

Abstract
In the last three decades, the understanding of the renin angiotensin system (RAS) has been changed by the discoveries of functional local systems, novel biologically active peptides, additional specific receptors, alternative pathways of angiotensin (Ang) II generation, and new roles for enzymes and precursor components other than those in Ang II synthesis. In this regard, the discovery that Ang-(1-7) opposes the pressor, proliferative, pro-fibrotic, and pro-inflammatory effects mediated by Ang II has contributed to the realization that the RAS is composed of two axes. The first axis consists of the angiotensin-converting enzyme (ACE), with Ang II as the end product, and the angiotensin type 1 (AT1) receptor as the main effector mediating the biological actions of Ang II. The second axis results from ACE2-mediated hydrolysis of Ang II, leading to the production of Ang-(1-7), with the Mas receptor as the main effector conveying the vasodilatory, anti-proliferative, anti-fibrotic, and anti-inflammatory effects of Ang-(1-7). Experimental and clinical studies have shown that both axes of the RAS may take part in the pathogenesis of liver diseases. In this manuscript, we summarize the current evidence regarding the role of RAS in hepatic cirrhosis and its complications, including hemodynamic changes and hepatorenal syndrome. The therapeutic potential of the modulation of RAS molecules in liver diseases is also discussed.
Keywords: Renin angiotensin system, Angiotensin II, Angiotensin-(1-7), Hepatic cirrhosis, Liver fibrosis, Hepatorenal syndrome
Core tip: This Editorial reports recent advances on the understanding of the renin angiotensin system in regard to the role of the two main and counter-regulatory mediators, Angiotensin II and Angiotensin-(1-7), in liver diseases and in their main complications. Experimental and clinical findings so far show that Angiotensin-(1-7) by binding to Mas receptor opposes Angiotensin II actions mediated by AT1 receptors in liver tissue, by eliciting anti-inflammatory, anti-oxidative and anti-fibrotic effects. This knowledge may help in paving the way for the development of novel treatments for liver diseases and their complications.
INTRODUCTION
The renin angiotensin system (RAS) is classically conceived as a hormonal system mainly responsible for blood pressure control and hydroelectrolyte balance[1]. In this context, renin, the first enzyme of the classic RAS, is produced in the juxtaglomerular cells of the afferent renal arteriole in response to glomerular hypoperfusion, reduced sodium intake, and increased activity of the sympathetic nervous system[2]. Next, circulating renin cleaves its substrate angiotensinogen to form the decapeptide angiotensin I (Ang I), which is converted by angiotensin-converting enzyme (ACE) to the major active component of the RAS, angiotensin II (Ang II). The major biological actions of Ang II are mediated by angiotensin type 1 (AT1) receptors[3]. The excessive activity of ACE-Ang II-AT1 arm frequently contributes to several pathophysiological changes including excessive renal sodium reabsorption, abnormal vascular smooth muscle cell contraction, disproportionately high aldosterone secretion and inappropriate cardiovascular responses[4]. Additionally, pro-inflammatory, pro-thrombotic and pro-fibrotic pathways are stimulated by AT1 receptor activation[5].
On the other hand, recent advances have changed our understanding of the RAS. These have included the discovery of functional local systems, novel biologically active peptides, additional specific receptors, alternative pathways of Angiotensin peptides generation, and new roles for enzymes and precursor components other than Ang II synthesis[4,6]. Especially relevant for the reconceptualization of the RAS was the identification of the heptapeptide Ang-(1-7)[7], the ACE homologue enzyme responsible for the conversion of Ang II into Ang-(1-7), ACE2[8,9], and the Mas receptor, a G-protein coupled receptor which mediates the main effects of Ang-(1-7)[10]. Also important, a considerable amount of evidence supports a counter-regulatory role for Ang-(1-7). This peptide opposes several Ang II AT1 receptor-mediated effects, including vasoconstriction, cell proliferation, inflammation and tissue fibrosis[11-13]. Considering the opposite roles of the two main mediators of the RAS, Ang II and Ang-(1-7), independent research groups have proposed a new view of the RAS. In this model, the RAS can be envisioned as a dual function system in which the vasoconstrictor/proliferative or vasodilator/anti-proliferative actions are primarily driven by the balance between two axes of the RAS, the classical one formed by ACE-Ang II-AT1 and the counter-regulatory comprising ACE2-Ang-(1-7)-Mas[4,12-15]. In general, these axes present opposite effects in physiological and pathological states, including liver diseases[16]. It is well-established that RAS blockers, ACE inhibitors and angiotensin receptor antagonists (ARAs), can inhibit the ACE-Ang II-AT1 arm, but also stimulate the activity of ACE2-Ang-(1-7)-Mas axis[15,17,18]. Both agents have been broadly used in the clinical practice for congestive heart failure, hypertension, chronic kidney disease[19] and seem to exert beneficial effects on liver diseases[20]. Table 1 displays the main opposite effects of both RAS axes.
Table 1.
Main and opposite actions of both renin angiotensin system axes
| Organ/tissue | ACE-Ang II- AT1 axis | ACE2-Ang-(1-7)-Mas axis |
| Blood vessels | Vasoconstriction | Vasodilation |
| Heart | Hypertrophic, arrithmogenic | Anti-hypertrophic, anti-arrithmogenic |
| Kidney | Inflammation, fibrosis | Anti-inflammatory, anti-fibrogenic |
| Lung | Alergic, fibrosis | Anti-alergic, anti-fibrosis |
| Brain | Ischemia | Reduction of ischemia |
| Adipose tissue | Increase insulin resistance | Decrease insulin resistance |
ACE: Angiotensin-converting enzyme; Ang II: Angiotensin II, AT1: Angiotensin type 1 receptor; Ang-(1-7): Angiotensin-(1-7); Mas: Angiotensin-(1-7) receptor.
In this editorial, we summarize recent evidence on the role of both RAS axes in liver diseases and their complications. Furthermore, the general idea that the final RAS effect represents a balance between the “friend”, ACE2-Ang-(1-7)-Mas axis, and the “foe”, ACE-Ang II-AT1 axis, is discussed in the context of liver diseases and potential therapeutic strategies.
RAS IN PHYSIOLOGICAL AND PATHOPHYSIOLOGICAL CONDITIONS INVOLVING THE LIVER
The RAS was classically described as a circulating hormonal system that plays a pivotal role in the maintenance of blood pressure and fluid homeostasis[21]. However, this view changed when the concept of local RAS was introduced. The concept of “local RAS” was first based on the discoveries of RAS components in unlikely places, such as renin, an enzyme originally described in the kidney, found in the brain. New hypotheses about local functions of the RAS were raised based on the tissue-based synthesis of Ang II with independent actions. Lately, contemporary concepts of local RAS have become function-oriented[22].
The local angiotensin-generating system is important for regulating tissue/organ functions with clinical implications via autocrine, paracrine or intracrine actions[21]. The local hepatic RAS is not well-defined, although studies about RAS involvement in hepatic diseases have indicated a role for this system in the liver[21]. Ang II is a pro-inflammatory, pro-oxidant, and pro-thrombotic agent that interferes in several steps of intracellular insulin signaling. In sharp contrast, Ang-(1-7) enhances glucose tolerance, insulin sensitivity, insulin-stimulated glucose uptake as well as decreases triglyceride and cholesterol levels and reduces abdominal fat mass. Furthermore, Ang-(1-7) has been demonstrated to decrease liver gluconeogenesis and the Mas receptor is an essential component of the insulin receptor signaling pathway[23].
