

Abstract
The relationship between stress challenges and adverse health outcomes, particularly for the development of affective disorders, is now well established. The highly conserved neuroimmune mechanisms through which responses to stressors are transcribed into effects on males and females have recently garnered much attention from researchers and clinicians alike. The use of animal models, from mice to guinea pigs to primates, has greatly increased our understanding of these mechanisms on the molecular, cellular, and behavioral levels, and research in humans has identified particular brain regions and connections of interest, as well as associations between stress-induced inflammation and psychiatric disorders. This review brings together findings from multiple species in order to better understand how the mechanisms of the neuroimmune response to stress contribute to stress-related psychopathologies, such as major depressive disorder, schizophrenia, and bipolar disorder.
Keywords: depression, guinea pig, inflammatory mediator, microglia, neuroimmune response, nonhuman primate, sickness, stress reactivity
Abstract
Actualmente está bien establecida la relación entre los efectos del estrés y los resultados adversos sobre la salud, especialmente para el desarrollo de los trastornos afectivos. Los mecanismos neuroinmunes, muy bien conservados entre las especies, y a través de los cuales las respuestas a los estresores se traducen en efectos sobre hombres y mujeres, han generado recientemente gran atención tanto para los investigadores como para los clínicos. El empleo de modelos animales, desde ratones y cobayos, hasta primates, ha mejorado enormemente nuestra comprensión acerca de estos mecanismos a nivel molecular, celular y conductual. La investigación en humanos ha identificado regiones y conexiones cerebrales de interés, como también asociaciones entre la inflamación producida por el estrés y los trastornos psiquíatricos. Esta revisión reúne hallazgos en múltiples especies para una mejor comprensión de cómo contribuyen los mecanismos de la respuesta neuroinmune a las psicopatologías relacionadas con el estrés como el trastorno depresivo mayor, la esquizofrenia y el trastorno bipolar.
Abstract
Les effets nocifs du stress sur la santé sont maintenant bien connus, en particulier en ce qui concerne le développement des troubles affectifs. Les mécanismes neuro-immunitaires très bien conservés parmi les espèces et par lesquels les réponses aux facteurs de stress se répercutent sur les hommes et les femmes ont récemment suscité une attention particulière des chercheurs et des cliniciens. L'utilisation de modelès animaux, de la souris au cobaye et jusqu'aux primates, a considérablement amélioré notre compréhension de ces mécanismes aux niveaux moléculaire, cellulaire et comportemental. La recherche chez l'homme a permis d'identifier des régions cérébrales particulières et des connexions intéressantes, ainsi que des associations entre l'inflammation induite par le stress et les troubles psychiatriques. Cet article fait la synthèse des données de nombreuses espèces afin de mieux comprendre comment les mécanismes de la réponse neuro-immunitaire au stress contribuent aux psychopathologies liées au stress, comme les troubles dépressifs caractérisés, la schizophrénie et les troubles bipolaires.
Introduction
Stressful life experiences have the amazing ability to synchronize organism-wide physiological responses toward the common goal of surviving threat. In this way, the major stress-responsive systems are often regarded by evolutionary biologists as essential adaptive mechanisms that ultimately promote survival. However, as stress challenges become greater in magnitude or more protracted in length, the toll on the host organism can be quite severe, and the adaptive value of mobilizing physiological processes toward survival exacts a long-term cost. This essential framework for understanding the relationship between stress challenges and their impact on organism health was articulated by Hans Selye, who noted that nearly all stress challenges were followed by a “syndrome of being sick.”1 Unbeknownst to Selye, this prescient view of the relationship between stressful experiences and their sickness-like outcomes, in today's vernacular, implies that immune processes are probably essential mediators of the link between stress and adverse health outcomes. With this in mind, the present review takes the position that neuroimmune mechanisms of stress are highly conserved across species and serve as a likely mechanistic bridge between classic stress-responsive systems and stress-related pathologies (Figure 1).
Figure 1. A multispecies approach toward understanding neuroimmune mechanisms of stress and their relation to human affective disorders. The neuroimmune consequences of stress are highly conserved across mammalian species, yet vary within species as a function of sex, age, and past history of stress. Mouse and rat models are commonly used to examine basic molecular and cellular components of the stress response. The use of guinea pigs is advantageous as they are born highly precocious and form a strong attachment to their mother, thereby creating a highly tractable model of maternal separation during early life. Findings from guinea pigs can be applied to generate hypotheses to test on nonhuman primates, such as rhesus macaques, which will then inform basic research and clinical applications in humans that can guide therapeutic approaches. Rat photo courtesy of Dr Lisa M. Savage; guinea pig photo was contributed by Dr Michael B. Hennessy; photo showing troop of rhesus macaques was courtesy of the California National Primate Research Center; human photo courtesy of Anastacia Kudinova.

To support this position, we (i) provide a succinct overview of neuroimmune mechanisms of stress that have been largely established in preclinical (mouse and rat) models; (ii) extend these findings to other mammalian systems (guinea pigs and nonhuman primates); and (iii) translate these findings to the human condition. The intent here is not to provide a comprehensive summary, but rather to succinctly link together commonalities across models/species that argue in favor of viewing neuroimmune consequences of stress as highly conserved across taxonomic orders. At the same time (and perhaps ironically), we argue that key features of the stress challenge (nature, intensity, and duration of threat), as well as individual subject characteristics (sex, age, stress history), will be critical for delineating individualized therapeutic approaches for the future. In doing so, we hope to provide guidance on significant gaps in our knowledge that remain to be filled and some possible pathways for the future. Although we focus largely on the link between stress and negative affective states, such as depression and anxiety, the same processes are probably involved in risk for other forms of psychopathology as well.
