

Abstract
Clinical Relevance
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) including aspirin are of intensive use nowadays. These drugs exert their activity via the metabolism of arachidonic acid (AA) by cyclooxygenase inhibition. Though beneficial for health in some instances, both unspecific and specific cyclooxygenase inhibitor activity interfere with AA metabolism producing also proinflammatory lipids that may promote cancer.
Materials and Methods
This review is based on available literature on clinical uses, biochemical investigations, molecular medicine, pharmacology, toxicity, and epidemiology-clinical studies on NSAIDs and other drugs that may be used accordingly, which was collected from electronic (SciFinder, Medline, Science Direct, and ACS among others) and library searches of books and journals.
Results
Relevant literature supports the notion that NDSAID use may also promote proinflammatory biochemical events that are also related to precancerous predisposition. Several agents are proposed that may be employed in immediate future to supplement and optimize treatment with NSAIDs. In this way serious side effects arising from promotion of inflammation and cancer, especially in chronic NSAID users and high risk groups of patients, could be avoided.
1. Introduction
1.1. Inflammation Route via Arachidonic Acid Metabolism
Inflammation is driven by complex metabolic pathways, with arachidonic acid (AA) as one important molecule of origin. AA metabolism is fundamental for both promotion and inhibition of inflammatory processes. Several enzymes are involved in this regulation of inflammation, cyclooxygenases 1 and 2 [1], lipoxygenases [1], cytochrome P 450 (CYP) epoxygenases and ω-hydroxylases [2], and also the nonenzymatic processes of AA metabolism like the free radical-catalyzed peroxidation [3]. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) have been designed to decrease above all the classical symptoms of pain and tumefaction, but in the meantime it is known that they cause proinflammatory effects, too. Aspirin targets the COX-1 pathway, whereas the classical NSAIDs target mainly the COX-2 pathway by inhibiting prostaglandin E2 (PGE2) formation [4]. The anti-inflammatory effect is due to the inhibition of vasodilatation and to the shortening of mast cell and other immune cells recruitment. Aspirin however acetylates also the COX-2 isoenzyme but due to slight sequence variations this evidently consumes less binding energy for arachidonic acid to become bound and be further metabolized [5].
1.2. Cyclooxygenase (COX) Activity
Early findings on aspirin inhibitory mode of action on prostaglandin (PG) synthesis led to the initial discovery of cyclooxygenase (COX) [6, 7]. This enzyme, now called COX-1, is central to AA catabolism to end up producing PGI2, also known as prostacyclin, with clear antithrombogenic [8, 9] and cytoprotective to gastric mucosa [10, 11] physiological functions. In 1991, a 64% sequence homologue to COX-1 enzyme was discovered [12, 13] that was inducible in a number of cells to certain proinflammatory stimuli [11] and inhibited in its expression by corticosteroids [7]. This is the enzyme now termed as COX-2. In this area, the variation of severity of side effects caused by different anti-inflammatory drugs including aspirin was puzzling. Particularly, stomach side effects by aspirin led to the development of safer drugs like meloxicam, nimesulide, and etodolac, which are now well accepted as selective COX-2 inhibitors [7]. Although both COX isoenzymes catalyze the oxygenation of arachidonate, COX-2 shows a more diverse substrate selectivity compared to COX-1. For example, COX-2 apart from arachidonic acid oxygenates in the same efficiency 2-arachidonylglycerol (endocannabinoid) [14]. The most evident difference between COX isoenzymes is in their expression in tissue distribution. Unlike COX-1, which is ubiquitous and constitutively expressed throughout the gastrointestinal system, the kidneys, the vascular smooth muscle, and the platelets, COX-2 is constitutively expressed in endothelial cells, brain, and kidneys and is variably induced in its expression by distinct inflammatory stimuli and neoplastic conditions [15, 16]. Unexplained antipyretic and analgesic effects of acetaminophen, phenacetin, and dipyrone, without evident COX-1 or COX-2 inhibition, were made clear by the discovery of yet another COX isoenzyme termed COX-3 that when expressed showed selective inhibition to these agents [17].
1.3. How NSAIDs May Cause Side Effects
NSAIDs, by inhibiting cyclooxygenase enzyme activity, even by different means, may all share to a greater or lesser extent a similar kind of side effects [7]. However, these side effects may be both (a) specific to the NSAID type and (b) cell type specific. Side effects depend on the specific inhibition of prostanoid synthesis due to the agent inducing the COX inhibition and the type of targeted tissue [15]. Prostanoid synthesis alteration contributes to disturbance of homeostasis [7] that may be cell specific, giving an end organ specific toxicity [18–22] (Figure 1).
Figure 1.
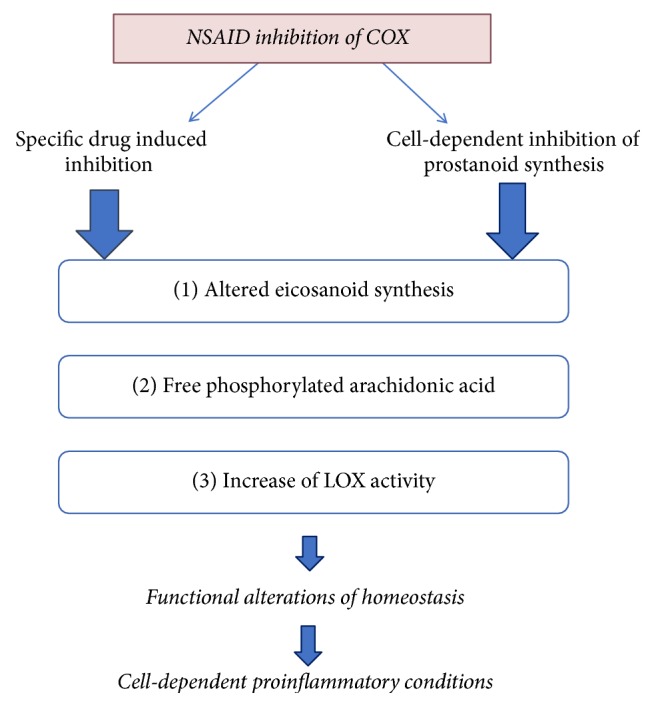
Levels of possible side effects of NSAIDs [18]. Drug- and cell-specific inhibition of COX isoenzymes [15] and respective prostanoids results in alteration of homeostasis [7] and in promotion of proinflammatory conditions [18–22].
1.3.1. The Mechanisms by Which Aspirin Induces Proinflammatory Effects
Acetylation of serine (Ser-530) of COX-1 even by low aspirin concentrations and in a few minutes results in the inhibition of prostaglandin E2 (PGE2) formation and the inhibition of platelet function (anticoagulant activity) [23]. This acetylating reaction irreversibly inactivates COX-1 activity [24]. As a consequence, related tissue and blood pressure homeostasis depending on PGE2 formation may be affected [7, 25]. An example is the kidney normal function that depends on PGE2 synthesis. Renin is secreted by PGE2 formation and angiotensin II stimulation is mainly mediated by PGE2 production by COX-1, but also by COX-2 [25]. Acetylation by aspirin is also occurring on the COX-2 isoform in almost the same manner due to the structural homology between COX-1 and COX-2 isoenzymes. Tyrosine residues (tyr-385) to a greater extent and (tyr-348) to a lesser extent are critical for this acetylation event of COX-2 by aspirin [26]. These tyrosine residues constitute a hydrogen binding network that is critical for the precise positioning and the relative reactivity rendering the closeness of Ser-530 with the acetyl group of aspirin feasible. Arginine 120 (Arg-120) just below Ser-530 in the active sites of COX, however, makes the difference of arachidonic acid binding ability between COX-1 and COX-2 isoenzymes (Figure 2). In the COX-1 case of binding of arachidonic acid, an ionic bond is formed between Arg-120 and the carboxylate of arachidonate. In COX-2 case however, instead of an ionic bond, a hydrogen bond is formed with Arg-120 and arachidonate thus conferring less to the binding energy needed for the molecule to become bound [27, 28]. During the acetylation event in COX-1, the arachidonic acid is irreversibly inhibited from binding when Arg-120 makes the Ser-530 acetylation efficient by forming a weak ionic bond with the carboxylate of aspirin. Conversely, the acetylation event in COX-2 does not irreversibly inhibit its activity but just lowers the arachidonic acid binding ability to the enzyme's active site (Figure 2).
Figure 2.

(a) Aspirin acetylation of COX-1 irreversibly inhibits arachidonic acid to become bound, whereas (b) acetylation of COX-2 leads to the formation of lipoxins [27–30, 37].
The acetylation event by aspirin, shown to produce also the aspirin-triggered lipotoxins by transcellular (cell-to-cell) interactions [29, 30], is also supported by clinical evidence. Direct clinical evidence shows that among the derived eicosanoids produced by aspirin acetylation are the lipoxin A4 (LXA4), which is vasodilatory, and the leukotrienes C4 and D4 (LTC4, LTD4), which are potent vasoconstrictors. These eicosanoids have been shown to be generated under aspirin treatment in the atherosclerotic lumen of blood vessels [31]. Also, in aspirin intolerance, excessive amounts of LTC4 have been isolated from nasal secretions and bronchial biopsies [32]. These leukotrienes produced are implicated to severe gastrointestinal [33] and severe cardiovascular side effects [34] as they constitute important mediators of inflammation, ischemia [35], and bronchoconstriction [36].
1.3.2. The Transcellular Biosynthesis of Eicosanoid Derivatives: Crossover Pathways
The transcellular biosynthesis of lipoxins requires interactions between LOX isoenzymes (LOX-LOX interactions) and can promote generation of leukotrienes (LTs) by endothelial cells [37]. When COX-2 is acetylated by aspirin, interaction with 5-lipoxygenase (5-LOX) occurs that triggers the transcellular biosynthesis of 15-R epimers of lipoxins [29, 38]. The derived eicosanoids by the acetylation of COX-2 in close association with 5-LOX are of the type of “S” conformation [39, 40]. Hereby, it has to be emphasized that in all cases arachidonic acid is first transformed to unstable precursor intermediate molecules ending up to many “S” conformations after aspirin treated COX-2 [30, 41]. Therefore, even 15-S-HETE formation cannot be excluded even by aspirin-induced specific 15-R-HETE formation as it may occur also in a nonenzyme dependent fashion [41]. During aspirin-triggered lipoxin synthesis, the precursor LTA4 (that is also of “S” conformation) may be formed by 5-LOX and serve as substrate for leukotriene synthesis of LTB4 that is formed prior to LTC4 and LTD4 [35] (Figure 3(a)). Apart from the absolute belief of 15-R-HETE being a sole product, as derived by the acetylation of COX-2, the presence of the double dioxygenated product, 5S-12S-DiHETE isolated in vivo, suggests further transcellular metabolic events that show further eicosanoid synthesis by 5- and 12-LOX interactions [31]. Research during that time may have identified generation of byproducts via enzymatic conversion of LTA4 [30, 42, 43]. Similar latter results indicate the possibility of 5S-15S-DiHETE to be formed in vivo by the acetylated COX-2 activity and 5-LOX-15-LOX interactions having 5S-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-S-HETE) as a substrate [44]. As LOX activity is not inhibited by aspirin, through the unstable intermediate formed by the acetylated COX-2, LOX isoenzymes continue the eicosanoid synthesis to produce active compounds [30, 40]. Further, all S-conformations produced during COX-2 acetylation and interaction with lipoxygenases can be relatively good substrates for 5-hydroxyeicosanoid dehydrogenase (5-HEDH) to produce 5-OXO-ETE [45]. Thus, another important proinflammatory mediator may be also formed (Figure 3(a)). Under oxidative conditions 5-HEDH transforms (5-S-HETE) to 5-OXO-ETE [46] as under normal conditions 5-HEDM is inactive because it requires NADP+ as a cofactor. Under stress conditions, however, NADPH oxidase activity has been described for its serious role in mediating inflammation [47].