Liver diseases are major causes of morbidity and mortality worldwide. The most common liver diseases are hepatitis B and hepatitis C virus infections, alcoholic liver disease and nonalcoholic fatty liver disease (NAFLD). Without proper treatment, all types of chronic hepatitis will progress into end-stage liver diseases, including cirrhosis, liver failure and hepatocellular carcinoma, which ultimately lead to death[20,24]. The pathological characteristics of chronic liver diseases include enhanced fibrosis, oxidative stress and inflammatory markers. These processes are associated with sinusoidal capillarization and increased hepatic vascular resistance, eventually resulting in portal hypertension. Edema, ascites, hyperdynamic circulation and hepatorenal syndrome can occur because of compensatory mechanisms attempting to restore hepatic function[20]. The RAS is associated with all these processes. Furthermore, the local (i.e., hepatic) RAS, in addition to the systemic RAS, is thought to contribute to the pathophysiology of liver diseases.
For instance, Ang II caused a rapid and pronounced rise in portal pressure in an experimental model of cirrhosis[25]. In line with this result, the plasma concentration of Ang II was elevated in patients with cirrhosis, while losartan, an AT1 receptor antagonist, was capable of reducing the hepatic venous pressure gradient in patients with moderate to severe portal hypertension[26]. Altogether, these data point to the involvement of Ang II in cirrhosis-related portal hypertension. Ang II is thought to exert its vasoconstrictive effects on the postsinusoidal venules[21].
Ang II effects in the liver go far beyond vasoconstriction. For instance, by activating AT1 receptors, Ang II can induce hepatic stellate cell proliferation and up-regulate the expression of transforming growth factor (TGF)-b1 in vitro, indicating that Ang II plays an important role in the development of liver fibrosis[27]. Hepatic fibrosis is a dynamic process commonly resulting from different causes of chronic hepatic injury. Fibrosis is a complex process that involves several cell types and mediators, including cytokines, chemokines and growth factors, leading to a disruption of homeostatic mechanisms in the liver. Ultimately, hepatic tissue remodeling depends on the balance between collagen degradation and synthesis. Several mediators are involved in this process, including the RAS components. Notably, Ang II acts as pro-inflammatory and pro-fibrotic mediator, while Ang-(1-7) appears to exert opposite effects in liver tissue[28], comparable to heart and kidney effects[15]. Corroborating these findings, both the AT1 receptor antagonist Candesartan and the ACE inhibitor Perindopril significantly attenuated fibrosis and the associated pathological markers in an animal model of fibrosis[27]. The fibrogenic effect of Ang II can also be mediated by Kupffer cells, specialized phagocytic cells located in the liver that are known to be actively involved in the fibrotic process. AT1 receptors are expressed in Kupffer cells, as are renin and ACE[21]. The presence of RAS components in Kupffer cells and in the liver itself (where AT2 receptors and, obviously, angiotensinogen can also be found)[21] corroborates the hypothesis of the existence of a local RAS in the liver. Accordingly, experimental hepatic fibrosis was associated with RAS activation, characterized by increased levels of plasma renin activity, Ang I, Ang II and Ang-(1-7). Additionally, treatment with the Mas receptor antagonist, A-779, worsened hepatic fibrosis, thus suggesting a protective role for endogenous Ang-(1-7)[29].
NAFLD is the most common chronic liver disease worldwide and an important risk factor for non-alcoholic steatohepatitis, type 2 diabetes and cardiovascular diseases[30,31]. Ang II actions are associated with a series of deleterious effects that together contribute to the spectrum of histological changes observed in NAFLD and its progressive form, non-alcoholic steatohepatitis. The deleterious effects of Ang II include the stimulation of insulin resistance, de novo lipogenesis, mitochondrial dysfunction, reactive oxygen species generation, and pro-inflammatory cytokine production as well as the activation of hepatic stellate cells to trigger fibrogenesis[31,32]. Accordingly, experimental studies and clinical trials have shown that either the inhibition of the classical arm (composed by ACE-Ang II-AT1)[33-37] or the activation of the counter-regulatory arm (ACE2-Ang-(1-7)-Mas)[38-42] is beneficial in NAFLD and associated syndromes.
EXPERIMENTAL AND CLINICAL EVIDENCE ON THE ROLE OF RAS IN CIRRHOSIS
Cirrhosis is the end stage of progressive hepatic fibrosis, mainly characterized by liver architecture disruption due to fibrous scars and development of regenerating tissue. Fibrosis leads to significant changes in hepatic perfusion, enhanced portal blood flow resistance as well as liver dysfunction[43,44]. The leading causes of liver fibrosis are chronic viral hepatitis B and C, alcohol use and steatohepatitis related to obesity. These disorders have accounted for a significant increase in the incidence of cirrhosis and the deaths of at least 800000 people worldwide annually[45]. Although the pathophysiology of hepatic fibrosis remains not fully clear, current views have postulated that cirrhosis might be potentially reversible, particularly in a compensated stage, thus making the search for drug targets a scientific goal of highest priority[46].
Emerging evidence has supported the existence of RAS not only in the circulation but also in several organs such as heart, kidney and liver[47]. Locally, the classical RAS axis components, especially through AT1 receptor signaling, have been implicated in the modulation of cell growth and proliferation, generation of reactive oxygen species, apoptosis, hormone secretion, inflammatory and pro-fibrogenic processes in response to physiological and pathophysiological stimuli[48]. Accordingly, an up-regulation of RAS components including angiotensinogen, renin, ACE, Ang II and AT1 receptors has been reported in experimental and clinical liver injury studies, pointing out a role for this system in hepatic fibrosis and cirrhosis[28,47,49-51]. For instance, elevated serum levels of ACE were reported as a marker of fibrosis in patients diagnosed with chronic hepatitis B[52], whereas increased expression of ACE and AT1 receptors was found in human cirrhotic liver autopsies and biopsies[50]. In line with these studies, the beneficial effects of ACE inhibitors, such as Captopril, were reported on the analysis of liver biopsies of subjects with hepatitis C virus-related fibrosis[53]. In vitro approaches involving human culture-activated hepatic stellate cells, the main cells involved in the pro-fibrogenic process in the liver, showed that they increase their proliferation and acquire contractile properties in response to Ang II through AT1 receptors[54]. Culture-activated hepatic stellate cells from human cirrhotic livers also expressed high levels of active renin and ACE and secreted Ang II, indicating that locally generated components of the classical RAS axis contribute to tissue fibrosis in human liver[47]. There is evidence that Ang II effects on hepatic stellate cells are mediated, at least in part, by the activation of NADPH oxidase. Ang II, by means of phosphorylation of p47phox, a regulatory subunit of NADPH oxidase, induces reactive oxygen species formation and stimulates DNA synthesis, cell migration, pro-collagen alpha1 mRNA expression, and secretion of TGF-β1 and inflammatory cytokines, all of which contribute to hepatic fibrosis. Mice lacking p47phox exhibited attenuated liver injury and fibrosis associated with decreased hepatic concentration of TGF-β1 and inflammatory cytokines (TNF-α, IL-1β, IL-8 and MCP-1) following two weeks of bile duct ligation[55]. Moreover, blockade of Ang II activity by Lisinopril (an ACE inhibitor), Losartan (AT1 receptor antagonist) or N-acetylcysteine and diphenylene iodonium (NADPH oxidase inhibitors) prevented RAS pro-fibrogenic effects[47,54,55].