The first step in understanding neuroimmune mechanisms of stress and their eventual role in stress-related health outcomes is to define what is meant by “neuroimmune mechanisms.” Historically, most studies have focused on intercellular signaling factors, such as cytokines, chemokines, and the many species of prostaglandins, whose primary roles were initially defined within an immunologically relevant context. Demonstration that such intercellular mediators are often invoked by stress challenges in which there is no apparent tissue damage or other immunological insult suggests a natural role for these agents in mediating stress outcomes. However, the identification and interpretation of such findings can be quite difficult because there are literally dozens of inflammatory signaling factors that can be simultaneously regulated; cytokines often have redundant biological action with one another; and nearly all cytokines have pleiotropic functions. Even more troublesome is the fact that actions of cytokines can manifest as generally pro- or anti-inflammatory, depending upon the presence of other signaling molecules and the context in which they are induced.
In addition to these conceptual issues surrounding the identification and interpretation of cytokine action, there are also technical considerations surrounding the assessments of cytokines and other immune mediators. Because immune-related factors are expressed at appreciably low quantities in the normal central nervous system (CNS), the techniques to detect and measure such cytokines, particularly in early studies, have often been inadequate to yield precise results. For instance, many studies have utilized gross tissue dissections (or micropunches) that aggregate cytokine measures over large anatomical areas and across cell types within the dissected tissue, which can significantly influence outcomes and interpretation.2 A second major technical consideration lies in the use of quantification procedures that allow for signal amplification (reverse transcription polymerase chain reaction) versus those that do not (in situ hybridization), and the more general juxtaposition of studies that measure protein versus messenger RNA (mRNA). The availability of well-validated antibodies and the use of appropriate immunohistochemical controls for target specificity and cell type have also been a historical problem.3 Finally, deploying standard neuroscientific approaches (cannulation, lesions, etc) for manipulation of inflammatory signaling in the CNS can significantly alter sensitivity to later stress challenges.4 Thus, one must carefully consider the technical approach employed by studies as a key constraint in interpreting experimental outcomes.5-7
With that said, several inflammatory mediators have emerged as highly stress-responsive, and their physiological impact has been delineated clearly. For instance, numerous studies have indicated that interleukin 1β ip is rapidly increased in key limbic structures (paraventricular nucleus [PVN]; amygdala) in response to stress challenges that involve application of an aversive/noxious stimulus such as footshock,7-10 but not in response to social stress challenges.11 In contrast, social stressors appear to increase release of another proinflammatory cytokine, IL-6, in both plasma and brain of mice, thereby contributing to stress phenotypes.12,13 Importantly, recent evidence suggests that early exposure to maternal separation in male rats may change cytokine reactivity to later social stress challenges incurred during adulthood.14 Moving beyond some of the classic cytokines (such as IL-1β, IL-6, tumor necrosis factor a [TNF-α]), recent evidence suggests that chemokines are also dynamically altered by stress challenges.8,15,16 As a separate class of immune-related molecules, these stress-induced chemokines probably play a key role in structural changes to the cytoskeleton of microglia that allows for the motility and retraction of processes,17,18 in recruitment of other intrinsic microglia to the site of release,19,20 and in potentially inducing passage of monocytes across the blood-brain barrier.21,22 Indeed, genetic ablation of the chemokine CX3CL1 (fractalkine) was recently shown to prevent microglial activation associated with chronic unpredictable stress.23 In addition, prostaglandins have emerged as rapid, stress-sensitive inflammatory mediators, particularly within the cortex.24 Given the role that prostaglandins play as final common mediators of the febrile response, prostaglandins are also likely mediators of stress-induced fever responses.25 Thus, a multitude of inflammatory signaling families are mobilized by stressful experiences and significantly impact CNS function.