Figure 3.

Metabolic events that follow acetylation of COX-2 and further transcellular activities: (a) eicosanoid production by crossover pathways of acetylated COX-2 and LOX isoenzymes [31, 35, 37–44]. (b) Eicosanoid production by free arachidonic acid [3, 44–46, 53, 54, 61].
5-S-HETE is on its own a potent proinflammatory mediator [48] to induce stress conditions together with potent proinflammatory leukotrienes LTB4 and the cysteinyl leukotrienes LTC4 and LTD4. The metabolic tendency of 5-LOX to produce the 5-OXO-ETE derivative coupled with LTB4 is important for inflammation and cancer [48, 49], which has not been linked adequately as a serious proinflammatory condition due to NSAID use. The 5-S-HETE metabolite, when accumulating, is a potent neutrophil activator [50]. The 5-OXO-ETE derivative, however, has been shown to be a 100 times more potent neutrophil activator than its precursor [51]. As synthetic 5-OXO-HETE derivatives prove to be even more potent than 5-OXO-ETE, the native derivatives also traced in vivo may be further implicated in the promotion of chronic inflammation and cancer [49, 52]. 5-OXO-HETE acts proinflammatorily via the OXO-ETE receptor that is known to promote eosinophil and other inflammatory cells migration [48]. 5-OXO-ETE is formed in neutrophil microsomes under the presence of NADP+- and Ca2+- dependent translocation of 5-LOX to the nuclear membrane to act on arachidonic acid bound by the nuclear membrane accessory protein 5-LOX activating protein (FLAP). The neutrophil microsomes under reduced conditions prefer, as well as the 5-S-HETE, to produce the 5-OXO-ETE derivative lipid using the 6-transanalogue of LTB4 as a substrate [46].
Free activated arachidonic acid not bound to COX-2 may be also used by 5-LOX to produce leukotrienes during inflammation in vivo [42]. Furthermore, it may be utilized by the P450 metabolism to produce 20-HETE [53] and by nonenzymatic conversion to form PGF2 isoprostanes [3, 54] (Figure 3(b)).
2. Materials and Methods
The literature study was conducted from scientific journals and books and electronic sources such as SciFinder, Science Direct, Medline, and Google Scholar, covering the period from January 1945 to the end of December 2016.
3. Results
3.1. Nonaspirin NSAIDs
3.1.1. Traditional NSAID Clinical Side Effects
Traditional NSAIDs during clinical practice vary on the degree of causing vascular side effects. Increased risk is noticed by high doses of diclofenac and ibuprofen due to the increased myocardial infarction events recorded, whereas increased doses of naproxen have substantially smaller risk [55], suggesting differential inhibition of activity of COX-2. Acute myocardial infarction risk is potentiated in patients with coronary artery disease by high and low doses of diclofenac and rofecoxib and other NSAIDs, but not with naproxen even when administered in high doses [34]. The risk for renal disease development is tightly correlated with acetaminophen overuse [56]. Acetaminophen provides a unique example of cell-specific COX inhibition that may negatively affect the prostanoid synthesis in tumor cells by altering the levels of PGE2 [57, 58]. Upon NSAID inhibition of COX activity by traditional COX-2 inhibitors like diclofenac, an alternate housekeeping COX-1-like activity, of a third COX isoenzyme may be also inhibited by concurrent use of acetaminophen [28]. Also, this drug's specific COX-2-like inhibition may affect homeostatic mechanisms of the central nervous system, the gastrointestinal system, and the renal system [15].
3.1.2. Proinflammatory Mechanisms Caused by Traditional NSAIDs
In general, for traditional nonselective COX inhibitors, the mechanism of drug generated myocardial pathology [34] may be due to prostacyclin and other prostanoid inhibition that depends on the degree of COX-2 inhibition [7]. The constitutive COX-2 isoenzyme that is inhibited plays an important role in the regulation of salt, volume, and blood pressure maintenance [59] by providing the appropriate prostaglandins to regulate the renin-angiotensin system [60]. Apart from prostanoid synthesis diminishment, free hydrolyzed arachidonic acid from c Phospolipase A2 (cPLA2) may be utilized by LOX isoenzymes to produce increased amounts of proinflammatory leukotrienes [61] and toxic metabolites like 5-S-HETE and 5-OXO-ETE as seen with the aspirin acetylation of COX-2 [45, 46] (Figure 3(b)).
3.2. “More Selective” COX-2 Inhibitors
3.2.1. Proinflammatory Mechanisms by “More Selective” COX-2 Inhibitors
The clear distinction between COX-1 and COX-2 inhibitors cannot be defined fully [62]. The term selective COX-2 inhibitor requires further examination as it is oversimplified and therefore the term “more selective” is used in this article. To identify proinflammatory effects caused by COX-2 more selective inhibitors, the interrelationships between COX-1 and COX-2 catalytic functions have to be taken into account. COX-1 and COX-2 have similar binding sites for NSAIDs other than aspirin that block arachidonic acid metabolism. Naproxen, for example, due to its smaller molecular size occupies easily the hydrophobic COX-1 binding site of arachidonic acid where an isoleucine is at position 523. Celecoxib is a larger molecule that naproxen cannot occupy the COX-1 binding site for arachidonic acid. Instead it occupies in an easier manner the COX-2 binding site where a smaller valine instead of isoleucine is at position 523 [63]. Thus due to small structural differences between the two COX isoenzyme active sites NSAIDs show greater or lower selectivity for COX-1 and COX-2 resulting in greater or lower relative inhibition of arachidonic acid metabolism. Relatively increased inhibition of COX-2 activity results in relative diminishment of prostacyclin inhibition which is a known cardioprotective prostanoid [64]. Recent epidemiologic studies come to directly associate the use of a large number of individual NSAIDs with hospital administration for heart failure [65]. Looking at a different metabolic pathway, more selective COX inhibitory NSAIDs may block the metabolism of 20-HETE to PGF2α and other mediators during P450 metabolism of arachidonic acid thereby resulting in increased accumulation of 20-HETE [53] (Figure 3(b)). 20-HETE has also been shown to be a serious promoter of renal hypertension and to be implicated in an increased risk for renal [33] and cardiovascular diseases [34] such as myocardial infarction, hypertension, and heart failure that have also been observed but in a smaller scale with the administration of nonaspirin traditional NSAIDs [6, 7, 34] (Figure 3(b)). Arachidonic acid that remains not bound and oxygenized by COX isoenzymes may be used by p450 and 5-LOX dependent, as well as enzyme independent, metabolic pathways to produce proinflammatory metabolites [45, 46, 61] like LTB4, LTC4, and 5-OXO-ETE as in aspirin acetylation's case (Figure 3(b)).
3.2.2. “More Selective” COX-2 Inhibitor Clinical Side Effects
Withdrawal of rofecoxib (a similar agent to celecoxib with increased vascular side effects) from clinical use is perhaps the best example to account for side effects by a more selective COX-2 inhibitor [53, 66]. Clearly, increased myocardial infarction events are associated with more selective COX-2 inhibitor use although nonselective inhibitors of cyclooxygenase are not excluded from being potent risk factors for the development of cardiovascular episodes [34, 62, 67].
Coadministration of aspirin in clinical practice is recommended for certain groups of patients taking vast amounts of NSAIDs as a thrombolytic agent for cardioprotection [63]. These patients are at increased risk from thrombotic events by taking selective COX-2 inhibitor NSAIDs to treat inflammatory disorders [68]. Guidelines however state that aspirin use may not always be an efficient protection [62]. As for traditional NSAIDs, the more selective COX-2 inhibition may also contribute to a subsequent surplus of arachidonic acid that can be used by lipoxygenases (Figure 3(b)). As already described 5-LOX may be an important mediator enzyme for inflammation and cancer producing more proinflammatory leukotrienes LTC4 [69], LTB4 [46, 61, 70], and 5-OXO-ETE [45, 49].
3.3. Hypersensitivity Response
Eosinophils among other stimuli are also driven by LTC4, which is induced by NSAID use (Figures 3(a) and 3(b)), and are essential mediator cells in the production of allergic inflammation [71]. Various types of NSAIDs are warranted for causing respiratory intolerance [72]. By proinflammatory mediator generation they have been implicated to produce allergic and inflammatory reactions as well as ischemia at the level of lung mucosa leading to asthma [73, 74]. NSAID-induced gastrointestinal injury is mediated by increased LTB4 synthesis, too. LTB4 level is elevated in arthritis treated with NSAIDs [75] (Figures 3(a) and 3(b)). Indomethacin may cause acute gastropathy, and the induced overproduction of tumor necrosis factor α (TNFα) has also been implicated in the pathogenesis of disease state [76]. Complementarily, indomethacin to a greater extent than ibuprofen causes renal dysfunctional abnormalities in preterm neonates, and unfortunately both are the drugs of choice for patent ductus arteriosus failure [77]. The immune response in urticaria provides another good example for NSAID driven side effect [78]. Urticaria is the clinical term of a heterogenous group of diseases characterized by wheels and flares of skin's vascular inflammation. Aspirin and other more selective to COX-2 NSAIDs (rofecoxib) and traditional NSAIDs (nimesulide, acetaminophen) cause the aspirin acute intolerant urticaria that in some cases may lead to the aspirin chronic urticaria [79] that has a sound basis of autoimmunity [78]. As it has been shown that in chronic cases of urticaria a specific set of IgE autoantibodies directed against thyroid peroxidase may constitute a novel pathogenetic mechanism, this may be serious for chronic NSAID users [80]. Selective to COX-2 NSAIDs and aspirin have been reported to be implicated in hypersensitivity responses and excess of histamine release, and this may be extended to various hypersensitivity immune disorders [81–83]. However, further studies are needed to investigate the possible IgE elevation in urticaria events that is caused by “more selective” NSAIDs.