A recent meta-analysis revised the effectiveness of RAS inhibitors in randomized controlled trials in patients with liver fibrosis and reported significant reduction in fibrosis score and area[56]. A significant decrease in serum fibrosis markers, including TGF-b1, collagen I, IV, and matrix metalloproteinase-2 (MMP2), after treatment with RAS inhibitors was also reported in clinical studies with hepatic fibrosis or cirrhotic patients[57].
Over the past decades, several studies investigated the role of the classical RAS arm as well as the mechanisms underlying its deleterious effects on liver function by employing different models of liver fibrosis, including bile duct ligation, carbon tetrachloride (CCl4) treatment or continuous Ang II infusion[27,49,58-61]. For instance, both ACE and AT1 receptor genes were up-regulated in rats submitted to bile duct ligation. Additionally, Ang II induced an increase in mRNA expression of TGF-β1 in culture-activated hepatic stellate cells from rats. This finding supports the concept that RAS, especially the interaction of Ang II with AT1 receptors, plays a pivotal role in liver fibrosis development, mainly through the activation of hepatic stellate cells[27,49]. Accordingly, infusion of Ang II (25 ng/kg per hour) in bile duct-ligated rats via a subcutaneous pump significantly increased hepatic fibrosis by enhancing inflammation, TGF-β1 concentration, collagen deposition, lipid peroxidation product and phosphorylation of c-Jun and p42/44 mitogen-activated protein kinase (JAK2 and MAPK)[59]. By employing in vivo and in vitro approaches, Granzow et al[61] showed that Ang II, acting at AT1 receptors, promotes pro-fibrotic processes by phosphorylation of JAK2 and subsequent RhoA/Rho-kinase activation in rodents and human liver. Pharmacological inhibition of JAK2 prevented liver fibrosis in rats, indicating that inhibition of this pathway might be a promising therapy for this condition. Moreover, AT1 receptor-deficient mice presented less liver fibrosis than wild type mice following bile duct ligation or CCl4 administration. The protective effect of the absence of the AT1 receptor was associated with a significant decrease in inflammatory mediators, TGF-β1, lipid peroxidation products and phosphorylation of JAK2 and p42/44 MAPK, suggesting that the blockade of AT1 receptor signaling might also be a promising approach to treat liver fibrosis[58,60,62]. Indeed, blocking Ang II activity either by ACE inhibition or AT1 receptor antagonism prevented liver injury and fibrosis in different experimental models[16,28,48]. A more recent study revealed that the treatment with Captopril, an ACE inhibitor, accelerates liver regeneration in mice following partial hepatectomy[63]. The beneficial effects of Captopril were potentially due to reduction of the inflammatory response, paving the way for the hypothesis that blocking the classical RAS arm might not only prevent but also reverse severe liver damage[63].
More recently, it has been proposed that liver fibrosis and hepatic cirrhosis depend on the balance between the classical (ACE-Ang II-AT1 receptor) and the counter-regulatory (ACE2-Ang-(1-7)-Mas receptor) RAS axes[16,28,64-67]. In cirrhotic patients and rats with liver fibrosis, the balance between both RAS axes also accounts for hemodynamic changes, including splanchnic vasodilatation and portal hypertension[62,66].
An elegant study showed widespread parenchymal expression of ACE2 in the liver of rats submitted to bile duct ligation as well as in cirrhotic patients, providing the first evidence of a potential role of the counter-regulatory RAS axis in chronic liver disease[68]. Similar findings were found with the progression of liver fibrosis induced by CCl4 administration in rats[65]. Importantly, as liver fibrosis progresses, liver tissue expression of ACE2 and plasma levels of Ang-(1-7) increase[16,29,64,66,67]. Both RAS axes, the counter-regulatory and the classical, seem to be simultaneously activated in patients with liver cirrhosis and experimental models of chronic liver disease[16,29,64,66,67]. This finding supports the concept that the activation of the counter-regulatory RAS axis is a compensatory mechanism to counteract the deleterious effects of Ang II-AT1 receptor-mediated actions[16,29,64,66,67]. Accordingly, in vivo and in vitro inhibition of ACE under conditions of liver injury up-regulated the mRNA expression of ACE2 and Mas receptor, contributing to liver protection[65]. It is worth mentioning that ACE2 activity seems to be important as an endogenous negative regulator of RAS in chronic, but not acute liver injury, primarily by promoting the conversion of Ang II into Ang (1-7). This statement is supported by the fact that ACE2 knockout mice only presented increased hepatic fibrosis 21 d after bile duct ligation or following chronic administration of CCl4. On the other hand, no differences were found between ACE2 knockout mice and wild type littermates when animals were subjected to acute liver injury. Moreover, genetic ablation of ACE2 in one-year-old mice resulted in spontaneous inflammatory cell infiltration and mild liver fibrosis[69].
In this scenario, components of the counter-regulatory RAS axis may be regarded as promising targets for the development of novel anti-fibrotic therapies for chronic liver diseases. Our group has previously demonstrated that pharmacological blocking of Mas receptor with its antagonist A-779 aggravated bile duct ligation-induced liver fibrosis, which was associated with elevation in hepatic hydroxyproline and TGF-β1 concentrations[29]. Conversely, infusion of Ang-(1-7) markedly attenuated hepatic fibrosis in bile duct ligated rats, decreased hydroxyproline content and down-regulated key genes involved in liver fibrosis and angiogenesis such as collagen 1A1, α-SMA (smooth muscle actin), VEGF (vascular endothelial growth factor) and CTGF (connective tissue growth factor)[64]. In line with these findings, culture hepatic stellate cells treated with Ang-(1-7) or the Mas receptor agonist AVE 0991 expressed less α-SMA and hydroxyproline, while treatment with the Mas receptor antagonist A779 induced opposite effects[70]. Cultured hepatic stellate cells express Mas receptor and binding of Ang-(1-7) with Mas inhibits Ang II-induced phosphorylation of extracellular signal-regulated kinase (ERK)1/2, a classical pathway of tissue fibrosis[69].
Conversion of the pro-fibrotic peptide Ang II in the anti-fibrotic peptide Ang-(1-7) depends on ACE2 catalytic action, making this enzyme a very interesting therapeutic target for liver fibrosis[65,69]. A recent study investigated the long-term therapeutic effect of recombinant ACE2 by employing a liver-specific adeno-associated viral genome 2 serotype 8 vector (rAAV2/8-ACE2) with a liver-specific promoter in chronic liver disease models, including bile duct ligation and CCl4 administration[71]. The rAAV2/8-ACE2 therapy promoted a rapid up-regulation of hepatic ACE2 and an attenuation of liver fibrosis. These findings were associated with reduction in hepatic Ang II levels concomitant with increased concentrations of Ang-(1-7) in liver tissue. Also revealed were reductions in NADPH oxidase activity, oxidative stress, ERK1/2 and p38 phosphorylation without unwanted systemic effects[71].
