. Roberto, D. Earley, D. F. Bruhn, S. E. Brink, S. P. O'Connell, M. E. Delwiche, and F. S. Colwell. 2001. Attached and unattached bacterial communities in a 120-meter corehole in an acidic, crystalline rock aquifer. Appl. Environ. Microbiol. 67:2095-2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madigan, M. T., J. M. Martinko, and J. Parker. 2003. Brock biology of microorganisms, 10th ed. Prentice-Hall, Inc., London, United Kingdom.
- 33.Miller, T. L., M. J. Wolin, Z. Hongxue, and M. P. Bryant. 1986. Characteristics of methanogens isolated from bovine rumen. Appl. Environ. Microbiol. 51:201-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ollivier, B., J. L. Cayol, B. K. C. Patel, M. Magot, M. L. Fardeau, and J. L. Garcia. 1997. Methanoplanus petrolearius sp. nov., a novel methanogenic bacterium from an oil-producing well. FEMS Microbiol. Lett. 147:51-56. [DOI] [PubMed] [Google Scholar]
- 35.Oremland, R. S. 1988. Biogeochemistry of methanogenic bacteria, p. 641-705. In A. J. B. Zehnder (ed.), Biology of anaerobic microorganisms. John Wiley & Sons, New York, N.Y.
- 36.Oremland, R. S., and D. G. Capone. 1988. Use of “specific” inhibitors in biogeochemistry and microbial ecology. Adv. Microb. Ecol. 10:285-383. [Google Scholar]
- 37.Page, R. D. M. 2004. TreeView 1.6.6. http://taxonomy.zoology.gla.ac.uk/rod/rod.html.
- 38.Pesaro, M., and F. Widmer. 2002. Identification of novel Crenarchaeota and Euryarchaeota clusters associated with different depth layers of a forest soil. FEMS Microbiol. Ecol. 42:89-98. [DOI] [PubMed] [Google Scholar]
- 39.Pombo, S. A., J. Kleikemper, M. H. Schroth, and J. Zeyer. Field-scale isotopic labeling of phospholipid fatty acids from acetate-degrading sulfate-reducing bacteria. FEMS Microbiol. Ecol., in press. [DOI] [PubMed]
- 40.Pombo, S. A., O. Pelz, M. H. Schroth, and J. Zeyer. 2002. Field-scale C-13-labeling of phospholipid fatty acids (PLFA) and dissolved inorganic carbon: tracing acetate assimilation and mineralization in a petroleum hydrocarbon-contaminated aquifer. FEMS Microbiol. Ecol. 41:259-267. [DOI] [PubMed] [Google Scholar]
- 41.Raskin, L., L. K. Poulsen, D. R. Noguera, and B. E. Rittmann. 1994. Quantification of methanogenic groups in anaerobic biological reactors by oligonucleotide probe hybridization. Appl. Environ. Microbiol. 60:1241-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raskin, L., J. M. Stromley, B. E. Rittmann, and D. A. Stahl. 1994. Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl. Environ. Microbiol. 60:1232-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Röling, W. F. M., B. M. van Breukelen, M. Braster, B. Lin, and H. W. van Verseveld. 2001. Relationships between microbial community structure and hydrochemistry in a landfill leachate-polluted aquifer. Appl. Environ. Microbiol. 67:4619-4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroth, M. H., J. D. Istok, G. T. Conner, M. R. Hyman, R. Haggerty, and K. T. O'Reilly. 1998. Spatial variability in in situ aerobic respiration and denitrification rates in a petroleum-contaminated aquifer. Ground Water 36:924-937. [Google Scholar]
- 45.Schroth, M. H., J. Kleikemper, C. Bolliger, S. M. Bernasconi, and J. Zeyer. 2001. In situ assessment of microbial sulfate reduction in a petroleum-contaminated aquifer using push-pull tests and stable sulfur isotope analyses. J. Contam. Hydrol. 51:179-195. [DOI] [PubMed] [Google Scholar]
- 46.Sigler, W. V., C. H. Nakatsu, Z. J. Reicher, and R. F. Turco. 2001. Fate of the biological control agent Pseudomonas aureofaciens TX-1 after application to turfgrass. Appl. Environ. Microbiol. 67:3542-3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snodgrass, M. F., and P. K. Kitanidis. 1998. A method to infer in situ reaction rates from push-pull experiments. Ground Water 36:645-650. [Google Scholar]
- 48.Stahl, D. A., and R. I. Amann. 1991. Development and application of nucleic acid probes, p. 205-248. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, N.Y.