4. Discussion
4.1. Alleviating NSAID Associated Proinflammatory Activity
As already described, NSAID side effects occur primarily due to the inhibition of cyclooxygenases that metabolize arachidonic acid and synthesize prostaglandins with beneficial activities under normal conditions [7, 62]. An example of a current therapeutic way to overcome NSAID side effects is the combination of aspirin coadministration in patients receiving “more selective” COX-2 inhibitors in order to avoid thrombotic vascular events, although this may not always be sufficiently protective [62]. Scientific evidence remains to be clarified by large epidemiological and meta-analysis studies to establish safety standards for patients in high risk of developing serious cardiovascular side effects [84]. This process is both time and cost demanding. For example, results from large and recent epidemiologic studies in Europe clarify that heart failure is associated with increased NSAID usage. This increased risk of heart failure is dose dependent and associated with traditional and more selective to COX-2 inhibitors [65]. In all respects, the optimum selection of NSAID coadministration requires deep scientific knowledge to identify the bottom end of prostaglandin synthesis and inhibition with subsequent imbalance of homeostasis (Figure 1).
Better chances to optimize treatment of NSAIDs with relatively high and low COX-2 inhibitory activities can perhaps be conferred by supplementary agents that may interfere with COX in a different manner.
Arachidonic acid hydrolyzed by cPLA2 (phospholipase A2), if not metabolized by cyclooxygenases, remains an available substrate to be used in other catabolic pathways: (1) the lipoxygenase, (2) the P450 epoxygenase, and (3) the nonenzymatic synthesis leading to isoprostane (Figure 3(b)).
It has been described that by inhibition of COX activity the increase of cysteinyl leukotriene family (CysLT) potent proinflammatory lipid mediators is feasible [63]. Human studies on aspirin intolerance support this hypothesis. When PGE2 levels are decreased by inhibition of COX-1, altered prostanoid production, combined with increased enzymatic expression like the LTC4 synthase expression, leads to increased leukotriene synthesis producing the disease state [32, 85, 86].
A way to circumvent proinflammatory leukotrienes (LTB4 and LTC4) production by an overwhelming 5-LOX activity may be the already developed specific LOX inhibitors [87, 88]. These may block the undesirable side effects of both LTB4 and LTC4 (a cysteinyl derived leukotriene) [89]. Furthermore, the use of selective agonists of cysteinyl leukotriene receptor 1 (CysLTR1) is referred to circumvent leukotriene-associated pathologies probably by inhibition of cytosolic Ca2+ [90]. Also the inhibition of CysLTR by other agents may provide suitable pharmacologic activity. The use of either leukotriene biosynthesis inhibitors or leukotriene receptor antagonists [91] may also help to minimize NSAID side effects. Since LTC4 production is blocked by inactivation of CysLT1 receptor, selective CysLT1 antagonists may be applied [92]. Also, CysLT1 receptor antagonists to reduce eosinophilia may be of therapeutic value [91].
NSAIDs activity as already discussed (Figures 3(a) and 3(b)) may favor 5-LOX to catalyze the formation of LTA4 and 5-S-hydroperoxy-6,8,11,14-eicosatetraenoic acid (5-HpETE), which is rapidly reduced to 5S-hydroxy-6E,8Z,11Z,14Z eicosatetraenoic acid (5-S-HETE). This metabolic intermediate is oxidized by 5-hydroxyeicosanoid dehydrogenase (5-HEDH) to produce 5-OXO-ETE [46]. 5-HEDH activity is inhibited by 5-hydroxy-fatty acids [45]. Recent advances in the formation of 5-OXO-ETE receptor antagonists [93] may also help to prevent hypersensitivity reactions by COX inhibitors. Conversion of 5-S-HETE into 5-OXO-ETE is highly reversible in the presence of NADPH and alleviation of oxidant conditions. The use of antioxidants may favor restoration of 5-OXO-ETE side effects via the 5-OXO-ETE receptor in asthma, cancer, and cardiovascular conditions [48, 94].
Apart from LOX favored metabolism during COX inhibition, oxidized arachidonic acid may sustain nonenzymatic conversion to form prostaglandin F2 compounds (PGF2-isoprostanes). Isoprostanes are very readily formed in biological fluids [3] from oxidized arachidonic acid and through endoperoxide intermediates [54]. Modulation of inflammation by NSAIDs as a natural way of treatment is a very natural way of treatment is by the concurrent prolonged use of naturally derived d-α tocopheryl acetate [95, 96]. Asthmatic and atherosclerotic patients seem to benefit by natural-source d-α-tocopheryl acetate as this is shown to reduce allergen-induced F2-isoprostane formation [95, 97].
Arachidonic acid, once liberated from membrane phospholipases and not being metabolized further by cyclooxygenases due to NSAID inhibition, may be efficiently metabolized by isoforms of the cytochrome P450 (CYP) family to form 20-hydroxyeicosatetraenoic acid (20-HETE) [98, 99]. 20-HETE promotes coagulation of platelets, thus shortening the time of bleeding, and its synthesis is being increased by rofecoxib, suggesting serious cardiovascular side effects for this drug [53].
In order to identify agents that may inhibit undesired 20-HETE synthesis by NSAID-COX inhibition, the experimental model of spontaneous hypertensive rats provides significant clinical information. Agents that induce heme oxygenase reduce the renal formation of 20-HETE and also decrease hypertension [100, 101]. In clinical research, heme oxygenase inducers are of increasing interest to overcome spontaneous reactions that lead to kidney failure [102].
Heme oxygenase-1 (HO-1), which is expressed in all tissues, receives electrons from NADPH by P450 enzyme fractions due to CRP microsomal protein mediator and P450 protein-to-protein interactions [103]. This may prove to be important clinically, since under severe hypoxia there may be a way to circumvent 5-OXO-ETE accumulation by expenditure of NADPH to NADP+ to reform 5-HETE [104] and to deprive arachidonic acid reserves to form 20-HETE by P450 enzymes at the same time [105] (Figure 6). In this respect, attention is drawn on the induction of HO-1 by naturally derived agents like the endogenous haloamines of taurine, that is, N-chlorotaurine (NCT) and N-bromotaurine (NBrT), also termed as small molecule NSAIDS [106, 107]. These haloamines have been shown to downregulate the production of Cox-2 derived PGE2 [108] in a way independent of COX-2. NCT exerts its anti-inflammatory activity in rheumatoid arthritis synoviocytes by inhibiting IL-β induced production of PGE2 by decreasing COX-2 isoenzyme expression leaving COX-1 expression unaltered [109]. However, at lower cytotoxic concentrations both NCT and NBrT decrease PGE2 synthesis without affecting COX-2 expression [108]. Haloamines of taurine (NBrT and NCT) at present state can be administered locally in cases of cutaneous body cavities and organ infection and inflammation to inactivate microbes, minimize inflammation, and reduce pain and other symptoms [110–113].
Figure 6.

Proposed mechanism of heme oxygenase inducer application to overcome accumulation of toxic metabolites [102, 104, 105] induced by NSAIDs. Induction of heme oxygenase by several agents [100, 101, 108] and receival of electrons via P450 [103, 105] may result in decreased formation of 20-HETE and 5-OXO-ETE toxic metabolite accumulation.
Another target for NSAID minimization of side effects on the cardiovascular system may be the maintenance of low levels of nitric oxide (NO) that are essential for cardioprotection [114] (Figure 4). NO at normal levels inhibits thromboxane synthase and activates prostacyclin synthase [115]. LOX activity leading to increased LTB4 and LTC4 formation may create a surplus of reactive oxygen species (ROS) and especially superoxide [63]. In such a case, cardioprotective levels of nitric oxide may be consumed by ROS to form peroxynitrite, a prostacyclin synthase inhibitor and thromboxane receptor stimulator [116]. The overall effect caused by more selective COX-2 inhibitors is a low level of prostacyclin and high levels TXA2 [63], promoting a platelet activating thrombosis event. Restoration of nitric oxide levels is said to be achieved by consumption of certain doses of taurine (2-aminoethanesulfonic acid), which may act as an antioxidant on a diseased vascular state and as a prooxidant in an otherwise normal vasculature being at risk from NSAID use [117]. Also, taurine derivatives NCT and NBrT are known to reduce excessive nitric oxide formation [118]. Curcumin, a natural antioxidant consumption, may also be of help for NSAID users, especially for those being at risk. Curcumin modulates arachidonic acid release from cellular membranes by blocking the hydrolysis event by cytosolic phospholipase A2, and it inhibits the 5-LO catalytic functions and nitric oxide synthase activity [119]. During COX-1 and COX-2 inhibition by NSAIDs, generation of NO and ROS is not suppressed under inflammatory stimulation, whereas cPLA2 activity is increased under inflammatory conditions. A probable direct synergy with its function with 12- and 15-LOX isoenzymes to produce NO and ROS, via a cPLA2. 12- and 15-LOX pathway are suggested [120]. The use of 12-/15-LOX inhibitors may be beneficial, especially in neurodegenerative diseases where NO activity is a major proinflammatory mediator [120, 121].
Figure 4.

Possible target points of supplementary agent's use to alleviate NSAID promotion of proinflammatory and cancerous conditions.
Finally, recent scientific effort is focusing on the trials of new cyclooxygenase inhibitors [122] in order to overcome undesirable cyclooxygenase metabolism of arachidonic acid in inflammation and cancer. These compounds have lower isoform selectivity to COX than the NSAID “more specific” cyclooxygenase inhibitors (coxibs), which may result in reduced side effects.
4.2. NSAIDs and Risk for Cancer
4.2.1. Studies with Clinical Evidence for Cancer Development
Epidemiologic studies provide contradictory results on cancer risk development by NSAIDS that may be due to the cell-specific activity of producing prostanoids and the specificity on COX inhibition by a particular NSAID (Figures 1 and 2 and Table 1).
Table 1.