RAS IN COMPLICATIONS OF LIVER DISEASES
Hemodynamic changes
Hemodynamic changes including portal hypertension and hyperdynamic circulation are the main cause of morbidity and mortality in patients with cirrhosis. Hemodynamic disorders can have widespread impact on the body according to the severity of the cirrhosis[72]. Portal hypertension and hyperdynamic circulation are characterized by elevated cardiac output and low systemic vascular resistance. Arterial vasodilation in the splanchnic circulation and the resulting decrease in systemic vascular resistance are associated with portal hypertension in cirrhosis. Compensatory mechanisms following the reduction of systemic vascular resistance lead to hyperdynamic circulation. The effective arterial blood volume and the circulating levels of RAS components and antidiuretic hormone remain normal at early stages of the disease, even with reduced systemic vascular resistance. Nevertheless, hyperdynamic circulation is insufficient to correct the effective arterial hypovolemia when the disease progresses and arterial vasodilation increases, resulting in arterial hypotension and consequent activation of the circulating RAS and the sympathetic nervous system and secretion of antidiuretic hormone. Maintenance of arterial pressure is a result of vasoconstrictive effects of Ang II in extra-splanchnic vascular areas, as the splanchnic circulation is resistant to Ang II, noradrenaline and vasopressin effects[66].
Evaluating patients at different stages of cirrhosis, one study showed that the circulating RAS is not activated at early stages of the disease. In contrast, patients at the advanced stages of cirrhosis presented an activation of peripheral and splanchnic RAS, and a metabolic deviation toward the RAS vasodilator axis in the splanchnic circulation. Furthermore, these authors observed a positive correlation between the Ang-(1-7)/Ang II ratio and cardiac output as well as a negative correlation between Ang-(1-7)/Ang II ratio and systemic vascular resistance, concluding that the final effect of the RAS may reflect a balance between the two opposing axes[66]. In this regard, the positive effects observed with blockade of the classical RAS arm are due, at least in part, to activation of the RAS counter-regulatory axis. Indeed, the hemodynamic effects of Ang-(1-7) has already been demonstrated in rats, in which Ang-(1-7) produced a significant increase in cardiac index (30%) and a decrease in total peripheral resistance[73]. However, chronic treatment with propranolol (a β-adrenergic receptor antagonist) in cirrhotic patients resulted in marked changes in the precursors of the RAS cascade, i.e., Renin and Ang I, with inhibition of both RAS arms at splanchnic and peripheral circulation. The chronic use of propranolol produced hemodynamic changes that were probably able to control the hyperdynamic circulation of cirrhotic patients. These effects were associated with overall RAS inhibition instead of changes in the balance between the two RAS arms[74] (Figure 1).
Figure 1.
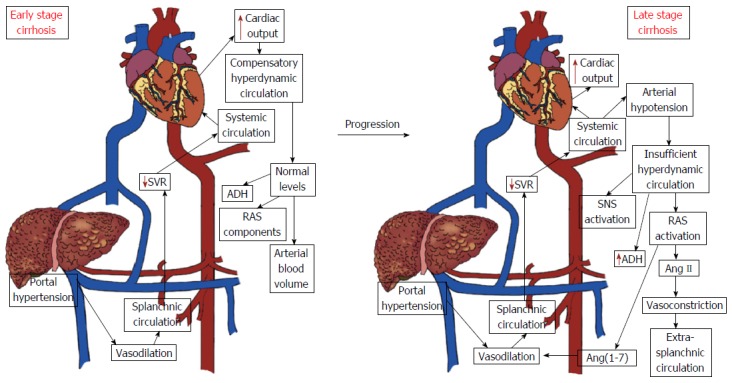
Hemodynamic changes in early and advanced stages of liver cirrhosis. The early phase of cirrhosis is characterized by elevated cardiac output and low systemic vascular resistance without changes in the circulating levels of renin angiotensin system (RAS) components and antidiuretic hormone (ADH). However, as the disease progresses, activation of the circulating RAS and of the sympathetic nervous system and secretion of the antidiuretic hormone occur in response to persistent arterial hypotension. Legend: Ang II: Angiotensin II; Ang-(1-7): Angiotensin (1-7); SNS: Sympathetic nervous system; SVR: Systemic vascular resistance.
Hepatorenal syndrome
Hepatorenal syndrome (HRS) has been defined as progressive renal failure that occurs in patients with chronic liver disease and advanced hepatic failure in the absence of any apparent clinical cause for renal insufficiency[75-77]. HRS represents the final stage of a process that gradually reduces renal blood flow and the glomerular filtration rate (GFR) due to marked renal vasoconstriction[75-77]. Despite the severity of renal failure, no significant histological abnormalities are found in the kidneys.
The pathophysiology of HRS is still poorly understood. The progressive reduction in systemic vascular resistance leads to effective arterial hypovolemia. To maintain arterial pressure within normal limits in this setting, there is activation of systemic vasoconstrictor systems, including the RAS, sympathetic nervous system and in late stages, nonosmotic hypersecretion of vasopressin. Although these systems help to maintain blood pressure, they have a negative influence on kidney function, leading to the retention of sodium and free water, and at late stages of the disease, producing an intense kidney vasoconstriction, which in turn decreases the glomerular filtration rate resulting in HRS. Indeed, hypoperfusion of the kidney due to the exaggerated action of renal vasoconstrictors has been considered the hallmark of HRS[76,78] (Figure 2).
Figure 2.
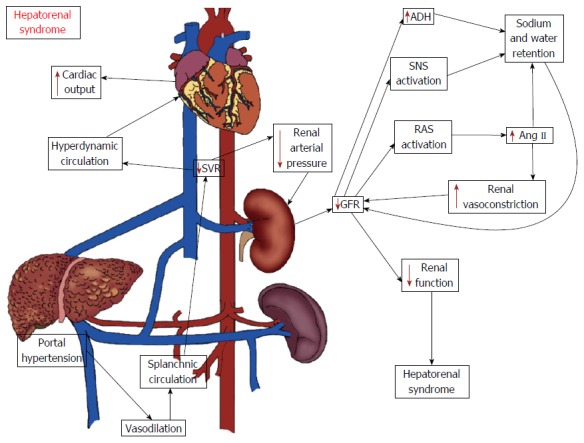
Potential mechanisms of hepatorenal syndrome. The hemodynamic changes associated with advanced stages of cirrhosis may lead to reductions in renal blood flow and in the glomerular filtration rate (GFR) as a compensatory mechanism to the low systemic vascular resistance and arterial hypotension. The kidney hypoperfusion is the hallmark of hepatorenal syndrome (HRS) and occurs as a consequence of the activation of systemic vasoconstrictor factors, including the classical axis of the renin angiotensin system (RAS), sympathetic nervous system and antidiuretic hormone (ADH). The increase in kidney vasoconstriction negatively influences its function, ultimately leading to HRS. Legend: Ang II: Angiotensin II; SNS: Sympathetic nervous system; SVR: Systemic vascular resistance.
The activation of the classical RAS arm, ACE-Ang II-AT1 receptor, is one of the main factors responsible for renal vasoconstriction in HRS[16,20]. Plasma renin activity and Ang II levels are increased in HRS[79]. Ang II infusion stimulates renal vasoconstriction and there is an inverse correlation between renal hypoperfusion and activation of the classical RAS in cirrhotic patients[80,81]. It has been considered that at the early stages of hepatic injury, the renal effects of Ang II can represent a compensatory mechanism against the decrease in renal blood flow[76,78,82]. However, the continuous, uncontrolled and exacerbated action of Ang II may progressively compromise renal function. Pereira and colleagues have previously shown that bile duct ligated rats exhibited increased circulating levels of Ang II and Ang-(1-7) even at early stages of hepatic damage[29]. Bile duct ligation is an experimental that leads to hepatic fibrosis and HRS[83]. However, it is difficult to know whether the changes in the components of the RAS preceded or were caused by the decline in renal function. The liver, or perhaps the kidney, can produce Ang peptides, which, in turn, act either as systemic hormones or as locally generated factors. Accordingly, Paizis and colleagues[68] detected an up-regulation of ACE2, the main enzyme responsible for Ang-(1-7) synthesis[84], in liver tissue from cirrhotic patients and bile duct ligated rats. Herath et al[64] showed increased expression of the Mas receptor in experimental biliary fibrosis, suggesting a role for ACE2-Ang-(1-7)-Mas arm in liver injury.