A growing number of studies have also examined cellular manifestations of neuroinflammation, with microglia emerging as highly reactive to stress challenges. Early studies examining microglial activation established that administration of minocycline, a putative microglial inhibitor, blocked the induction of central, but not peripheral, IL-1β by footshock.8,26 Other studies have shown that chronic stress exposure drives proliferation of microglia27 and increases both the density and activational state of microglia within certain brain structures.23,28-30 Stress challenges also alter the expression of receptors expressed on the cell surface of microglia that are both indicative of microglial activation state and positively coupled to cytokine expression within microglia.8,31 Indeed, dynamic alterations in cell surface receptors on microglia probably accounts, at least in part, for certain priming/sensitization effects incurred by stress, including more rapid cytokine responses produced by later injection of lipopolysaccharide (LPS).32,33 Together, these findings provide multiple avenues by which stress challenges impact microglia and strongly support the notion that microglial (re)activity may be a key culprit in mediating stress-dependent changes in behavior,34 cognition,35-37 and potentially multiple forms of psychopathology.38,39
The importance of inflammatory phenotype to behavioral and mood-related changes incurred by stress has been strengthened in recent years through the use of adoptive transfer studies. These clever studies show that repeated exposure to various forms of repeated social stress in mice increases microglial activation and expression/release of cytokines. Intriguingly, when circulating monocytes are then extracted from previously stressed mice and transferred to other mice in which the existing monocytes/lymphocytes have been depleted, the recipient mice display behavioral and mood tendencies that reflect those of the original stressed host.40-42 These findings, combined with recent evidence showing that circulating monocytes may transit into the CNS as a result of stressor exposure and actively influence mood state,15,43 add a new dimension to our understanding of bidirectional communication between the brain and the peripheral immune system.44 Though this provocative area of research is still in its infancy, the emerging body of evidence provides, for the first time, causal evidence for the notion that non-neuronal cells (microglia) may be responsible for encoding stress-dependent changes in mood regulation, particularly for negative affective states like depression and anxiety.45,46
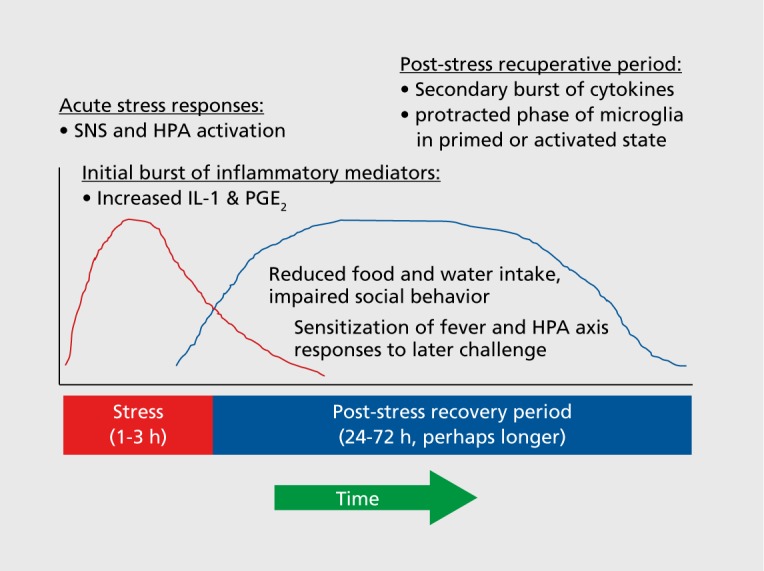
A key issue that must be considered in understanding the relation between stress challenges and their neuroimmune consequences is timing. In laboratory models, acute stress challenges or the individual bouts of daily, intermittent chronic stress procedures are typically elaborated across a 30-min to 3-hour window of stress exposure (Figure 2). The central questions requiring consideration here are (i) How rapidly are neuroimmune signaling agents induced? (ii) How long do such changes persist? (iii) Are there downstream neuroimmune effects that cascade or coalesce into subsequent neuroimmune alterations? and (iv) What are the functional outcomes that can be tied to individual components of such neuroimmune cascades? Although the discussion above did not address these issues by indicating the time point at which individual changes were observed, recent studies examining the time course for neuroimmune responses provide some guidance. For instance, induction of IL-1β gene expression is prevalent and significant within 30 min of stress onset, 6,7 with IL-1β protein responses peaking shortly thereafter (60 to 120 minutes after stress onset).47 In contrast, most studies examining microglial morphology, proliferation, or activational state tend to show changes that are prevalent 24 hours or more after stress termination, 28,29 by which point many cytokine changes have largely resolved. Though perhaps correlational at this stage, one interpretation of the timing of these events is that the expression and release of intercellular mediators (cytokines, chemokines, prostaglandins) have a nearly immediate impact upon behavioral changes observed in the post-stress recuperative period, while at the same time eliciting parallel changes in other aspects of neuroimmune function that persist for days to weeks after cessation of stress. Thus, it will be critical for future studies to conceptually discriminate between those neuroimmune changes that represent acute, activational responses to an individual bout of stress, versus those neuroimmune changes that reflect the aggregate influence of a repeated history of stress across a more protracted period of time.
Figure 2. Stress-related neuroinflammation mediates the post-stress recuperative period. Acute stress induces activation of the sympathetic nervous system and HPA axis, leading to release of inflammatory factors, such as cytokines (eg, IL-1), chemokines (eg, CCL2/MCP1), and prostaglandins (eg, PGE2). These factors regulate various features of the post-stress recuperative period, in which the subject displays reduced food and water intake, impaired social behavior, and often sickness-like responses. This constellation of behavioral changes probably represents a recuperative motivational state that promotes recovery after intensely stressful experiences. CCL2/MCP1, chemokine (C-C motif) ligand 2, also referred to as monocyte chemoattractant protein 1; HPA, hypothalamic-pituitary-adrenal; IL, interleukin; PGE2, prostaglandin; SNS, sympathetic nervous system.