Cancer risk development from NSAID use as recorded from various epidemiology studies.
| Type of cancer | Aspirin | Traditional nonaspirin NSAID | More selective COX-2 inhibitor |
|---|---|---|---|
| Proximal colon∗ | No effect on risk [123] Decreased risk in women [124] |
Reduced risk [123] | No data available (NDA) |
|
| |||
| Distal colon∗ | Decreased risk [123] No effect in women [124] |
Reduced risk [123] | NDA |
|
| |||
| Rectum∗ | Decreased risk [123] No effect in women [124] |
No effect on risk [123] | NDA |
|
| |||
| Nonmelanoma skin cancer (NMSC) | Decreased risk for BCCa SCCb [133] Slightly decreased risk for BCC SCC [134] |
Decreased risk for BCC SCC [133] Slightly decreased risk for BCC SCC [134] |
Decreased risk for BCC SCC [133] Stronger decreased risk for BCC SCC [134] |
|
| |||
| Melanoma skin cancer (MSC) | Slightly decreased risk [133] | Slightly decreased risk [133] | No effect on risk [133] |
|
| |||
| Breast ER/PR (+) | Highly increased risk by aspirin-only NSAID users [131] No effect on risk [127] |
No effect on risk by acetaminophen [131] | Increased risk [131] Decreased risk in HER2+ [127] ∗∗ |
|
| |||
| Breast ER/PR (−) | Highly increased risk by aspirin-only NSAID users [131] Increased risk [132] |
No effect on risk by acetaminophen [131] | Increased risk [131] Decreased risk in HER2+ [127] ∗∗ |
|
| |||
| Brain glioma | No effect on risk [144] | No effect on risk [144] | No effect on risk [144] |
|
| |||
| Brain meningioma | No effect on risk [144] | Slightly increased risk [144] | Slight increased risk [144] |
|
| |||
| Hepatocellular Carcinoma (HCC) | Decreases risk [142, 143] | Decreases risk [142] | Decreases risk [142] |
|
| |||
| Intrahepatic cholangiosarcoma (ICC) | Decreases risk in men [142] No effect on risk in women [142] |
No effect on risk by ibuprofen [142] | No effect on risk [142] |
|
| |||
| Prostate∗∗∗ | No effect on risk [138] | Increased risk with acetaminophen, even stronger for metastatic type [138] | Increased risk, even stronger for metastatic type [138] |
|
| |||
| Esophageal squamous cell carcinoma (ESCC) | Decreased risk [135, 136] | Slightly decreased risk [135, 136] | Slightly decreased risk [135, 136] |
|
| |||
| Esophageal adenocarcinoma (EA) | Decreased risk [135, 136] | Slightly decreased risk [135, 136] | Slightly decreased risk [135, 136] |
|
| |||
| Noncardia gastric carcinoma | Decreased risk [135, 136] | Slightly decreased risk [135, 136] | Slightly decreased risk [135, 136] |
|
| |||
| Cardia gastric carcinoma | No effect on risk [135, 136] | No effect on risk [135, 136] | No effect on risk [135, 136] |
Subjects using certain types of NSAIDs are being protected from colorectal cancer [123]. Specifically, aspirin users have better protection from the close to rectum and distal to colon cancer development, whereas the nonaspirin NSAID users are being more protected from the proximal to colon cancer development [123–125]. It should be mentioned, however, that anatomic locations in this type of cancer development are also tightly associated with the age, gender, and the race of patients [126]. Some studies indicate that breast cancer development risk is also lowered by NSAID use [127, 128], although this may be a small decrease of relative risk [129]. Moreover, type of NSAID, specific dose, and duration of treatment have not been yet identified [130]. Other studies, however, indicate increased risk for developing breast cancer irrespective of hormonal status (estrogen/progesterone (ER/PR) receptor positive/negative) and that the risk for developing ER/PR (−) breast cancer is raised in long term daily use aspirin users [131, 132]. Traditional NSAID use (diclofenac, etodolac, and meloxicam) is also associated with decreased risk in developing aggressive skin cancers, whereas “more selective” to COX-2 inhibitors were not found to be protective [133, 134]. Also, whilst aspirin and other NSAIDS may protect from developing esophageal and noncardia gastric carcinomas, these are nonprotective for the development of cardia gastric carcinoma [135, 136].
Epidemiological studies are controversial regarding a protective [137] or promoting [138] effect of NSAIDs for prostate cancer. In prostate cancer, genetic variation in the COX-2 gene is associated with increased risk [139]. Epidemiologic studies clearly indicate that acetaminophen and nonaspirin NSAIDs are associated with a significant risk of developing kidney cancer [140, 141]. Complementarily, whilst aspirin and other NSAID users have lower risk for developing hepatocellular carcinoma (HCC) [142, 143], men only gain protection from intrahepatic carcinoma (IHC) by taking aspirin and this did not account for any other NSAID use [142]. Aspirin and other NSAID users are not protected from developing brain tumors and the use of traditional and “more selective” COX inhibitors seems to increase the risk for developing meningiomas [144].
Although epidemiology data on cancer risk by NSAIDs are controversial, by comparison of results important indications may be drawn (Table 1). Inhibition of prostanoid synthesis due to aspirin acetylation on COX isoenzymes, although in a different manner [7, 26], may be protective of a variety of types of cancers. Acetylating of COX by aspirin was found to be protective of prostate cancer development in a subgroup of subjects having specific sequence variations within the COX-2 genome [139]. Also, risk reduction for colorectal cancer development with aspirin is related only to specific genotypes near the IL-6 genome [145]. Thereby genetic variation seems be an important parameter for the aspirin effect in cancer development. By contrast, aspirin use increases the risk for breast cancer development irrespective of hormonal influence [131]. Cell-dependent prostanoid formation is also another important serious parameter (Figure 1). Any given prostanoid forming cell selects a particular prostanoid as its major product [15]. Brain and mast cells, for example, preferably produce PGD2, and its formation provides the core for vital homeostasis mechanisms [146, 147].
4.2.2. What Are the Effects of NSAIDs on Cancer?
Traditional and “more selective” COX inhibitors preferentially bind on arachidonic acid's active site of respective isoenzymes. The degree to which COX inhibitors cause inhibition of COX-1, COX-2, or COX-3 depends on their selective preference for COX active sites [62, 63]. Under normal conditions, constitutive COX-1 maintains prostacyclin and PGE2 levels as well as other prostanoids in all tissues, whereas COX-2, when activated in an inducible way (i.e., during inflammation), produces prostacyclin and other prostanoids in most, but not all, organs [15]. From epidemiologic studies it is evident that some groups of subjects benefit from developing certain types of cancer by nonaspirin NSAIDs, for example, nonmelanoma skin cancers [134], whereas the same use of NSAIDs elevates the risk of developing some other types of cancer, for example, in the kidney [141]. The differential inhibition of COX-1, COX-2, and COX-3 isoenzymes results in differential beneficial activities and side effects [15] (Figure 1). For cancer, the situation is even more complicated as it may also depend on both the impact on COX and the subsequent synthesis inhibition of protective prostaglandins.
4.3. Mechanisms of Promotion of Cancer by the AA System and the NSAIDs
PGE2 association with tumorigenesis has been thoroughly investigated. Measurement of increased amounts of PGE2 in colorectal cancer has long been implicated to contribute to tumorigenesis [148], where apparently COX-2 and not COX-1 is overexpressed [149], and COX-2 expression is more essential for tumor development in the distal colon [150]. Nowadays, inducible PGE2 synthesis is implicated in many types of cancers [58, 151]. However, it should always be considered that PGE2 is a major component of tissue homeostasis under normal conditions [21, 58] and its constitutive synthesis may be impaired by the frequent use of all classes of NSAIDs [7].
Frequent use of NSAIDS leading to depletion of COX activity may favor the metabolism of arachidonic acid by the LOX pathways [15, 63] as discussed. There are several human and animal studies finely reviewed to support this hypothesis [18]. 5- and 12-LOX metabolic pathways are linked to carcinogenesis [152], and altered COX and LOX metabolism of arachidonic acid are a common factor in malignancy [35]. One major metabolite of 5-LOX, the LTB4, was shown to produce cancer predisposition by activating transcriptional factor NF-kB in hepatoma cells [153]. The cysteinyl leukotrienes (TLC4, LTD4, and LTE4) may also be oversynthesized as increased amounts of LTC4 and LTD4 and decreased amounts of PGE2 are detected in nasal secretions if patients with aspirin intolerance are treated with this medication [154] (Figure 3(a)). LTC4 induces the phosphorylation of NF-kB p65, activates the complex NF-kB p50-p66 [155], and via the CysLT2 receptor induces the phosphorylation of IkBa by involving protein kinase family enzymes [156]. Aspirin decreases the expression of Bcl-2 by blocking the IL6-IL6R-STAT3 signaling pathway [157]. The decreased expression of Bcl-2 may cause apoptosis by tumor necrosis factor apoptosis-inducing ligand and increased levels of TNFα expressed [158]. However, overproduced TNFα may not function proapoptotically but contribute to cell survival (Figure 5). TNFα bound to the tumor necrosis receptor 1 (TNFR-1), apart from other causalities, recruits TNFR-1-associated death domain protein (TRADD). TRADD in turn has a dual activity. When TRADD recruits the receptor interacting protein kinase- (RIPK-) and Fas-associated death domain protein (FADD), this finally results in apoptosis [159]. However, when TRADD recruits TNF receptor factor 2 (TRAF-2) and FADD, this results in activation of survival transcription factor NF-kB [160–162]. TNF-α also results in activation of the transcription factor AP-1 via the JNK signaling cascade, which subsequently increases cellular proliferation [163].
Figure 5.

Molecular pathways that may contribute to promoting cancer by NSAID use [155–158, 160–163]. LTC4 and TNFa may activate transcription factor NF-kB and increase cellular proliferation by concurrent pathways that otherwise induce apoptosis.
Furthermore, extensive 5-LOX activity from arachidonic acid accumulating from NSAID inhibition of COX may also lead to increased 5-OXO-ETE formation (Figures 3(a) and 3(b)). This may be of raised interest in oncology studies as 5-OXO-ETE lipid molecules seem to be required for cancer cell proliferation [48]. In prostate cancer cells, for example, 5-OXO-ETE and the 12-LOX metabolism are also important for tumor propagation [48, 88]. Marked expression of 5- and 12-LOX is being detected in prostate neoplasia in contrast to normal and benign epithelia [88]. The platelet 12-LOX overexpression in prostate cancer is also said to be a trigger for angiogenesis and tumor growth by enhancing avβ3 and avβ5 integrin expression [15]. Complementarily, by data drawn from a Gln261Arg polymorphism of the 12-LOX gene meta-analysis study, clearly enough, this polymorphism was shown to be a significant risk factor for increased susceptibility to at least five types of cancer, including prostate cancer, specifically in the Asian population [164].
4.4. Description of New Drugs and Their Possible Use for Alleviation of Cancer Predisposition with NSAIDs
Lipoxygenases are an emerging group of cancer targets as numerous studies indicate that 5-LOX and 15-LOX-1 are associated with the development of cancer via the NF-kB pathway [61]. Treatment with specific LOX inhibitors may be important as therapeutic option in order to overcome LOX overexpression [88]. Looking at the pathway of downstream production of 5-OXO-ETE, as a serious promoter of carcinogenesis, the novel synthesis of 5-OXO-ETE receptor antagonists may be another therapeutic option [93] (Figure 5). Recently we have discovered that the taurine derivative NBrT is a significant proliferative inhibitor leading to cell death among numerous cancer cell lines. Antiproliferative activity is enhanced on PC3 (prostate cancer), A549 (lung cancer), HeLa (cervical cancer), and MDA-MB231 (breast cancer) cell lines, (A. Kyriakopoulos et al. unpublished data). It would be a future interesting model to test the aspirin-induced proliferative ability of these particular cell lines and subsequently the inhibitory effect by the heme oxygenase inducers such as NBrT. The extensive antiproliferative effect on numerous human cancer cell lines by NBrT is in accordance with the recently demonstrated effect of G cycle arrest of glucocorticoid resistant cancer cells and the optimized concurrent anticancer effect of cisplatin with NBrT. NBrT has been considered in studies as a small molecule NSAID as it leads to decreased PGE2 levels independently of COX expression [106, 108, 165]. As cyclooxygenase [166] and lipoxygenase [61] metabolism have long ago been associated with tumorigenesis, a possible therapeutic intervention with heme oxygenase inducers (including taurine derivatives) may be of significance and should be tested in animal models. Stress conditions that may lead to renal disease [33, 53, 103] (under hypoxic conditions [60]) and possible cancer predisposition enhancement may be circumvented (Figure 6).