Although the renal effects of Ang II are well documented in patients with chronic liver disease, it is not yet clear whether Ang-(1-7) influences renal hemodynamics and renal tubular handling of sodium and free water. It has been previously shown that Ang-(1-7) exerts complex renal actions (see reference[85], for review). In vivo and in vitro administration of Ang-(1-7) can increase water reabsorption by acting at the distal nephron and by interacting with vasopressin V2 receptor[86-88]. In contrast, other studies reported that Ang-(1-7) has natriuretic and diuretic effects by inhibiting sodium reabsorption at proximal tubule[89-91]. Additionally, Ang-(1-7) has a vasodilator effect on pre-constricted rabbit afferent arterioles in vitro via Mas receptor and the release of nitric oxide, and attenuates pressor response to Ang II in rat renal vasculature[73,92,93]. However, the role of Ang-(1-7) in modifying renal vascular responses in HRS has not been investigated yet.
The treatment of HRS remains a significant challenge. HRS progresses rapidly. Therefore, liver transplantation evaluation should begin promptly to achieve ideal clinical conditions for successful transplantation[77]. Pharmacological and surgical interventions have not shown survival benefits but serve as temporary modalities to be used as a bridge to liver transplantation[77]. Therapy with systemic vasoconstrictors has been established as first line for HRS. The combination of terlipressin and albumin remains the standard of care for treating HRS, based on the available evidence[94]. Therefore, novel and alternative therapeutic approaches are needed to improve survival rate of HRS and to maintain patients in satisfactory clinical conditions for liver transplantation[77,94]. In this regard, evaluation of the role of molecules that modulate both RAS axes may be investigated in HRS[20].
CONCLUSION
RAS plays multiple roles in the pathophysiology of liver diseases. The classical RAS axis with its major mediator Ang II exerts pro-oxidant, fibrogenic, and pro-inflammatory actions in the liver. Conversely, the counter-regulatory RAS axis with its main effector Ang-(1-7) produces opposite actions in liver tissue, including anti-inflammatory, anti-oxidative and anti-fibrotic effects. Therefore, the balance between both RAS axes most likely affects the clinical and histopathological expression of liver diseases.
Pharmacological agents that inhibit Ang II formation (e.g., ACE inhibitors) or its binding to AT1 receptors (e.g., ARAs) have exhibited beneficial effects in chronic liver diseases. However, further studies are needed to incorporate them into clinical practice. Another relevant aspect to be better investigated is the elevation of circulating levels of Ang-(1-7) during chronic RAS inhibition. In particular, an altered balance between Ang II and Ang-(1-7) might be involved in the mechanisms of action of ACE inhibitors and AT1 receptor antagonists[95-97]. Therefore, these agents may not only blunt the effects of the classical RAS axis (“the foe”) but may also activate the counter-regulatory RAS axis (“the friend”). Most studies showing the therapeutic potential of ACE2-Ang-(1-7)-Mas axis are still pre-clinical. To date, Ang-(1-7) has only been administered in phase I/II studies as a putative anti-proliferative and anti-angiogenic agent to patients with advanced cancer refractory to standard treatment and as a hematopoietic agent to patients with multilineage cytopenias following chemotherapy[98,99]. These studies were very limited in scope but no dose-limiting toxicities have been reported. Therefore, further research on the contribution of the ACE2-Ang-(1-7)-Mas axis to the pathophysiology of liver diseases might lead to the development of pharmacological approaches. These new approaches may, in turn, result in the design of molecular or genetic methods to increase the expression of ACE2 and increased tissue levels of Ang-(1-7) and/or activation of the Mas receptor.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Brazil
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: Simões e Silva AC, Miranda AS, Rocha NP and Teixeira AL declare no conflict of interest related to this publication.
Peer-review started: February 8, 2017
First decision: March 3, 2017
Article in press: April 12, 2017
P- Reviewer: Kanda T, Lleo A S- Editor: Qi Y L- Editor: A E- Editor: Zhang FF
References
- 1.Hall JE, Guyton AC, Mizelle HL. Role of the renin-angiotensin system in control of sodium excretion and arterial pressure. Acta Physiol Scand Suppl. 1990;591:48–62. [PubMed] [Google Scholar]
- 2.Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiol Rev. 1990;70:1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- 3.Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88. doi: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- 4.Simões E Silva AC, Flynn JT. The renin-angiotensin-aldosterone system in 2011: role in hypertension and chronic kidney disease. Pediatr Nephrol. 2012;27:1835–1845. doi: 10.1007/s00467-011-2002-y. [DOI] [PubMed] [Google Scholar]
- 5.Kamo T, Akazawa H, Komuro I. Pleiotropic Effects of Angiotensin II Receptor Signaling in Cardiovascular Homeostasis and Aging. Int Heart J. 2015;56:249–254. doi: 10.1536/ihj.14-429. [DOI] [PubMed] [Google Scholar]
- 6.Santos RA, Ferreira AJ, Simões E Silva AC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol. 2008;93:519–527. doi: 10.1113/expphysiol.2008.042002. [DOI] [PubMed] [Google Scholar]
- 7.Santos RA, Brosnihan KB, Chappell MC, Pesquero J, Chernicky CL, Greene LJ, Ferrario CM. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension. 1988;11:I153–I157. doi: 10.1161/01.hyp.11.2_pt_2.i153. [DOI] [PubMed] [Google Scholar]
- 8.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 9.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 10.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, Gembardt F, Kellett E, Martini L, Vanderheyden P, et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111:1806–1813. doi: 10.1161/01.CIR.0000160867.23556.7D. [DOI] [PubMed] [Google Scholar]
- 12.Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertens Res. 2009;32:533–536. doi: 10.1038/hr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prestes TR, Rocha NP, Miranda AS, Teixeira AL, Simoes-E-Silva AC. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr Drug Targets. 2016 doi: 10.2174/1389450117666160727142401. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 14.Varagic J, Ahmad S, Nagata S, Ferrario CM. ACE2: angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr Hypertens Rep. 2014;16:420. doi: 10.1007/s11906-014-0420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simões E Silva AC, Teixeira MM. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res. 2016;107:154–162. doi: 10.1016/j.phrs.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 16.Grace JA, Herath CB, Mak KY, Burrell LM, Angus PW. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci (Lond) 2012;123:225–239. doi: 10.1042/CS20120030. [DOI] [PubMed] [Google Scholar]