Mechanisms underlying stress-related neuroinflammation
The multitude of inflammatory signaling pathways that appear to be activated by stress challenges raises questions regarding the mechanisms that control such neuroimmune changes. Here, we suggest that multiple upstream signaling pathways converge upon regulatory elements associated with neuroimmune signaling, thereby propagating various features of the neuroimmune response to stress. For ease of description, we will describe these pathways in three functional clusters (Figure 3), with full recognition that these pathways are not independent of one another. The first of these mechanisms falls into the general category of rapid neural signals. For instance, norepinephrine release during stress-associated activation of the sympathetic nervous system has long been regarded as a primary driver of neuroimmune consequences of stress.6,26 A wide range of pharmacological studies lend support to this notion, showing that administration of β-adrenergic blockers (eg, propranolol) block the induction of IL-1β incurred by stress, whereas β-adrenergic agonists (eg, isoproterenol) potently induce IL-1β expression.8,26,48 Consistent with this, previous treatment with desipramine (a norepinephrine-reuptake inhibitor) potentiated both basal and stress-induced IL-1β expression.26 Gross neurotoxic lesions of norepinephrine-containing cells via intracerebroventricular injection of the selective neurotoxin N-(2-chloroethyl)-N-ethyl-2 bromobenzylamine (DSP-4) blocked the induction of IL-1β in the hippocampus,48 whereas more focal lesions of the ventral noradrenergic bundle produced modest effects on IL-1 induction by stress in the PVN.6 Together, these studies underscore the importance of norepinephrine signaling as a key driver of neuroimmune consequences of stress. However, other studies have clearly tied microglial proliferative responses incurred by chronic stress to glutamate signaling, since chronic administration of MK801 effectively blocked this proliferative response,27 and N-Methyl-D-aspartate (NMDA)-receptor activation is positively coupled to inflammatory signaling pathways.49,50 Interestingly, considering that adenosine triphosphate (ATP) is co-packaged and released with many classic neurotransmitters (including norepinephrine, in particular), other groups have recently posited purinergic signaling as a putative mechanism controlling functional release of IL-1β (and potentially other neuroimmune factors) in response to stress.2,51,52
Figure 3. Signaling molecules involved in regulating stress-related neuroinflammation. Neuroimmune signaling molecules that control inflammatory response can be categorized into three types, as follows: (i) host-derived danger, damage, and disease signals—including cytokines, chemokines, and damage-associated molecular patterns (DAMPS)—that acutely activate inflammatory signaling pathways; (ii) rapid neural signals, such as norepinephrine and glutamate, that rapidly drive the stress response and cytokine production throughout the body; and (iii) neuroendocrine signals, particularly corticosteroids and progesterone, which may serve to constrain the inflammatory process and dampen production of inflammatory factors. AP-1 , activator protein 1 ; ATP, adenosine triphosphate; AR, adrenergic receptor; cAMP, cyclic adenosine monophosphate; CRE, cAMP-response element; CREB, cAMP-response-element binding protein; DAMPs, damage-associated molecular patterns; ERK, extracellular-signal-regulated kinase; GR, glucocorticoid receptor; HMGB1, high-mobility group box 1 ; HSPs, heat shock proteins; ICE, interleukin-1 -converting enzyme; IL, interleukin; MAPK, mitogen-activated protein kinase; MyD88, myeloid differentiation primary response gene 88; NFkB, nuclear factor kB; nGRE, negative glucocorticoid response element; NMDAR, N-methyl-d-aspartate receptor; PKA, protein kinase A; PKC, protein kinase C; TNF, tumor necrosis factor; TLR, Toll-like receptor; TRAF6, TNF receptor-associated factor 6.

Another emerging class of neuroimmune intermediaries falls into the category of what we describe as danger, damage, and disease signals (Figure 3).53 The basic premise is that our innate immune defense network is constructed to respond to general molecular motifs associated with pathogens or self-injury. DAMPs—damage-associated molecular patterns—are host-derived molecules that are released in response to tissue trauma or other insults and are thus capable of activating inflammatory signaling pathways. A variety of studies have convincingly demonstrated several families of DAMPs that are activated by exposure to intense stress challenges, including heat shock proteins (HSP72, in particular)54 and high-mobility group box 1 (HMGB1).55,56 When released, these DAMPs interact with cognate receptors that are positively coupled to inflammatory signaling pathways. This family of receptors is often referred to as pathogen recognition receptors (PRRs) because they recognize and bind to general molecular motifs associated with classes of bacteria and viruses, as well as to certain DAMPs that may be activated by stress and/or damage. Yet another intriguing twist is the recent notion that bacteria from the gut or other areas of the microbiome may be stress-sensitive, and perhaps even mobile in response to stress challenges, thereby incurring a mild form of endogenous infection.57-59 Thus, the DAMP->PRR pathways have emerged as important alternative pathways by which neuroimmune consequences of stress are primed and/or activated.60
The propagation of neuroimmune signaling cascades in response to stress is also constrained by other features of physiology. Perhaps the most logical of these counter-regulatory elements is the release of glucocorticoid (GC) hormone (corticosterone in rat and mouse; cortisol in guinea pigs and primates) as a result of hypothalamic-pituitary-adrenal-axis (HPA) activation that accompanies stress exposure. The anti-inflammatory action of GCs has been established for decades, with great progress being made in recent years in the cis- and trans regulatory elements that give rise to anti-inflammatory effects of GCs.61,62 Within stressful contexts, this was particularly evident in early studies where induction of central IL-1β was massively potentiated in adrenalectomized subjects, and normalized by replacement of corticosterone.10,47 Similar effects have been observed after pretreatment with metyrapone (a GC synthesis inhibitor).8,26 However, there appear to be certain instances in which GCs prime or modestly stimulate inflammation, a notion which is now challenging some of the central dogma on the relation between GC hormone signaling and inflammation.63 These latter, proinfiammatory-like effects of GCs probably represent a small proportion of the overall GC effects on inflammation and relate to very specific parameters of low GC dose, the timing of GC hormone exposure, and the specific tissue in which the GCs act.64-66 Gene expression profiling studies predict such effects,53,67 and so it is quite interesting to see these effects bearing out on functional responsiveness within the neuroimmune system. Furthermore, a recent study found that microglial activation was more profound after several weeks of repeated restraint, but not after an equivalent exposure to chronic variable stress.29 Although the reason for this difference has yet to be determined, the authors propose that HPA-axis habituation, and thus the lack of circulating GC hormone that accompanies repeated restraint, led to a disinhibition of stress-dependent microglial activation. Additional studies showed that pharmacological activation of the HPA axis (in the absence of an overt stress challenge) via corticotropin-releasing hormone or corticotropin injection led to an expected inhibition of cytokine expression in the PVN, though paradoxically, both peptides stimulated inflammatory signaling at the level of the adrenal gland.7 Yet another added complexity is that GCs are not the only hormone system engaged by stress exposure. Recent studies demonstrated that progesterone is also rapidly released in response to stress, even in male rats. This makes sense as progesterone is a precursor molecule in the biosynthesis of corticosterone, yet the possibility that progesterone may contribute to the anti-inflammatory influence of GCs has not been systematically tested.68