- 49.Straub, K. L., and B. E. E. Buchholz-Cleven. 2001. Geobacter bremensis sp. nov. and Geobacter pelophilus sp. nov., two dissimilatory ferric-iron-reducing bacteria. Int. J. Syst. Evol. Microbiol. 51:1805-1808. [DOI] [PubMed] [Google Scholar]
- 50.Stumm, W., and J. J. Morgan. 1981. Aquatic chemistry: an introduction emphasizing chemical equilibria in natural waters, 2nd ed. Wiley Interscience, New York, N.Y.
- 51.Utsumi, M., S. E. Belova, G. M. King, and H. Uchiyama. 2003. Phylogenetic comparison of methanogen diversity in different wetland soils. J. Gen. Appl. Microbiol. 49:75-83. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe, K., Y. Kodama, N. Hamamura, and N. Kaku. 2002. Diversity, abundance, and activity of archaeal populations in oil-contaminated groundwater accumulated at the bottom of an underground crude oil storage cavity. Appl. Environ. Microbiol. 68:3899-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe, K., K. Watanabe, Y. Kodama, K. Syutsubo, and S. Harayama. 2000. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude oil storage cavities. Appl. Environ. Microbiol. 66:4803-4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitman, W. B., T. L. Bowen, and D. R. Boone. 1992. The methanogenic bacteria, p. 739-745. In A. Balows, H. G. Truper, M. Dworkin, W. Harder, and K. H. Schleifer (ed.), The prokaryotes, vol. 1. Springer-Verlag, New York, N.Y. [Google Scholar]
- 55.Woods, D. D. 1936. Hydrogenlyases. IV. The synthesis of formic acid by bacteria. J. Biol. Chem. 77:515-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Worm, J., K. Gustavson, K. Garde, N. H. Borch, and M. Sondergaard. 2001. Functional similarity of attached and free-living bacteria during freshwater phytoplankton blooms. Aquat. Microb. Ecol. 25:103-111. [Google Scholar]
- 57.Wu, W. M., R. F. Hickey, M. K. Jain, and J. G. Zeikus. 1993. Energetics and regulations of formate and hydrogen metabolism by Methanobacterium formicicum. Arch. Microbiol. 159:57-65. [Google Scholar]
- 58.Wuebbles, D. J., and K. Hayhoe. 2002. Atmospheric methane and global change. Earth-Sci. Rev. 57:177-210. [Google Scholar]
- 59.Zarda, B., D. Hahn, A. Chatzinotas, W. Schönhuber, A. Neef, R. I. Amann, and J. Zeyer. 1997. Analysis of bacterial community structure in bulk soil by in situ hybridization. Arch. Microbiol. 168:185-192. [Google Scholar]
- 60.Zehnder, A. J. B. 1988. Biology of anaerobic microorganisms. John Wiley & Sons, New York, N.Y.
- 61.Zengler, K., H. H. Richnow, R. Rosselló-Mora, W. Michaelis, and F. Widdel. 1999. Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401:266-269. [DOI] [PubMed] [Google Scholar]
- 62.Zepp Falz, K., C. Holliger, R. Grosskopf, W. Liesack, A. N. Nozhevnikova, B. Müller, B. Wehrli, and D. Hahn. 1999. Vertical distribution of methanogens in the anoxic sediment of Rotsee (Switzerland). Appl. Environ. Microbiol. 65:2402-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zinder, S. H. 1993. Physiological ecology of methanogens, p. 128-206. In J. G. Ferry (ed.), Methanogenesis: ecology, physiology, biochemistry and genetics. Chapman & Hall, Inc., New York, N.Y.