- 17.Silveira KD, Barroso LC, Vieira AT, Cisalpino D, Lima CX, Bader M, Arantes RM, Dos Santos RA, Simões-E-Silva AC, Teixeira MM. Beneficial effects of the activation of the angiotensin-(1-7) MAS receptor in a murine model of adriamycin-induced nephropathy. PLoS One. 2013;8:e66082. doi: 10.1371/journal.pone.0066082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 19.Casas JP, Chua W, Loukogeorgakis S, Vallance P, Smeeth L, Hingorani AD, MacAllister RJ. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet. 2005;366:2026–2033. doi: 10.1016/S0140-6736(05)67814-2. [DOI] [PubMed] [Google Scholar]
- 20.Ahmadian E, Pennefather PS, Eftekhari A, Heidari R, Eghbal MA. Role of renin-angiotensin system in liver diseases: an outline on the potential therapeutic points of intervention. Expert Rev Gastroenterol Hepatol. 2016;10:1279–1288. doi: 10.1080/17474124.2016.1207523. [DOI] [PubMed] [Google Scholar]
- 21.Leung PS. The peptide hormone angiotensin II: its new functions in tissues and organs. Curr Protein Pept Sci. 2004;5:267–273. doi: 10.2174/1389203043379693. [DOI] [PubMed] [Google Scholar]
- 22.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 23.Moreira de Macêdo S, Guimarães TA, Feltenberger JD, Sousa Santos SH. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides. 2014;62:189–196. doi: 10.1016/j.peptides.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Wang FS, Fan JG, Zhang Z, Gao B, Wang HY. The global burden of liver disease: the major impact of China. Hepatology. 2014;60:2099–2108. doi: 10.1002/hep.27406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rockey DC, Weisiger RA. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: implications for regulation of portal pressure and resistance. Hepatology. 1996;24:233–240. doi: 10.1002/hep.510240137. [DOI] [PubMed] [Google Scholar]
- 26.Schneider AW, Kalk JF, Klein CP. Effect of losartan, an angiotensin II receptor antagonist, on portal pressure in cirrhosis. Hepatology. 1999;29:334–339. doi: 10.1002/hep.510290203. [DOI] [PubMed] [Google Scholar]
- 27.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34:745–750. doi: 10.1053/jhep.2001.28231. [DOI] [PubMed] [Google Scholar]
- 28.Pereira RM, dos Santos RA, da Costa Dias FL, Teixeira MM, Simões e Silva AC. Renin-angiotensin system in the pathogenesis of liver fibrosis. World J Gastroenterol. 2009;15:2579–2586. doi: 10.3748/wjg.15.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira RM, Dos Santos RA, Teixeira MM, Leite VH, Costa LP, da Costa Dias FL, Barcelos LS, Collares GB, Simões e Silva AC. The renin-angiotensin system in a rat model of hepatic fibrosis: evidence for a protective role of Angiotensin-(1-7) J Hepatol. 2007;46:674–681. doi: 10.1016/j.jhep.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–649. doi: 10.3109/07853890.2010.518623. [DOI] [PubMed] [Google Scholar]
- 31.Musso G, Cassader M, Cohney S, Pinach S, Saba F, Gambino R. Emerging Liver-Kidney Interactions in Nonalcoholic Fatty Liver Disease. Trends Mol Med. 2015;21:645–662. doi: 10.1016/j.molmed.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, Ma KL, Zhang Y, Wen Y, Wang GH, Hu ZB, Liu L, Lu J, Chen PP, Ruan XZ, et al. Lipid disorder and intrahepatic renin-angiotensin system activation synergistically contribute to non-alcoholic fatty liver disease. Liver Int. 2016;36:1525–1534. doi: 10.1111/liv.13131. [DOI] [PubMed] [Google Scholar]
- 33.Hirata T, Tomita K, Kawai T, Yokoyama H, Shimada A, Kikuchi M, Hirose H, Ebinuma H, Irie J, Ojiro K, et al. Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY) Int J Endocrinol. 2013;2013:587140. doi: 10.1155/2013/587140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, Hasegawa T, Tokusashi Y, Miyokawa N, Nakamura K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–1225. doi: 10.1002/hep.20420. [DOI] [PubMed] [Google Scholar]
- 35.Goh GB, Pagadala MR, Dasarathy J, Unalp-Arida A, Sargent R, Hawkins C, Sourianarayanane A, Khiyami A, Yerian L, Pai R, et al. Renin-angiotensin system and fibrosis in non-alcoholic fatty liver disease. Liver Int. 2015;35:979–985. doi: 10.1111/liv.12611. [DOI] [PubMed] [Google Scholar]
- 36.Orlic L, Mikolasevic I, Lukenda V, Anic K, Jelic I, Racki S. Nonalcoholic fatty liver disease and the renin-angiotensin system blockers in the patients with chronic kidney disease. Wien Klin Wochenschr. 2015;127:355–362. doi: 10.1007/s00508-014-0661-y. [DOI] [PubMed] [Google Scholar]
- 37.Pelusi S, Petta S, Rosso C, Borroni V, Fracanzani AL, Dongiovanni P, Craxi A, Bugianesi E, Fargion S, Valenti L. Renin-Angiotensin System Inhibitors, Type 2 Diabetes and Fibrosis Progression: An Observational Study in Patients with Nonalcoholic Fatty Liver Disease. PLoS One. 2016;11:e0163069. doi: 10.1371/journal.pone.0163069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao X, Yang FY, Xin Z, Xie RR, Yang JK. The ACE2/Ang-(1-7)/Mas axis can inhibit hepatic insulin resistance. Mol Cell Endocrinol. 2014;393:30–38. doi: 10.1016/j.mce.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 39.Feltenberger JD, Andrade JM, Paraíso A, Barros LO, Filho AB, Sinisterra RD, Sousa FB, Guimarães AL, de Paula AM, Campagnole-Santos MJ, et al. Oral formulation of angiotensin-(1-7) improves lipid metabolism and prevents high-fat diet-induced hepatic steatosis and inflammation in mice. Hypertension. 2013;62:324–330. doi: 10.1161/HYPERTENSIONAHA.111.00919. [DOI] [PubMed] [Google Scholar]
- 40.Santos SH, Andrade JM, Fernandes LR, Sinisterra RD, Sousa FB, Feltenberger JD, Alvarez-Leite JI, Santos RA. Oral Angiotensin-(1-7) prevented obesity and hepatic inflammation by inhibition of resistin/TLR4/MAPK/NF-κB in rats fed with high-fat diet. Peptides. 2013;46:47–52. doi: 10.1016/j.peptides.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 41.de Macedo SM, Guimarares TA, Andrade JM, Guimaraes AL, Batista de Paula AM, Ferreira AJ, Sousa Santos SH. Angiotensin converting enzyme 2 activator (DIZE) modulates metabolic profiles in mice, decreasing lipogenesis. Protein Pept Lett. 2015;22:332–340. doi: 10.2174/0929866522666150209125401. [DOI] [PubMed] [Google Scholar]
- 42.Cao X, Yang F, Shi T, Yuan M, Xin Z, Xie R, Li S, Li H, Yang JK. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis activates Akt signaling to ameliorate hepatic steatosis. Sci Rep. 2016;6:21592. doi: 10.1038/srep21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedman SL, Maher JJ, Bissell DM. Mechanisms and therapy of hepatic fibrosis: report of the AASLD Single Topic Basic Research Conference. Hepatology. 2000;32:1403–1408. doi: 10.1053/jhep.2000.20243. [DOI] [PubMed] [Google Scholar]