One major area of neglect in the literature has been the elaboration of sex differences in neuroimmune responses to stress. In a recent set of studies from our lab, we examined fluctuations in the IL-1β response to stress as a function of estrous stage in female rats.69 These studies reported relatively uniform IL-1β induction at all phases of the cycle except during metestrus—where basal IL-1β was moderately increased, and the IL-1β response to stress appeared to be blunted in the PVN. However, this effect requires further study to generalize the effects from mRNA to protein, to expand the analysis from the PVN to other stress-sensitive sites, to incorporate additional neuroimmune measures, and to replicate what was originally a slightly underpowered effect. Nevertheless, subsequent studies demonstrated that stress-induced IL-1β was potentiated in ovariectomized rats and restored by estradiol-replacement injections. However, because endogenous progesterone evinced a stress-induced surge, we cannot at this time attribute the “rescue” effect of estradiol replacement exclusively- to estradiol (versus an estradiol plus progesterone effect). What we do know, however, is that progesterone replacement alone did not recapitulate the effect of estrogen replacement. Nevertheless, few studies, if any, have examined how sex-specific gonadal steroids differentially affect sex differences in neuroimmune responses to stress in rodents. Thus, future studies need to give serious consideration to potential additive, cooperative, and synergistic effects of steroid hormones on stress-dependent changes in neuroimmune function.53
Social separation and inflammation: guinea pig and monkey models
Although the vast majority of studies on stress and inflammation have focused on rats and mice (as indicated in the above discussion), investigators have sometimes turned to alternative rodent species.70-72 Guinea pigs in particular offer a number of advantages, especially in developmental studies. Unlike rats and mice, guinea pigs are born in an advanced state of development. Brain regions are well defined, and the skull is calcified even in the first few days, which facilitates procedures such as implanting indwelling cannula and immunohistochemistry. Thermoregulation matures rapidly,73 so LPS- or stress-induced fever can be investigated early in preweaning pups. But for studies of stress-induced inflammatory responses during infancy, perhaps the greatest advantage stems indirectly from the fact that the pups are precocial. Guinea pigs are capable of coordinated behavior, including locomotion and ingestion, from shortly after birth.74 Maternal behavior is extremely passive. Licking of the pups, the primary active maternal behavior, is infrequent, particularly after about a week of age.75 There is no retrieval, no nest, and mothers simply accommodate nursing attempts initiated by the pups.76 As a result, mother-young proximity is maintained almost exclusively by the strong attraction or attachment that pups display for the mother. Indeed, pups exhibit evidence of the classic markers of attachment commonly used in primate studies—namely, approach, recognition, and preference for the attachment object; use of the attachment object as a secure base for exploration; and distressful responses to separation.77-79
Maternal separation in guinea pigs thus affords a compelling translational model for the effects of attachment disruption, a class of early stressors frequently linked to later psychopathology in humans as well as altered inflammatory activity.38,80,81
There are many similarities in the responses to maternal separation shown by young guinea pigs and infant monkeys.82 These include, for instance, increased HPA and sympathetic activity83,84 and central catecholamine turnover associated with stress.85,86 As is seen in some species of macaques,87 as well as in human infants,88 guinea pig pups also display a two-stage, active/passive behavioral response to separation. When placed alone in a novel cage, pups initially show “protest” by vocalizing in an apparent attempt to re-establish contact with the mother. But after about an hour of separation, the pups typically quiet and adopt a characteristic crouched stance, with closed eyes, extensive piloerection, and apparent disinterest in their surroundings (Figure 1).89 Neither the active nor passive stages are observed if the mother is placed in the novel cage with the pup,90 establishing that the mother's absence, and not just the novelty of the environment, is responsible for these responses. The passive, second stage of separation in guinea pig infants is reminiscent of the “despair” shown by monkeys separated for much longer periods91 and even the “anaclitic depression” that Spitz (1946) described in institutionalized children.
Although the nature of the passive stage of separated guinea pig pups can suggest depression to a comparative psychologist, it may just as readily suggest cytokine-induced sickness behavior to the psychoneuroimmunologist. Each of the components of the response—crouch, eye-close, piloerection—is characteristic of sick animals.92 Moreover, direct stimulation of sickness with LPS results in the same behavioral constellation of these three behaviors.90 It appears then that the stressor of separation in a novel environment initiates an inflammatory reaction that mediates the behavioral response. This claim is bolstered by findings that the separation procedure elicits a transitory fever93 and increased expression of the proinflammatory cytokine TNF-α in spleen,94 two physiological indicators of a sickness response. Behaviors of the second stage of separation can also be reduced by a variety of anti-inflammatory agents, including α-melanocyte-stimulating hormone (α-MSH),95 indomethacin,96 IL-10,97 and naproxen.98 Although other systems are also probably involved, it is clear that inflammatory mechanisms play a fundamental role in the depressive-like response of separated guinea pig pups. In the adult human literature, there is now overwhelming evidence for involvement of inflammation in depressive illness (as described below).99 The guinea pig results suggest that the particular form of depressive response shown by separated nonhuman—and perhaps human—primate infants may also be mediated, at least in part, by inflammatory factors.