5. Conclusion
Only scarce previous studies in the past have been focused on the avoidance of adverse effects of NSAID use [167]. Due to both older and recent research data on proinflammatory effects and cancer development in connection with NSAIDs, selective therapeutic targets and newer agents like the small molecule NSAIDs with an improved benefit-risk ratio become of interest. By all means, a thorough investigation of lipid metabolism under NSAID use is required, and this must be coupled with large scale epidemiological studies to provide valuable clinical information. For example, although accumulated data suggest a protective role of some COX inhibitors in the development of certain types of cancer, the predicted increased risk for other cancers by NSAID use is also equally important. Alterations of AA metabolism by NSAID, prompt further investigating the possible development of inflammation and cancer.
Acknowledgments
The authors thank N. Kotsalas MD and I. Panagiotopoulos, MD, for their aid with the schematic presentations and literature research. This manuscript has been solely supported by Nasco AD Biotechnology Laboratory. The authors also thank JPA Medical Co. for sponsorship of publication fees.
Conflicts of Interest
It is hereby declared that all authors have no conflicts of interest.
Authors' Contributions
A. M. Kyriakopoulos has written the manuscript with the assistance of M. Nagl. V. Zoumpourlis provided the unpublished cancer data. S. Baliou aided mostly with schematic presentations making her contribution adequate for coauthorship.
References
- 1.Capone M. L., Tacconelli S., Di Francesco L., Sacchetti A., Sciulli M. G., Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins and Other Lipid Mediators. 2007;82(1–4):85–94. doi: 10.1016/j.prostaglandins.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Capdevila J., Chacos N., Werringloer J., Prough R. A., Estabrook R. W. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(9):5362–5366. doi: 10.1073/pnas.78.9.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrow J. D., Harris T. M., Jackson Roberts L., II Noncyclooxygenase oxidative formation of a series of novel prostaglandins: analytical ramifications for measurement of eicosanoids. Analytical Biochemistry. 1990;184(1):1–10. doi: 10.1016/0003-2697(90)90002-q. [DOI] [PubMed] [Google Scholar]
- 4.Ouellet M., Riendeau D., Percival M. D. A high level of cyclooxygenase-2 inhibitor selectivity is associated with a reduced interference of platelet cyclooxygenase-1 inactivation by aspirin. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(25):14583–14588. doi: 10.1073/pnas.251543298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rieke C. J., Mulichak A. M., Garavito R. M., Smith W. L. The role of arginine 120 of human prostaglandin endoperoxide H synthase- 2 in the interaction with fatty acid substrates and inhibitors. Journal of Biological Chemistry. 1999;274(24):17109–17114. doi: 10.1074/jbc.274.24.17109. [DOI] [PubMed] [Google Scholar]
- 6.Vane J. R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biology. 1971;231(25):232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 7.Vane J. R. The fight against rheumatism: from willow bark to COX-1 sparing drugs. Journal of Physiology and Pharmacology. 2000;51(4):573–586. [PubMed] [Google Scholar]
- 8.Moncada S., Gryglewski R., Bunting S., Vane J. R. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263(5579):663–665. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- 9.Berenger F. P., Cano J.-P., Rolland P. H. Antithrombogenic endothelial cell defense. Basal characteristics in cultured endothelial cells and modulation by short-term and long-term exposure to isosorbide nitrates. Circulation Research. 1987;60(4):612–620. doi: 10.1161/01.RES.60.4.612. [DOI] [PubMed] [Google Scholar]
- 10.Whittle B. J. Inhibition of prostacyclin (PGI2) formation in the rat small-intestine and gastric mucosa by the ulcerogen, indomethacin [proceedings] British Journal of Pharmacology. 1978;64(3):p. 438P. [PMC free article] [PubMed] [Google Scholar]
- 11.Whittle B. J. R., Higgs G. A., Eakins K. E., Moncada S., Vane J. R. Selective inhibition of prostaglandin production in inflammatory exudates and gastric mucosa. Nature. 1980;284(5753):271–273. doi: 10.1038/284271a0. [DOI] [PubMed] [Google Scholar]
- 12.Simmons D. L., Levy D. B., Yannoni Y., Erikson R. L. Identification of a phorbol ester-repressible v-src-inducible gene. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(4):1178–1182. doi: 10.1073/pnas.86.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie W., Chipman J. G., Robertson D. L., Erikson R. L., Simmons D. L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(7):2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozak K. R., Rowlinson S. W., Marnett L. J. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. The Journal of Biological Chemistry. 2000;275(43):33744–33749. doi: 10.1074/jbc.m007088200. [DOI] [PubMed] [Google Scholar]
- 15.Clària J. Cyclooxygenase-2 biology. Current Pharmaceutical Design. 2003;9(27):2177–2190. doi: 10.2174/1381612033454054. [DOI] [PubMed] [Google Scholar]
- 16.Vernieri E., Gomez-Monterrey I., Milite C., et al. Design, synthesis, and evaluation of new tripeptides as COX-2 inhibitors. Journal of Amino Acids. 2013;2013:7. doi: 10.1155/2013/606282.606282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrasekharan N. V., Dai H., Roos K. L. T., et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnett B. P., Levy R. M. 5-Lipoxygenase metabolic contributions to NSAID-induced organ toxicity. Advances in Therapy. 2012;29(2):79–98. doi: 10.1007/s12325-011-0100-7. [DOI] [PubMed] [Google Scholar]
- 19.Bruno A., Tacconelli S., Patrignani P. Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic & Clinical Pharmacology & Toxicology. 2014;114(1):56–63. doi: 10.1111/bcpt.12117. [DOI] [PubMed] [Google Scholar]
- 20.DeCicco-Skinner K. L., Nolan S. J., Deshpande M. M., Trovato E. L., Dempsey T. A., Wiest J. S. Altered prostanoid signaling contributes to increased skin tumorigenesis in Tpl2 knockout mice. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0056212.e56212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagamachi M., Sakata D., Kabashima K., et al. Facilitation of Th1-mediated immune response by prostaglandin E receptor EP1. Journal of Experimental Medicine. 2007;204(12):2865–2874. doi: 10.1084/jem.20070773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sánchez-Borges M., Caballero-Fonseca F., Capriles-Hulett A., González-Aveledo L. Hypersensitivity reactions to nonsteroidal anti-inflammatory drugs: an update. Pharmaceuticals. 2010;3(1):10–18. doi: 10.3390/ph3010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth G. J., Stanford N., Majerus P. W. Acetylation of prostaglandin synthase by aspirin. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(8):3073–3076. doi: 10.1073/pnas.72.8.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tosco P., Lazzarato L. Mechanistic insights into cyclooxygenase irreversible inactivation by aspirin. ChemMedChem. 2009;4(6):939–945. doi: 10.1002/cmdc.200800438. [DOI] [PubMed] [Google Scholar]
- 25.Schnermann J. Cyclooxygenase-2 and macula densa control of renin secretion. Nephrology Dialysis Transplantation. 2001;16(9):1735–1738. doi: 10.1093/ndt/16.9.1735. [DOI] [PubMed] [Google Scholar]
- 26.Hochgesang G. P., Jr., Rowlinson S. W., Marnett L. J. Tyrosine-385 is critical for acetylation of cyclooxygenase-2 by aspirin. Journal of the American Chemical Society. 2000;122(27):6514–6515. doi: 10.1021/ja0003932. [DOI] [Google Scholar]
- 27.Rieke C. J., Mulichak A. M., Garavito R. M., Smith W. L. The role of arginine 120 of human prostaglandin endoperoxide H synthase-2 in the interaction with fatty acid substrates and inhibitors. The Journal of Biological Chemistry. 1999;274(24):17109–17114. doi: 10.1074/jbc.274.24.17109. [DOI] [PubMed] [Google Scholar]
- 28.Simmons D. L., Wagner D., Westover K. Nonsteroidal anti-inflammatory drugs, acetaminophen, cyclooxygenase 2, and fever. Clinical Infectious Diseases. 2000;31:S211–S218. doi: 10.1086/317517. [DOI] [PubMed] [Google Scholar]
- 29.Claria J., Serhan C. N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proceedings of the National Academy of Sciences. 1995;92(21):9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcus A. J., Hajjar D. P. Vascular transcellular signaling. Journal of Lipid Research. 1993;34(12):2017–2031. [PubMed] [Google Scholar]
- 31.Brezinski D. A., Nesto R. W., Serhan C. N. Angioplasty triggers intracoronary leukotrienes and lipoxin A4. Impact of aspirin therapy. Circulation. 1992;86(1):56–63. doi: 10.1161/01.cir.86.1.56. [DOI] [PubMed] [Google Scholar]
- 32.Sampson A. P., Cowburn A. S., Sladek K., et al. Profound overexpression of leukotriene C4 synthase in bronchial biopsies from aspirin-intolerant asthmatic patients. International Archives of Allergy and Immunology. 1997;113(1–3):355–357. doi: 10.1159/000237600. [DOI] [PubMed] [Google Scholar]
- 33.Patrignani P., Tacconelli S., Bruno A., Sostres C., Lanas A. Managing the adverse effects of nonsteroidal anti-inflammatory drugs. Expert Review of Clinical Pharmacology. 2011;4(5):605–621. doi: 10.1586/ecp.11.36. [DOI] [PubMed] [Google Scholar]
- 34.Varas-Lorenzo C., Riera-Guardia N., Calingaert B., et al. Myocardial infarction and individual nonsteroidal anti-inflammatory drugs meta-analysis of observational studies. Pharmacoepidemiology and Drug Safety. 2013;22(6):559–570. doi: 10.1002/pds.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D., DuBois R. N. Eicosanoids and cancer. Nature Reviews Cancer. 2010;10(3):181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Byrne P. M. Leukotrienes in the pathogenesis of asthma. Chest. 1997;111(2, supplement):27S–34S. doi: 10.1378/chest.111.2_supplement.27s. [DOI] [PubMed] [Google Scholar]
- 37.Claesson H.-E., Haeggström J. Human endothelial cells stimulate leukotriene synthesis and convert granulocyte released leukotriene A4 into leukotrienes B4, C4, D4 and E4. European Journal of Biochemistry. 1988;173(1):93–100. doi: 10.1111/j.1432-1033.1988.tb13971.x. [DOI] [PubMed] [Google Scholar]
- 38.Clària J., Lee M. H., Serhan C. N. Aspirin-triggered lipoxins (15-epi-LX) are generated bythe human lung adenocarcinoma cell line (A549) neutrophil interactions and are potent inhibitors of cell proliferation. Molecular Medicine. 1996;2(5):583–596. [PMC free article] [PubMed] [Google Scholar]
- 39.Borgeat P., Hamberg M., Samuelsson B. Transformation of arachidonic acid and homo-gamma-linolenic acid by rabbit polymorphonuclear leukocytes. Monohydroxy acids from novel lipoxygenases. The Journal of Biological Chemistry. 1976;251(24):7816–7820. [PubMed] [Google Scholar]