- 44.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kochanek KD, Murphy SL, Xu J, Tejada-Vera B. Deaths: Final Data for 2014. Natl Vital Stat Rep. 2016;65:1–122. [PubMed] [Google Scholar]
- 46.Kim G, Baik SK. Overview and recent trends of systematic reviews and meta-analyses in hepatology. Clin Mol Hepatol. 2014;20:137–150. doi: 10.3350/cmh.2014.20.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bataller R, Sancho-Bru P, Ginès P, Lora JM, Al-Garawi A, Solé M, Colmenero J, Nicolás JM, Jiménez W, Weich N, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125:117–125. doi: 10.1016/s0016-5085(03)00695-4. [DOI] [PubMed] [Google Scholar]
- 48.Warner FJ, Lubel JS, McCaughan GW, Angus PW. Liver fibrosis: a balance of ACEs? Clin Sci (Lond) 2007;113:109–118. doi: 10.1042/CS20070026. [DOI] [PubMed] [Google Scholar]
- 49.Paizis G, Cooper ME, Schembri JM, Tikellis C, Burrell LM, Angus PW. Up-regulation of components of the renin-angiotensin system in the bile duct-ligated rat liver. Gastroenterology. 2002;123:1667–1676. doi: 10.1053/gast.2002.36561. [DOI] [PubMed] [Google Scholar]
- 50.Ikura Y, Ohsawa M, Shirai N, Sugama Y, Fukushima H, Suekane T, Hirayama M, Ehara S, Naruko T, Ueda M. Expression of angiotensin II type 1 receptor in human cirrhotic livers: Its relation to fibrosis and portal hypertension. Hepatol Res. 2005;32:107–116. doi: 10.1016/j.hepres.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 51.Lubel JS, Herath CB, Burrell LM, Angus PW. Liver disease and the renin-angiotensin system: recent discoveries and clinical implications. J Gastroenterol Hepatol. 2008;23:1327–1338. doi: 10.1111/j.1440-1746.2008.05461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purnak T, Beyazit Y, Oztas E, Yesil Y, Efe C, Torun S, Celik T, Tenlik I, Kurt M, Ozaslan E. Serum angiotensin-converting enzyme level as a marker of fibrosis in patients with chronic hepatitis B. J Renin Angiotensin Aldosterone Syst. 2012;13:244–249. doi: 10.1177/1470320311434241. [DOI] [PubMed] [Google Scholar]
- 53.Corey KE, Shah N, Misdraji J, Abu Dayyeh BK, Zheng H, Bhan AK, Chung RT. The effect of angiotensin-blocking agents on liver fibrosis in patients with hepatitis C. Liver Int. 2009;29:748–753. doi: 10.1111/j.1478-3231.2009.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bataller R, Ginès P, Nicolás JM, Görbig MN, Garcia-Ramallo E, Gasull X, Bosch J, Arroyo V, Rodés J. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118:1149–1156. doi: 10.1016/s0016-5085(00)70368-4. [DOI] [PubMed] [Google Scholar]
- 55.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Q, Li N, Li F, Zhou Z, Han Q, Lv Y, Sang J, Liu Z. Therapeutic effect of renin angiotensin system inhibitors on liver fibrosis. J Renin Angiotensin Aldosterone Syst. 2016;17:1470320316628717. doi: 10.1177/1470320316628717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim G, Kim J, Lim YL, Kim MY, Baik SK. Renin-angiotensin system inhibitors and fibrosis in chronic liver disease: a systematic review. Hepatol Int. 2016;10:819–828. doi: 10.1007/s12072-016-9705-x. [DOI] [PubMed] [Google Scholar]
- 58.Kanno K, Tazuma S, Chayama K. AT1A-deficient mice show less severe progression of liver fibrosis induced by CCl(4) Biochem Biophys Res Commun. 2003;308:177–183. doi: 10.1016/s0006-291x(03)01357-3. [DOI] [PubMed] [Google Scholar]
- 59.Bataller R, Gäbele E, Parsons CJ, Morris T, Yang L, Schoonhoven R, Brenner DA, Rippe RA. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology. 2005;41:1046–1055. doi: 10.1002/hep.20665. [DOI] [PubMed] [Google Scholar]
- 60.Yang L, Bataller R, Dulyx J, Coffman TM, Ginès P, Rippe RA, Brenner DA. Attenuated hepatic inflammation and fibrosis in angiotensin type 1a receptor deficient mice. J Hepatol. 2005;43:317–323. doi: 10.1016/j.jhep.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 61.Granzow M, Schierwagen R, Klein S, Kowallick B, Huss S, Linhart M, Mazar IG, Görtzen J, Vogt A, Schildberg FA, et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology. 2014;60:334–348. doi: 10.1002/hep.27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grace JA, Klein S, Herath CB, Granzow M, Schierwagen R, Masing N, Walther T, Sauerbruch T, Burrell LM, Angus PW, et al. Activation of the MAS receptor by angiotensin-(1-7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology. 2013;145:874–884.e5. doi: 10.1053/j.gastro.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 63.Koh SL, Ager E, Malcontenti-Wilson C, Muralidharan V, Christophi C. Blockade of the renin-angiotensin system improves the early stages of liver regeneration and liver function. J Surg Res. 2013;179:66–71. doi: 10.1016/j.jss.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 64.Herath CB, Warner FJ, Lubel JS, Dean RG, Jia Z, Lew RA, Smith AI, Burrell LM, Angus PW. Upregulation of hepatic angiotensin-converting enzyme 2 (ACE2) and angiotensin-(1-7) levels in experimental biliary fibrosis. J Hepatol. 2007;47:387–395. doi: 10.1016/j.jhep.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Q, Xie Q, Shi CC, Xiang XG, Lin LY, Gong BD, Zhao GD, Wang H, Jia NN. Expression of angiotensin-converting enzyme 2 in CCL4-induced rat liver fibrosis. Int J Mol Med. 2009;23:717–723. doi: 10.3892/ijmm_00000185. [DOI] [PubMed] [Google Scholar]
- 66.Vilas-Boas WW, Ribeiro-Oliveira A, Pereira RM, Ribeiro Rda C, Almeida J, Nadu AP, Simões e Silva AC, dos Santos RA. Relationship between angiotensin-(1-7) and angiotensin II correlates with hemodynamic changes in human liver cirrhosis. World J Gastroenterol. 2009;15:2512–2519. doi: 10.3748/wjg.15.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang W, Miao J, Li P, Wang Y, Zhang Y. Up-regulation of components of the renin-angiotensin system in liver fibrosis in the rat induced by CCL4. Res Vet Sci. 2013;95:54–58. doi: 10.1016/j.rvsc.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paizis G, Tikellis C, Cooper ME, Schembri JM, Lew RA, Smith AI, Shaw T, Warner FJ, Zuilli A, Burrell LM, et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Osterreicher CH, Taura K, De Minicis S, Seki E, Penz-Osterreicher M, Kodama Y, Kluwe J, Schuster M, Oudit GY, Penninger JM, et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology. 2009;50:929–938. doi: 10.1002/hep.23104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lubel JS, Herath CB, Tchongue J, Grace J, Jia Z, Spencer K, Casley D, Crowley P, Sievert W, Burrell LM, et al. Angiotensin-(1-7), an alternative metabolite of the renin-angiotensin system, is up-regulated in human liver disease and has antifibrotic activity in the bile-duct-ligated rat. Clin Sci (Lond) 2009;117:375–386. doi: 10.1042/CS20080647. [DOI] [PubMed] [Google Scholar]