Most current research on attachment disruption and depression in humans focuses not on immediate effects, but rather on long-term vulnerability for developing depression engendered by early abuse, neglect, or prolonged separation. Whereas increased risk for later depressive illness has long been suspected for infants exposed to such forms of maltreatment,100 recent research has solidified this link and begun to provide glimpses of potential neurobiological mechanisms. The basic premise common to most hypotheses is one of sensitization. That is, the early stress of attachment disruption is seen as sensitizing underlying stress-responsive machinery so that, in later life, exposure to stressors with which other individuals would be able to effectively cope elicit disproportional, protracted, and inadequately regulated stress responses that lead to, or constitute the underlying basis of, the depressive episode.101 These “diathesis-stress” or “two-hit” models have most often emphasized effects on elements of the HPA axis and its control, including increases in central corticotropin-releasing factor secretion, amygdala activity, and GC resistance, as well as a reduction in HPA inhibition by the frontal cortex.80,101-103
However, recently, there also has been a proliferation of findings implicating inflammatory mechanisms in the sensitization process. Attachment disruption and other forms of early stress have repeatedly been found to be associated with increased markers of inflammation at later ages.104-109 From an evolutionary perspective, the increased inflammation may represent a remnant of an ontogenetic adaptation originating from a time when persistent early-life stress was predictive of a hazardous adult environment, in which injury was common and a robust innate immune system was adaptive.110 The process by which early stress enhances later inflammation remains unclear but may involve sensitization of resident microglia or increased transport of peripheral monocytes to the CNS,111,112 resulting in heightened central release of inflammatory mediators. The augmentation of inflammatory processes, in turn, may be driven by activation of, and alterations in, other stress-responsive systems, such as increased resistance to the suppressive action of GCs and elevated sympathetic activity.38,107,113
Results of guinea pig studies implicate inflammatory factors in long-term effects of early stress. Repeated separation increases, ie, sensitizes, both the depressive-like behavior and febrile response to later separations during both the preweaning period and beyond. Moreover, administration of the cyclooxygenase (COX)-inhibitor naproxen before the initial separation suppressed the sensitization response not only to the initial separation experience but also to separations that followed 1 and 10 days later.98 The sensitization that occurs appears to be related to some broader, depressive-like state rather than just the separation response, as previously separated guinea pigs also showed more immobility in the forced swim test, a measure and paradigm that is selectively sensitive to antidepressant medications.
In these studies, as is the case in the field more generally, sex differences have yet to be a major focal point. Although males and females have typically both been included in the guinea pig work, it has been in numbers too small to sufficiently power examination of male-female differences. However, as rodent models for the role of inflammation in stress-related disease are nowbecoming established, it is imperative that male-female differences be taken into account. This is particularly important for disorders in which sex differences are profound, such as major depression, for which women are about twice as likely as males to be afflicted.
Social separation in adult macaques
The above results argue that the guinea pig model has strong internal validity in that there is good evidence that attachment disruption in the form of maternal separation results in depressive-like behavior that sensitizes with repeated separation. The evidence also indicates that the sensitization involves inflammatory processes and reflects changes in an underlying state that is manifested in more than one depressive-like response. The external validity, ie, the generality of the results to humans, would, however, be bolstered if we could demonstrate that a similar experimental procedure produced similar results in a primate. Indeed, the guinea pig work was always intended as a complement to primate research; that is, as a way of doing the investigative work necessary to ultimately generate hypotheses that might then be tested in primate models. The challenge has been how this might be accomplished without proposing to repeat the prolonged separation of monkey infants from their mothers that was common in experiments of the 1950s and 1960s.
A possible solution arose from observations of the Behavioral Management Unit and others at the California National Primate Research Center. It was noted that when adult monkeys were brought from large outdoor social groups (Figure 1) to restricted indoor housing, as is commonplace for the beginning of experiments or veterinary care, a small proportion exhibited a hunched posture with apparent disinterest in their surroundings, a response that both is widely regarded as indicating a depressive-like reaction in macaques114 and which mimics the reaction observed in separated monkey infants. Furthermore, when recordings of adult male monkeys introduced to the restricted indoor housing with no human observer in the room were analyzed, an even larger percentage of animals were found to exhibit the depressive-like hunched posture.115 These findings suggest that presence of a human may evoke a defensive reaction incompatible with the depressive-like response. These findings are compatible with interpretation of these depressive-like consequences as a manifestation of sickness, since expression of sickness behaviors are easily perturbed in the face of threat.116 From a modeling standpoint, however, the observations suggested that the relatively simple procedure of transferring adult monkeys from outdoor social groups to indoor housing may provide a means of evaluating the general relevance of the guinea pig results for nonhuman primates.
Therefore a new model was developed in which adult male rhesus macaques were brought from half-acre outdoor social groups to standard indoor housing, either alone or together with an affiliative juvenile partner for 8 days on two occasions at an approximate 2-week interval. Behavior was filmed without a human observer present and blood samples were taken for cytokine analysis at the end of each 8-day period of indoor housing, as well as when the males were residing in the outdoor field cages. During the first separation, all monkeys of both groups exhibited the hunched posture, with an average of about a third of all observation time spent in this posture.117 During the second separation, time spent in the hunched posture increased (ie, sensitized), but only for those males housed alone. The increase in time spent in the hunched posture was accompanied by a decline in activity and environmental exploration.
There also were several effects on cytokine measures. The monkeys housed alone showed a relative decline from the first to the second period of indoor housing in LPS-induced expression of the anti-inflammatory cytokine IL-10, whereas those monkeys housed with a partner showed a relative increase. Furthermore, regardless of whether monkeys were alone or with a partner, indoor housing reduced sensitivity- of the proinflammatory cytokines IL-1 and TNF- to the suppressive action of GCs. It should be noted that both of these measures—response to LPS stimulation and GC resistance—were chosen because they appear responsive to early-life stress in humans.107 Finally, evidence for a coupling of the behavioral and cytokine findings was also observed. For monkeys tested alone, a large and significant positive correlation was observed between the number of seconds spent in the hunched posture during each period indoors and circulating levels of each of the three cytokines measured. Although many particulars of the experimental design and measures in the guinea pig and monkey studies differed, the broad similarity of results suggests some cross-species commonality or conservation of basic relations between social separation, inflammatory activity, and depressive-like behavior and its sensitization. The findings also suggest relatively simple rodent and monkey models that might be used to continue to disentangle the way in which earlylife stress and inflammation contribute to depressive illness and other negative affective states.