- 40.Borgeat P., Samuelsson B. Transformation of arachidonic acid by rabbit polymorphonuclear leukocytes. Formation of a novel dihydroxyeicosatetraenoic acid. The Journal of Biological Chemistry. 1979;254(8):2643–2646. [PubMed] [Google Scholar]
- 41.Borgeat P., Samuelsson B. Arachidonic acid metabolism in polymorphonuclear leukocytes: unstable intermediate in formation of dihydroxy acids. Proceedings of the National Academy of Sciences. 1979;76(7):3213–3217. doi: 10.1073/pnas.76.7.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zarini S., Gijón M. A., Ransome A. E., Murphy R. C., Sala A. Transcellular biosynthesis of cysteinyl leukotrienes in vivo during mouse peritoneal inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(20):8296–8301. doi: 10.1073/pnas.0903851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller A., Rechencq E., Kugel C., et al. Comparative biological activities of the four synthetic (5,6)-dihete isomers. Prostaglandins. 1989;38(6):635–644. doi: 10.1016/0090-6980(89)90046-4. [DOI] [PubMed] [Google Scholar]
- 44.Mulugeta S., Suzuki T., Hernandez N. T., Griesser M., Boeglin W. E., Schneider C. Identification and absolute configuration of dihydroxy-arachidonic acids formed by oxygenation of 5 S-HETE by native and aspirin-acetylated COX-2. Journal of Lipid Research. 2010;51(3):575–585. doi: 10.1194/jlr.M001719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel P., Cossette C., Anumolu J. R., et al. Substrate selectivity of 5-hydroxyeicosanoid dehydrogenase and its inhibition by 5-hydroxy-Delta 6-long-chain fatty acids. Journal of Pharmacology and Experimental Therapeutics. 2009;329(1):335–341. doi: 10.1124/jpet.108.143453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powell W. S., Gravelle F., Gravel S. Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. The Journal of Biological Chemistry. 1992;267(27):19233–19241. [PubMed] [Google Scholar]
- 47.Qin L., Crews F. T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. Journal of Neuroinflammation. 2012;9, article 5 doi: 10.1186/1742-2094-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell W. S., Rokach J. Biochemistry, biology and chemistry of the 5-lipoxygenase product 5-oxo-ETE. Progress in Lipid Research. 2005;44(2-3):154–183. doi: 10.1016/j.plipres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Grant G. E., Rokach J., Powell W. S. 5-Oxo-ETE and the OXE receptor. Prostaglandins and Other Lipid Mediators. 2009;89(3-4):98–104. doi: 10.1016/j.prostaglandins.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Flaherty J. T., Jacobson D., Redman J. Mechanism involved in the mobilization of neutrophil calcium by 5-hydroxyeicosatetraenoate. Journal of Immunology. 1988;140(12):4323–4328. [PubMed] [Google Scholar]
- 51.Powell W. S., Gravel S., MacLeod R. J., Mills E., Hashefi M. Stimulation of human neutrophils by 5-oxo-6,8,11,14-eicosatetraenoic acid by a mechanism independent of the leukotriene B4 receptor. Journal of Biological Chemistry. 1993;268(13):9280–9286. [PubMed] [Google Scholar]
- 52.Snyder N. W. Investigations of bio-activity, disposition and metabolism of lipids through liquid chromatography mass spectrometry [Ph.D. thesis] University of Pennsylvania; 2013. http://repository.upenn.edu/cgi/viewcontent.cgi?article=2087&context=edissertations. [Google Scholar]
- 53.Liu J. Y., Li N., Yang J., et al. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39):17017–17022. doi: 10.1073/pnas.1011278107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrow J. D., Hill K. E., Burk R. F., Nammour T. M., Badr K. F., Roberts L. J., II A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(23):9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kearney P. M., Baigent C., Godwin J., Halls H., Emberson J. R., Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. British Medical Journal. 2006;332(7553):1302–1305. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y.-G., Lin C.-L., Dai M.-S., et al. Risk of acute kidney injury and long-term outcome in patients with acetaminophen intoxication: a nationwide population-based retrospective cohort study. Medicine. 2015;94(46) doi: 10.1097/md.0000000000002040.e2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Botting R. M. Mechanism of action of acetaminophen: is there a cyclooxygenase 3? Clinical Infectious Diseases. 2000;31(5):S202–S210. doi: 10.1086/317520. [DOI] [PubMed] [Google Scholar]
- 58.Nakanishi M., Rosenberg D. W. Multifaceted roles of PGE2 in inflammation and cancer. Seminars in Immunopathology. 2013;35(2):123–137. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris R. C., McKanna J. A., Akai Y., Jacobson H. R., Dubois R. N., Breyer M. D. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. The Journal of Clinical Investigation. 1994;94(6):2504–2510. doi: 10.1172/jci117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris R. C. Macula densa signalling—a potential role of cyclooxygenase-2 (COX-2)? Nephrology Dialysis Transplantation. 2000;15(10):1504–1506. doi: 10.1093/ndt/15.10.1504. [DOI] [PubMed] [Google Scholar]
- 61.Wisastra R., Dekker F. J. Inflammation, cancer and oxidative lipoxygenase activity are intimately linked. Cancers. 2014;6(3):1500–1521. doi: 10.3390/cancers6031500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antman E. M., Bennett J. S., Daugherty A., Furberg C., Roberts H., Taubert K. A. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American heart association. Circulation. 2007;115(12):1634–1642. doi: 10.1161/circulationaha.106.181424. [DOI] [PubMed] [Google Scholar]
- 63.Antman E. M., DeMets D., Loscalzo J. Cyclooxygenase inhibition and cardiovascular risk. Circulation. 2005;112(5):759–770. doi: 10.1161/CIRCULATIONAHA.105.568451. [DOI] [PubMed] [Google Scholar]
- 64.Bing R. J., Lomnicka M. Why do cyclo-oxygenase-2 inhibitors cause cardiovascular events? Journal of the American College of Cardiology. 2002;39(3):521–522. doi: 10.1016/S0735-1097(01)01749-1. [DOI] [PubMed] [Google Scholar]
- 65.Arfè A., Scotti L., Varas-Lorenzo C., et al. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: nested case-control study. British Medical Journal. 2016;354 doi: 10.1136/bmj.i4857.i4857 [DOI] [PubMed] [Google Scholar]
- 66.Dieppe P. A., Ebrahim S., Martin R. M., Jüni P. Lessons from the withdrawal of rofecoxib. British Medical Journal. 2004;329(7471):867–868. doi: 10.1136/bmj.329.7471.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGettigan P., Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. The Journal of the American Medical Association. 2006;296(13):1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- 68.Schafer A. My doctor recently recommended that i take low-dose aspirin to minimize the risk of a heart attack or stroke. I routinely take ibuprofen for arthritis pain. Do i need both, and is it safe to combine the two? Health News. 2000;6:p. 10. [PubMed] [Google Scholar]
- 69.Steinhilber D. 5-Lipoxygenase: a target for antiinflammatory drugs revisited. Current Medicinal Chemistry. 1999;6(1):71–85. [PubMed] [Google Scholar]
- 70.Poulin S., Thompson C., Thivierge M., et al. Cysteinyl-leukotrienes induce vascular endothelial growth factor production in human monocytes and bronchial smooth muscle cells. Clinical and Experimental Allergy. 2011;41(2):204–217. doi: 10.1111/j.1365-2222.2010.03653.x. [DOI] [PubMed] [Google Scholar]
- 71.Stone K. D., Prussin C., Metcalfe D. D. IgE, mast cells, basophils, and eosinophils. Journal of Allergy and Clinical Immunology. 2010;125(2, supplement 2):S73–S80. doi: 10.1016/j.jaci.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grigiene G., Norkuniene J., Kvedariene V. The time delay between drug intake and bronchospasm for nonsteroidal antiinflammatory drugs sensitive patients. World Allergy Organization Journal. 2010;3(12):266–270. doi: 10.1097/WOX.0b013e3181fdfc5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alvarez C., Amaral M. M., Langellotti C., Vermeulen M. Leukotriene C4 prevents the complete maturation of murine dendritic cells and modifies interleukin-12/interleukin-23 balance. Immunology. 2011;134(2):185–197. doi: 10.1111/j.1365-2567.2011.03478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poulin S., Thompson C., Thivierge M., et al. Cysteinyl-leukotrienes induce vascular endothelial growth factor production in human monocytes and bronchial smooth muscle cells. Clinical & Experimental Allergy. 2011;41(2):204–217. doi: 10.1111/j.1365-2222.2010.03653.x. [DOI] [PubMed] [Google Scholar]
- 75.Hudson N., Balsitis M., Everitt S., Hawkey C. J. Enhanced gastric mucosal leukotriene B4 synthesis in patients taking non-steroidal anti-inflammatory drugs. Gut. 1993;34(6):742–747. doi: 10.1136/gut.34.6.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santucci L., Fiorucci S., Giansanti M., Brunori P. M., Di Matteo F. M., Morelli A. Pentoxifylline prevents indomethacin induced acute gastric mucosal damage in rats: role of tumour necrosis factor alpha. Gut. 1994;35(7):909–915. doi: 10.1136/gut.35.7.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allegaert K., de Hoon J., Debeer A., Gewillig M. Renal side effects of non-steroidal anti-inflammatory drugs in neonates. Pharmaceuticals. 2010;3(2):393–405. doi: 10.3390/ph3020393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Napoli D. C., Freeman T. M. Autoimmunity in chronic urticaria and urticarial vasculitis. Current Allergy and Asthma Reports. 2001;1(4):329–336. doi: 10.1007/s11882-001-0044-0. [DOI] [PubMed] [Google Scholar]
- 79.Asero R. Chronic urticaria with multiple NSAID intolerance: is tramadol always a safe alternative analgesic? Journal of Investigational Allergology and Clinical Immunology. 2003;13(1):56–59. [PubMed] [Google Scholar]
- 80.Shin Y. S., Suh D., Yang E., Ye Y., Park H. Serum specific IgE to thyroid peroxidase activates basophils in aspirin intolerant urticaria. Journal of Korean Medical Science. 2015;30(6):705–709. doi: 10.3346/jkms.2015.30.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsieh C. W., Lee J. W., Liao E. C., Tsai J. J. A disease marker for aspirin-induced chronic urticaria. International Journal of Molecular Sciences. 2014;15(7):12591–12603. doi: 10.3390/ijms150712591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Venturini Díaz M., San Juan De La Parra S., Segura Arazuri N. Selective immediate hypersensitivity to etoricoxib. Journal of Investigational Allergology and Clinical Immunology. 2008;18(6):485–487. [PubMed] [Google Scholar]