- 71.Mak KY, Chin R, Cunningham SC, Habib MR, Torresi J, Sharland AF, Alexander IE, Angus PW, Herath CB. ACE2 Therapy Using Adeno-associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol Ther. 2015;23:1434–1443. doi: 10.1038/mt.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim MY, Baik SK, Lee SS. Hemodynamic alterations in cirrhosis and portal hypertension. Korean J Hepatol. 2010;16:347–352. doi: 10.3350/kjhep.2010.16.4.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sampaio WO, Nascimento AA, Santos RA. Systemic and regional hemodynamic effects of angiotensin-(1-7) in rats. Am J Physiol Heart Circ Physiol. 2003;284:H1985–H1994. doi: 10.1152/ajpheart.01145.2002. [DOI] [PubMed] [Google Scholar]
- 74.Vilas-Boas WW, Ribeiro-Oliveira A, Ribeiro Rda C, Vieira RL, Almeida J, Nadu AP, Simões e Silva AC, Santos RA. Effect of propranolol on the splanchnic and peripheral renin angiotensin system in cirrhotic patients. World J Gastroenterol. 2008;14:6824–6830. doi: 10.3748/wjg.14.6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cárdenas A, Gines P. Hepatorenal syndrome. Clin Liver Dis. 2006;10:371–385, ix-x. doi: 10.1016/j.cld.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 76.Angeli P, Merkel C. Pathogenesis and management of hepatorenal syndrome in patients with cirrhosis. J Hepatol. 2008;48 Suppl 1:S93–S103. doi: 10.1016/j.jhep.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 77.Shah N, Silva RG, Kowalski A, Desai C, Lerma E. Hepatorenal syndrome. Dis Mon. 2016;62:364–375. doi: 10.1016/j.disamonth.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 78.Arroyo V, Fernandez J, Ginès P. Pathogenesis and treatment of hepatorenal syndrome. Semin Liver Dis. 2008;28:81–95. doi: 10.1055/s-2008-1040323. [DOI] [PubMed] [Google Scholar]
- 79.Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Ginès P, Moreira V, Milicua JM, Jiménez W, Arroyo V. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42:439–447. doi: 10.1002/hep.20766. [DOI] [PubMed] [Google Scholar]
- 80.Laragh JH, Cannon PJ, Bentzel CJ, Sicinski AM, Meltzer JI. Angiotensin II, norepinephrine, and renal transport of electrolytes and water in normal man and in cirrhosis with ascites. J Clin Invest. 1963;42:1179–1192. doi: 10.1172/JCI104803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bernardi M, Trevisani F, Gasbarrini A, Gasbarrini G. Hepatorenal disorders: role of the renin-angiotensin-aldosterone system. Semin Liver Dis. 1994;14:23–34. doi: 10.1055/s-2007-1007295. [DOI] [PubMed] [Google Scholar]
- 82.Aliaga L, Zozoya JM, Omar M, Mediavilla JD, Prieto J. Interrelationships between systemic hemodynamics, urinary sodium excretion, and renin-angiotensin system in cirrhosis. Acta Gastroenterol Belg. 1995;58:213–221. [PubMed] [Google Scholar]
- 83.Pereira RM, dos Santos RA, Oliveira EA, Leite VH, Dias FL, Rezende AS, Costa LP, Barcelos LS, Teixeira MM, Simoes e Silva AC. Development of hepatorenal syndrome in bile duct ligated rats. World J Gastroenterol. 2008;14:4505–4511. doi: 10.3748/wjg.14.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383:45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simões e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013;169:477–492. doi: 10.1111/bph.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Santos RA, Simões e Silva AC, Magaldi AJ, Khosla MC, Cesar KR, Passaglio KT, Baracho NC. Evidence for a physiological role of angiotensin-(1-7) in the control of hydroelectrolyte balance. Hypertension. 1996;27:875–884. doi: 10.1161/01.hyp.27.4.875. [DOI] [PubMed] [Google Scholar]
- 87.Simóes-e-Silva AC, Baracho NC, Passaglio KT, Santos RA. Renal actions of angiotensin-(1-7) Braz J Med Biol Res. 1997;30:503–513. doi: 10.1590/s0100-879x1997000400012. [DOI] [PubMed] [Google Scholar]
- 88.Magaldi AJ, Cesar KR, de Araújo M, Simões e Silva AC, Santos RA. Angiotensin-(1-7) stimulates water transport in rat inner medullary collecting duct: evidence for involvement of vasopressin V2 receptors. Pflugers Arch. 2003;447:223–230. doi: 10.1007/s00424-003-1173-1. [DOI] [PubMed] [Google Scholar]
- 89.Andreatta-van Leyen S, Romero MF, Khosla MC, Douglas JG. Modulation of phospholipase A2 activity and sodium transport by angiotensin-(1-7) Kidney Int. 1993;44:932–936. doi: 10.1038/ki.1993.334. [DOI] [PubMed] [Google Scholar]
- 90.DelliPizzi AM, Hilchey SD, Bell-Quilley CP. Natriuretic action of angiotensin(1-7) Br J Pharmacol. 1994;111:1–3. doi: 10.1111/j.1476-5381.1994.tb14014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lara LS, Vives D, Correa JS, Cardozo FP, Marques-Fernades MF, Lopes AG, Caruso-Neves C. PKA-mediated effect of MAS receptor in counteracting angiotensin II-stimulated renal Na+-ATPase. Arch Biochem Biophys. 2010;496:117–122. doi: 10.1016/j.abb.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 92.Ren Y, Garvin JL, Carretero OA. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension. 2002;39:799–802. doi: 10.1161/hy0302.104673. [DOI] [PubMed] [Google Scholar]
- 93.Botelho-Santos GA, Sampaio WO, Reudelhuber TL, Bader M, Campagnole-Santos MJ, Souza dos Santos RA. Expression of an angiotensin-(1-7)-producing fusion protein in rats induced marked changes in regional vascular resistance. Am J Physiol Heart Circ Physiol. 2007;292:H2485–H2490. doi: 10.1152/ajpheart.01245.2006. [DOI] [PubMed] [Google Scholar]
- 94.Gifford FJ, Morling JR, Fallowfield JA. Systematic review with meta-analysis: vasoactive drugs for the treatment of hepatorenal syndrome type 1. Aliment Pharmacol Ther. 2017;45:593–603. doi: 10.1111/apt.13912. [DOI] [PubMed] [Google Scholar]
- 95.Simões e Silva AC, Diniz JS, Pereira RM, Pinheiro SV, Santos RA. Circulating renin Angiotensin system in childhood chronic renal failure: marked increase of Angiotensin-(1-7) in end-stage renal disease. Pediatr Res. 2006;60:734–739. doi: 10.1203/01.pdr.0000246100.14061.bc. [DOI] [PubMed] [Google Scholar]
- 96.Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens. 1996;14:799–805. doi: 10.1097/00004872-199606000-00017. [DOI] [PubMed] [Google Scholar]
- 97.Kocks MJ, Lely AT, Boomsma F, de Jong PE, Navis G. Sodium status and angiotensin-converting enzyme inhibition: effects on plasma angiotensin-(1-7) in healthy man. J Hypertens. 2005;23:597–602. doi: 10.1097/01.hjh.0000160217.86597.b6. [DOI] [PubMed] [Google Scholar]
- 98.Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and pharmacokinetic study of angiotensin-(1-7), an endogenous antiangiogenic hormone. Clin Cancer Res. 2009;15:7398–7404. doi: 10.1158/1078-0432.CCR-09-1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodgers KE, Oliver J, diZerega GS. Phase I/II dose escalation study of angiotensin 1-7 [A(1-7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother Pharmacol. 2006;57:559–568. doi: 10.1007/s00280-005-0078-4. [DOI] [PubMed] [Google Scholar]