Neuroimmune response to stress: human literature
The key question, of course, is how well these animal models translate to humans and help us better understand the human stress response and risk for psychopathology. Psychosocial stress is a well-established risk factor for the development of various forms of psycho-pathology in humans, including major depressive disorder (MDD), posttraumatic stress disorder (PTSD), and other anxiety disorders.118-121 Similar to findings from animal models of stress, evidence from human studies shows that exposure to stressors provokes neurochemical changes, including changes in levels of inflammation, that are frequently associated with psychopathology, and which may help to explain the mechanisms by which stress increases risk for psychiatric disorders, including MDD.38,122 Neuroimmune effects of exposure to stressful events can persist beyond the immediate impact and potentiate an individual's response to future stressors, thus increasing risk for future psychopathology.105,107,123 Indeed, among individuals with MDD, those who experienced childhood adversity had higher circulating levels of C-reactive protein (CRP) than individuals with no history of childhood trauma.124,125 Additionally, adults who experienced childhood adversity exhibited higher levels of circulating IL-6 levels in response to an acute stressor.105 Finally, there is preliminary evidence that a haplotype in IL-33 moderated the link between women's history of childhood abuse and their risk for depression in adulthood.126
Across a variety of stressors, ranging from low socioeconomic status to traumatic events, mammalian immune cells show an immediate conserved transcriptional response to adversity, which involves increased expression of inflammatory genes and decreases in the genes underlying antiviral responses.113,127-131 Interestingly the inflammatory component of the leukocyte conserved transcriptional response to adversity may be driven by stress-induced upregulation of myelopoiesis and could contribute to development of negative health outcomes associated with adverse social conditions, including psychiatric disorders.132 Overall, one of the pathways for the effects of exposure to stressors on psychopathology risk seems to be an increase in immediate and long-term expression of immune -related genes, outcomes that appear to recapitulate effects observed in preclinical (animal) models. Immune-related transcripts involved in the cellular stress response have been shown to be upregulated in the prefrontal cortex (PFC) of individuals with schizophrenia, indicating that these genes may play an important role in chronic psychopathology. Importantly, these observed transcriptome changes were not a result of acute immune system activation, as there were no differences in markers of acute inflammation or responses to infections between individuals with schizophrenia and controls.133 Similarly, in the hippocampus, general upregulation of inflammatory transcripts was found in patients with MDD, bipolar disorder, and schizophrenia, though the authors reported no overlap in specific genes across disorders.134 This suggests that there are common immune-related gene changes in psychiatric disorders, but that there are also changes in expression that are differentially regulated between diseases. For example, circulating cytokines and upregulation of immune-related genes have been found to occur in patients undergoing a first episode of psychosis, whereas those with comorbid depression displayed a unique expression profile, suggesting separable transcriptional phenotypes.135 Additionally, increased mRNA levels of chemokine (C-C motif) ligand 24 (CCL24) were found in circulating leukocytes among participants with MDD compared with individuals with bipolar personality disorder and healthy controls. In contrast, C-C chemokine receptor type 6 (CCR6) was expressed consistently less among MDD patients than in controls.136 These immune targets may represent easily testable biomarkers of disease. Another example can be found in common variants in long-range enhancer elements that modulate transcriptional response to activation of GC receptors in human blood cells, thereby increasing the risk for later psychopathology, including MDD and schizophrenia.137 Moreover, those functional genetic variants were associated with overgeneralized amygdala reactivity, suggesting that individual differences in the leukocyte's immediate transcriptional reactivity to stress may influence an individual's neurophysiological response to stress.
Epigenetic alterations in genes related to immune function are one of the plausible mechanisms underlying the lasting neuroimmune effects of stress exposure and increased risk for psychopathology. One of the first epigenome-wide association studies (EWAS) that examined DNA methylation among participants with a history of MDD identified tryptophan metabolism-related genes as one of the top three functional clusters in individuals with no history of MDD,138 one of which was a cytokine-induced reduction in tryptophan, the primary serotonin precursor often implicated in MDD.139 There is also evidence that methylation levels of the IL-6 and CRP genes are inversely related to circulating levels of IL-6 and CRP among individuals with a history of MDD.138 Similarly, immune-related genes were shown to be over-represented among unmethylated genes among individuals with PTSD.138,140 Furthermore, hypomethylation of immune-related genes among PTSD-affected individuals was linked to increased peripheral levels of inflammatory cytokines, which were, in turn, related to history of childhood abuse and life stress.141 Overall, these findings suggest that exposure to stressors, perhaps particularly early-life stress, could result in immune-associated epigenetic changes that increase an individual's susceptibility to psychiatric disorders.