- 83.Altrichter S., Peter H.-J., Pisarevskaja D., Metz M., Martus P., Maurer M. IgE mediated autoallergy against thyroid peroxidase—a novel pathomechanism of chronic spontaneous urticaria? PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0014794.e14794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White W. B. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49(3):408–418. doi: 10.1161/01.HYP.0000258106.74139.25. [DOI] [PubMed] [Google Scholar]
- 85.Cowburn A. S., Sladek K., Soja J., et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. The Journal of Clinical Investigation. 1998;101(4):834–846. doi: 10.1172/jci620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Varghese M., Lockey R. F. Aspirin-exacerbated asthma. Allergy, Asthma and Clinical Immunology. 2008;4(2):75–83. doi: 10.1186/1710-1492-4-2-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pergola C., Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opinion on Therapeutic Patents. 2010;20(3):355–375. doi: 10.1517/13543771003602012. [DOI] [PubMed] [Google Scholar]
- 88.Matsuyama M., Yoshimura R., Mitsuhashi M., et al. Expression of lipoxygenase in human prostate cancer and growth reduction by its inhibitors. International Journal of Oncology. 2004;24(4):821–827. [PubMed] [Google Scholar]
- 89.Martel-Pelletier J., Lajeunesse D., Reboul P., Pelletier J.-P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Annals of the Rheumatic Diseases. 2003;62(6):501–509. doi: 10.1136/ard.62.6.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shimbori C., Shiota N., Okunishi H. Effects of montelukast, a cysteinyl-leukotriene type 1 receptor antagonist, on the pathogenesis of bleomycin-induced pulmonary fibrosis in mice. European Journal of Pharmacology. 2011;650(1):424–430. doi: 10.1016/j.ejphar.2010.09.084. [DOI] [PubMed] [Google Scholar]
- 91.Lee E., Robertson T., Smith J., Kilfeather S. Leukotriene receptor antagonists and synthesis inhibitors reverse survival in eosinophils of asthmatic individuals. American Journal of Respiratory and Critical Care Medicine. 2000;161(6):1881–1886. doi: 10.1164/ajrccm.161.6.9907054. [DOI] [PubMed] [Google Scholar]
- 92.Montuschi P., Peters-Golden M. L. Leukotriene modifiers for asthma treatment. Clinical and Experimental Allergy. 2010;40(12):1732–1741. doi: 10.1111/j.1365-2222.2010.03630.x. [DOI] [PubMed] [Google Scholar]
- 93.Gore V., Patel P., Chang C.-T., et al. 5-Oxo-ETE receptor antagonists. Journal of Medicinal Chemistry. 2013;56(9):3725–3732. doi: 10.1021/jm400480j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Erlemann K.-R., Rokach J., Powell W. S. Oxidative stress stimulates the synthesis of the eosinophil chemoattractant 5-oxo-6,8,11,14-eicosatetraenoic acid by inflammatory cells. The Journal of Biological Chemistry. 2004;279(39):40376–40384. doi: 10.1074/jbc.m401294200. [DOI] [PubMed] [Google Scholar]
- 95.Hoskins A., Roberts J. L., II, Milne G., Choi L., Dworski R. Natural-source d-α-tocopheryl acetate inhibits oxidant stress and modulates atopic asthma in humans in vivo. Allergy. 2012;67(5):676–682. doi: 10.1111/j.1398-9995.2012.02810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reddanna P., Krishna Rao M., Channa Reddy C. Inhibition of 5-lipoxygenase by vitamin E. FEBS Letters. 1985;193(1):39–43. doi: 10.1016/0014-5793(85)80075-2. [DOI] [PubMed] [Google Scholar]
- 97.Roberts L. J., II, Oates J. A., Linton M. F., et al. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radical Biology and Medicine. 2007;43(10):1388–1393. doi: 10.1016/j.freeradbiomed.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lasker J. M., Chen W. B., Wolf I., Bloswick B. P., Wilson P. D., Powell P. K. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of CYP4F2 and CYP4A11. The Journal of Biological Chemistry. 2000;275(6):4118–4126. doi: 10.1074/jbc.275.6.4118. [DOI] [PubMed] [Google Scholar]
- 99.Sacerdoti D., Gatta A., McGiff J. C. Role of cytochrome P450-dependent arachidonic acid metabolites in liver physiology and pathophysiology. Prostaglandins and Other Lipid Mediators. 2003;72(1-2):51–71. doi: 10.1016/S1098-8823(03)00077-7. [DOI] [PubMed] [Google Scholar]
- 100.Sacerdoti D., Escalante B., Abraham N. G., Mcgiff J. C., Levere R. D., Schwartzman M. L. Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science. 1989;243(4889):388–390. doi: 10.1126/science.2492116. [DOI] [PubMed] [Google Scholar]
- 101.Levere R. D., Martasek P., Escalante B., Schwartzman M. L., Abraham N. G. Effect of heme arginate administration on blood pressure in spontaneously hypertensive rats. Journal of Clinical Investigation. 1990;86(1):213–219. doi: 10.1172/JCI114686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nath K. A., Balla G., Vercellotti G. M., et al. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. Journal of Clinical Investigation. 1992;90(1):267–270. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spencer A. L. M., Bagai I., Becker D. F., Zuiderweg E. R. P., Ragsdale S. W. Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. Journal of Biological Chemistry. 2014;289(43):29836–29858. doi: 10.1074/jbc.m114.582783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Higashimoto Y., Sakamoto H., Hayashi S., et al. Involvement of NADPH in the interaction between heme oxygenase-1 and cytochrome P450 reductase. The Journal of Biological Chemistry. 2005;280(1):729–737. doi: 10.1074/jbc.M406203200. [DOI] [PubMed] [Google Scholar]
- 105.Roman R. J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiological Reviews. 2002;82(1):131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 106.Logotheti S., Khoury N., Vlahopoulos S. A., et al. N-bromotaurine surrogates for loss of antiproliferative response and enhances cisplatin efficacy in cancer cells with impaired glucocorticoid receptor. Translational Research. 2016;173:58–73.e2. doi: 10.1016/j.trsl.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 107.Rao P. P. N., Kabir S. N., Mohamed T. Nonsteroidal anti-inflammatory drugs (NSAIDs): progress in small molecule drug development. Pharmaceuticals. 2010;3(5):1530–1549. doi: 10.3390/ph3051530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Olszanecki R., Kurnyta M., Biedroń R., Chorobik P., Bereta M., Marcinkiewicz J. The role of heme oxygenase-1 in down regulation of PGE2 production by taurine chloramine and taurine bromamine in J774.2 macrophages. Amino Acids. 2008;35(2):359–364. doi: 10.1007/s00726-007-0609-x. [DOI] [PubMed] [Google Scholar]
- 109.Kontny E., Maśliński W., Marcinkiewicz J. Anti-inflammatory activities of taurine chloramine: implication for immunoregulation and pathogenesis of rheumatoid arthritis. Advances in Experimental Medicine and Biology. 2003;526:329–340. doi: 10.1007/978-1-4615-0077-3_41. [DOI] [PubMed] [Google Scholar]
- 110.Gottardi W., Nagl M. N-chlorotaurine, a natural antiseptic with outstanding tolerability. Journal of Antimicrobial Chemotherapy. 2010;65(3):399–409. doi: 10.1093/jac/dkp466. [DOI] [PubMed] [Google Scholar]
- 111.Gottardi W., Debabov D., Nagl M. N-chloramines: a promising class of well-tolerated topical antiinfectives. Antimicrobial Agents and Chemotherapy. 2013;57(3):1107–1114. doi: 10.1128/aac.02132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim C., Cha Y.-N. Taurine chloramine produced from taurine under inflammation provides anti-inflammatory and cytoprotective effects. Amino Acids. 2014;46(1):89–100. doi: 10.1007/s00726-013-1545-6. [DOI] [PubMed] [Google Scholar]
- 113.Marcinkiewicz J., Kontny E. Taurine and inflammatory diseases. Amino Acids. 2014;46(1):7–20. doi: 10.1007/s00726-012-1361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jones S. P., Bolli R. The ubiquitous role of nitric oxide in cardioprotection. Journal of Molecular and Cellular Cardiology. 2006;40(1):16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 115.Wade M. L., Fitzpatrick F. A. Nitric oxide modulates the activity of the hemoproteins prostaglandin I2 synthase and thromboxane A2 synthase. Archives of Biochemistry and Biophysics. 1997;347(2):174–180. doi: 10.1006/abbi.1997.0348. [DOI] [PubMed] [Google Scholar]
- 116.Bachschmid M., Thurau S., Zou M.-H., Ullrich V. Endothelial cell activation by endotoxin involves superoxide/NO-mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. The FASEB Journal. 2003;17(8):914–916. doi: 10.1096/fj.02-0530fje. [DOI] [PubMed] [Google Scholar]
- 117.Bircan F. S., Balabanli B., Turkozkan N., Ozan G. Effects of taurine on nitric oxide and 3-nitrotyrosine levels in spleen during endotoxemia. Neurochemical Research. 2011;36(11):1978–1983. doi: 10.1007/s11064-011-0521-3. [DOI] [PubMed] [Google Scholar]
- 118.Park E., Quinn M. R., Wright C. E., Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. Journal of Leukocyte Biology. 1993;54(2):119–124. doi: 10.1002/jlb.54.2.119. [DOI] [PubMed] [Google Scholar]
- 119.Hong J., Bose M., Ju J., et al. Modulation of arachidonic acid metabolism by curcumin and related β-diketone derivatives: effects on cytosolic phospholipase A2, cyclooxygenases and 5-lipoxygenase. Carcinogenesis. 2004;25(9):1671–1679. doi: 10.1093/carcin/bgh165. [DOI] [PubMed] [Google Scholar]
- 120.Chuang D. Y., Simonyi A., Kotzbauer P. T., Gu Z., Sun G. Y. Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. Journal of Neuroinflammation. 2015;12, article 199 doi: 10.1186/s12974-015-0419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brown G. C. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochemical Society Transactions. 2007;35(5):1119–1121. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- 122.Sharma S. K., Al-Hourani B. J., Wuest M., et al. Synthesis and evaluation of fluorobenzoylated di- and tripeptides as inhibitors of cyclooxygenase-2 (COX-2) Bioorganic and Medicinal Chemistry. 2012;20(7):2221–2226. doi: 10.1016/j.bmc.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 123.Ruder E. H., Laiyemo A. O., Graubard B. I., Hollenbeck A. R., Schatzkin A., Cross A. J. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. American Journal of Gastroenterology. 2011;106(7):1340–1350. doi: 10.1038/ajg.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mahipal A., Anderson K. E., Limburg P. J., Folsom A. R. Nonsteroidal anti-inflammatory drugs and subsite-specific colorectal cancer incidence in the Iowa Women's Health Study. Cancer Epidemiology Biomarkers & Prevention. 2006;15(10):1785–1790. doi: 10.1158/1055-9965.epi-05-0674. [DOI] [PubMed] [Google Scholar]
- 125.Smalley W. E., DuBois R. N. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Advances in Pharmacology. 1997;39:1–20. doi: 10.1016/s1054-3589(08)60067-8. [DOI] [PubMed] [Google Scholar]
- 126.Nelson R. L., Dollear T., Freels S., Persky V. The relation of age, race, and gender to the subsite location of colerectal carcinoma. Cancer. 1997;80(2):193–197. doi: 10.1002/(sici)1097-0142(19970715)80:2<193::aid-cncr4>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 127.Dierssen-Sotos T., Gómez-Acebo I., de Pedro M., et al. Use of non-steroidal anti-inflammatory drugs and risk of breast cancer: the Spanish Multi-Case-Control (MCC) study. BMC Cancer. 2016;16, article 660 doi: 10.1186/s12885-016-2692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Harris R. E., Namboodiri K. K., Farrar W. B. Nonsteroidal antiinflammatory drugs and breast cancer. Epidemiology. 1996;7(2):203–205. doi: 10.1097/00001648-199603000-00017. [DOI] [PubMed] [Google Scholar]
- 129.Khuder S. A., Mutgi A. B. Breast cancer and NSAID use: a meta-analysis. British Journal of Cancer. 2001;84(9):1188–1192. doi: 10.1054/bjoc.2000.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Agrawal A., Fentiman I. S. NSAIDs and breast cancer: a possible prevention and treatment strategy. International Journal of Clinical Practice. 2008;62(3):444–449. doi: 10.1111/j.1742-1241.2007.01668.x. [DOI] [PubMed] [Google Scholar]
- 131.Friis S., Thomassen L., Sørensen H. T., et al. Nonsteroidal anti-inflammatory drug use and breast cancer risk: a Danish cohort study. European Journal of Cancer Prevention. 2008;17(2):88–96. doi: 10.1097/cej.0b013e3282b6fd55. [DOI] [PubMed] [Google Scholar]
- 132.Marshall S. F., Bernstein L., Anton-Culver H., et al. Nonsteroidal anti-inflammatory drug use and breast cancer risk by stage and hormone receptor status. Journal of the National Cancer Institute. 2005;97(11):805–812. doi: 10.1093/jnci/dji140. [DOI] [PubMed] [Google Scholar]
- 133.Johannesdottir S. A., Chang E. T., Mehnert F., Schmidt M., Olesen A. B., Saørensen H. T. Nonsteroidal anti-inflammatory drugs and the risk of skin cancer: a population-based case-control study. Cancer. 2012;118(19):4768–4776. doi: 10.1002/cncr.27406. [DOI] [PubMed] [Google Scholar]
- 134.Reinau D., Surber C., Jick S. S., Meier C. R. Nonsteroidal anti-inflammatory drugs and the risk of nonmelanoma skin cancer. International Journal of Cancer. 2015;137(1):144–153. doi: 10.1002/ijc.29357. [DOI] [PubMed] [Google Scholar]
- 135.Farrow D. C., Vaughan T. L., Hansten P. D., et al. Use of aspirin and other nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiology Biomarkers and Prevention. 1998;7(2):97–102. [PubMed] [Google Scholar]
- 136.Wang W. H., Huang J. Q., Zheng G. F., Lam S. K., Karlberg J., Wong B. C. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. CancerSpectrum Knowledge Environment. 2003;95(23):1784–1791. doi: 10.1093/jnci/djg106. [DOI] [PubMed] [Google Scholar]
- 137.Vidal A. C., Howard L. E., Moreira D. M., Castro-Santamaria R., Andriole G. L., Freedland S. J. Aspirin, NSAIDs, and risk of prostate cancer: results from the REDUCE study. Clinical Cancer Research. 2015;21(4):756–762. doi: 10.1158/1078-0432.CCR-14-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Veitonmäki T., Murtola T. J., Määttänen L., et al. Prostate cancer risk and nonsteroidal antiinflammatory drug use in the Finnish prostate cancer screening trial. British Journal of Cancer. 2014;111(7):1421–1431. doi: 10.1038/bjc.2014.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shahedi K., Lindström S., Zheng S. L., et al. Genetic variation in the COX-2 gene and the association with prostate cancer risk. International Journal of Cancer. 2006;119(3):668–672. doi: 10.1002/ijc.21864. [DOI] [PubMed] [Google Scholar]
- 140.Choueiri T. K., Je Y., Cho E. Analgesic use and the risk of kidney cancer: a meta-analysis of epidemiologic studies. International Journal of Cancer. 2014;134(2):384–396. doi: 10.1002/ijc.28093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cho E., Curhan G., Hankinson S. E., et al. Prospective evaluation of analgesic use and risk of renal cell cancer. Archives of Internal Medicine. 2011;171(16):1487–1493. doi: 10.1001/archinternmed.2011.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Petrick J. L., Sahasrabuddhe V. V., Chan A. T., et al. NSAID use and risk of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: the liver cancer pooling project. Cancer Prevention Research. 2015;8(12):1156–1162. doi: 10.1158/1940-6207.capr-15-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sahasrabuddhe V. V., Gunja M. Z., Graubard B. I., et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. Journal of the National Cancer Institute. 2012;104(23):1808–1814. doi: 10.1093/jnci/djs452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bannon F. J., O'Rorke M. A., Murray L. J., et al. Non-steroidal anti-inflammatory drug use and brain tumour risk: a case-control study within the Clinical Practice Research Datalink. Cancer Causes and Control. 2013;24(11):2027–2034. doi: 10.1007/s10552-013-0279-9. [DOI] [PubMed] [Google Scholar]
- 145.Nan H., Hutter C. M., Lin Y., et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA. 2015;313(11):1133–1142. doi: 10.1001/jama.2015.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tanaka K., Ogawa K., Sugamura K., Nakamura M., Takano S., Nagata K. Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. The Journal of Immunology. 2000;164(5):2277–2280. doi: 10.4049/jimmunol.164.5.2277. [DOI] [PubMed] [Google Scholar]
- 147.Jowsey I. R., Thomson A. M., Flanagan J. U., et al. Mammalian class Sigma glutathione S-transferases: catalytic properties and tissue-specific expression of human and rat GSH-dependent prostaglandin D2 synthases. Biochemical Journal. 2001;359(3):507–516. doi: 10.1042/0264-6021:3590507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rigas B., Goldman I. S., Levine L. Altered eicosanoid levels in human colon cancer. The Journal of Laboratory and Clinical Medicine. 1993;122(5):518–523. [PubMed] [Google Scholar]
- 149.Kutchera W., Jones D. A., Matsunami N., et al. Prostaglandin H synthase 2 is expressed abnormally in human colon cancer: evidence for a transcriptional effect. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(10):4816–4820. doi: 10.1073/pnas.93.10.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nakanishi M., Menoret A., Tanaka T., et al. Selective PGE2 suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prevention Research. 2011;4(8):1198–1208. doi: 10.1158/1940-6207.capr-11-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Markosyan N., Chen E. P., Ndong V. N., et al. Deletion of cyclooxygenase 2 in mouse mammary epithelial cells delays breast cancer onset through augmentation of type 1 immune responses in tumors. Carcinogenesis. 2011;32(10):1441–1449. doi: 10.1093/carcin/bgr134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pidgeon G. P., Lysaght J., Krishnamoorthy S., et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer and Metastasis Reviews. 2007;26(3-4):503–524. doi: 10.1007/s10555-007-9098-3. [DOI] [PubMed] [Google Scholar]
- 153.Zhao Y., Wang W., Wang Q., Zhang X., Ye L. Lipid metabolism enzyme 5-LOX and its metabolite LTB4 are capable of activating transcription factor NF-κB in hepatoma cells. Biochemical and Biophysical Research Communications. 2012;418(4):647–651. doi: 10.1016/j.bbrc.2012.01.068. [DOI] [PubMed] [Google Scholar]
- 154.Picado C. Aspirin intolerance and nasal polyposis. Current Allergy and Asthma Reports. 2002;2(6):488–493. doi: 10.1007/s11882-002-0089-8. [DOI] [PubMed] [Google Scholar]
- 155.Lee K. S., Kim S. R., Park H. S., et al. Cysteinyl leukotriene upregulates IL-11 expression in allergic airway disease of mice. Journal of Allergy and Clinical Immunology. 2007;119(1):141–149. doi: 10.1016/j.jaci.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 156.Thompson C., Cloutier A., Bossé Y., et al. Signaling by the cysteinyl-leukotriene receptor 2: involvement in chemokine gene transcription. Journal of Biological Chemistry. 2008;283(4):1974–1984. doi: 10.1074/jbc.m608197200. [DOI] [PubMed] [Google Scholar]
- 157.Raza H., John A., Benedict S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. European Journal of Pharmacology. 2011;668(1-2):15–24. doi: 10.1016/j.ejphar.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 158.Kim K. M., Song J. J., An J. Y., Kwon Y. T., Lee Y. J. Pretreatment of acetylsalicylic acid promotes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by down-regulating BCL-2 gene expression. The Journal of Biological Chemistry. 2005;280(49):41047–41056. doi: 10.1074/jbc.m503713200. [DOI] [PubMed] [Google Scholar]
- 159.Baud V., Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends in Cell Biology. 2001;11(9):372–377. doi: 10.1016/S0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 160.Zhang S. Q., Kovalenko A., Cantarella G., Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12(3):301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 161.Devin A., Cook A., Lin Y., Rodriguez Y., Kelliher M., Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIPmediates IKK activation. Immunity. 2000;12(4):419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 162.Devin A., Lin Y., Yamaoka S., Li Z., Karin M., Liu Z. The α and β subunits of IκB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Molecular and Cellular Biology. 2001;21(12):3986–3994. doi: 10.1128/mcb.21.12.3986-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chadee D. N., Yuasa T., Kyriakis J. M. Direct activation of mitogen-activated protein kinase kinase kinase MEKK1 by the Ste20p homologue GCK and the adapter protein TRAF2. Molecular and Cellular Biology. 2002;22(3):737–749. doi: 10.1128/mcb.22.3.737-749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shan D., Shen K., Zhu J., et al. The polymorphism (Gln261Arg) of 12-lipoxygenase and cancer risk: a meta-analysis. International Journal of Clinical and Experimental Medicine. 2015;8(1):488–495. [PMC free article] [PubMed] [Google Scholar]
- 165.Kyriakopoulos A., Logotheti S., Marcinkiewicz J., Nagl M. N-chlorotaurine and N-bromotaurine combination regimen for the cure of valacyclovir-unresponsive herpes zoster comorbidity in a multiple sclerosis patient. International Journal of Medical and Pharmaceutical Case Reports. 2016;7(2):1–6. doi: 10.9734/ijmpcr/2016/25476. [DOI] [Google Scholar]
- 166.Dempke W., Rie C., Grothey A., Schmoll H.-J. Cyclooxygenase-2: a novel target for cancer chemotherapy? Journal of Cancer Research and Clinical Oncology. 2001;127(7):411–417. doi: 10.1007/s004320000225. [DOI] [PubMed] [Google Scholar]
- 167.Scheiman J. M., Hindley C. E. Strategies to optimize treatment with NSAIDs in patients at risk for gastrointestinal and cardiovascular adverse events. Clinical Therapeutics. 2010;32(4):667–677. doi: 10.1016/j.clinthera.2010.04.009. [DOI] [PubMed] [Google Scholar]