Complementing these findings, researchers have recently begun exploring neural mechanisms underlying immune activation after stress exposure in humans via structural and functional imaging. For example, a functional magnetic resonance imaging study that examined changes in mood and neural activity after an in vivo immune challenge found that immune-induced mood decline was associated with increased activity in the subgenual anterior cingulate cortex (sACC) during emotional face processing.142 Inflammation-induced mood deterioration was also associated with decreased functional connectivity of the sACC with the amygdala, medial PFC, nucleus accumbens, and superior temporal sulcus. Additionally, exposure to an acute social stressor has been associated with increased circulating levels of IL-6 and TNF-α, along with increased activity in the dorsal ACC (dACC) during a social rejection task.143 These findings suggest that stress-induced effects on inflammation may increase activity in brain regions associated with emotion processing, while decreasing connectivity with regions involved in emotion regulation. This increased sensitivity to social stressors and decreased emotion regulation may, in turn, increase the risk for various psychiatric disorders, including MDD.144
There is growing evidence that inflammation may also impact corticostriatal reward circuitry, which underlies symptoms of anhedonia that are common in various psychiatric disorders.145 Specifically, there is evidence that increased circulating levels of CRP, a common inflammatory marker, were associated with decreased connectivity between ventral striatum and ventromedial PFC and decreased dorsal striatal to ventromedial PFC connectivity among participants with MDD.146 Notably, the association between the differences in connectivity and symptoms of anhedonia and motor slowing were significantly predicted by participants peripheral CRP levels. Similarly, depressive symptoms after exposure to an in vivo immune challenge was contingent, at least in part, on a reduction in ventral striatum activity in response to anticipated rewards.147 These findings parallel the results of neuroimaging studies showing that administration of interferon-α, a potent proinflammatory cytokine, led to reduced activity in the ventral striatum during a hedonic reward task.148 Moreover, positron emission tomography was utilized to show an interferon-α-induced increase in18 F-dopa (radiolabeled dopamine precursor) uptake, but decreased18 F-dopa turnover, in the basal ganglia, which correlated with increased depressive symptoms.148 Together, these findings suggest that inflammation may adversely impact motivation and goal-directed behavior by decreasing activation and connectivity of brain regions involved in processing of rewarding stimuli and psychomotor speed, plausibly though the modulation of dopamine function.
The recent understanding of the above interactions between stress and inflammation has given rise to research that applied these findings to both new and longstanding treatment approaches. For instance, there is a growing body of research that suggests that cognitive behavior therapy (CBT) reduces inflammation in the context of improving disturbed sleep and depressed mood.149,150 Other treatment approaches, such as mindfulness meditation and yoga, are also associated with decreased stress-induced inflammation.151,152 Finally, there is evidence in human clinical populations that targeting inflammation directly may help to alleviate symptoms of psychopathology, including MDD, PTSD, and schizophrenia, but only in a subgroup of patients who exhibit increased initial levels of inflammatory markers.153-158 Specifically, meta-analyses have supported the beneficial use of anti-inflammatory medication in schizophrenia-affected individuals, particularly those who are in the early stages of this disorder.158,159 Although preliminary results of the anti-inflammatory therapy in the treatment of psychiatric disorders are promising, it is important to identify specific subgroups that would benefit the most from such treatment.160 For example, one study examined the potential antidepressant effect of the TNF-α inhibitor infliximab in patients with treatment-resistant depression who were otherwise healthy. Infliximab is a monoclonal antibodydirected against TNF-α that prevents this cytokine from binding to its receptor via immunoneutralization. Interestingly, the anti-inflammatory therapy outperformed placebo, but only in patients with high peripheral levels of CRP before treatment (>5 mg/L).161 Baseline levels of inflammation could, therefore, serve as a biomarker of an individual's likelihood of responding to anti-inflammatory therapies. Intriguingly, among participants with a baseline CRP value greater than 5 mg/L, anti-inflammatory therapy led to a reduction in a variety of symptoms, including sad mood, psychomotor retardation, anhedonia, anxiety, and suicidal ideation, all of which are linked to the neurocircuits typically targeted by inflammatory cytokines.161 Overall, therefore, anti-inflammatory therapy may be a promising treatment for specific subgroups of patients with a variety of psychiatric disorders, such as those with elevated circulating inflammatory markers. The identification of, and ability to detect, specific biomarkers that can identify individuals who would benefit from anti-inflammatory therapy is a critical step in delivering individualized therapy.
Despite recent advances made in understanding the role of inflammatory processes in human psychopathology, direct evidence examining potential sex differences is only recently being addressed. Historically, men and women have been known to display pronounced biological and psychological differences in responses to stress, with females typically displaying nearly double the GC response relative to males.162 Stress reactivity is also not constant across developmental epoch or across hormonal cycles; women who are between the ages of puberty and menopause typically exhibit lower HPA-axis and autonomic nervous system responses to acute psychosocial stressors than older women. However, the intensity of responses increases in the luteal phase and after menopause.163 What is striking is that women have higher rates of stress-related psychiatric disorders that have been strongly linked to inflammation, including MDD and anxiety disorders.164 This sex difference in depression is evident across Western and non-Western cultures.165 The sex-specific epidemiological pattern of psychiatric disorders highlights the role of sex hormones in stress reactivity, since many of these sex differences emerge in puberty. Interestingly, there is also evidence of sexual dimorphism in the susceptibility of women and men to immune-related disorders. For instance, the prevalence of many inflammatory conditions, including autoimmune diseases is significantly higher in women,166 whereas men are more likely to suffer from infectious diseases.167 Notably, young women were reported to exhibit higher peripheral levels of IL-6 than men after mental or physical stress.168 Clearly, more research is needed in this area, as it may be that sex differences in stress reactivity, including inflammatory and, potentially, neuroimmune responses to stress, could provide much needed insight into the vast sex differences in the rate of stress-related psychiatric disorders.
Conclusion
The goal of this review was to highlight the neuroimmune mechanisms underlying the response to stress with an emphasis on extending findings from animal models toward the human experience. Research on rodents has served as a starting point for understanding the molecular and cellular responses to stress, allowing for a greater understanding of how inflammatory factors, such as cytokines, chemokines, and prostaglandins, ultimately influence brain function. Importantly, microglia have emerged as a key interface between stress-related signals and neuroimmune consequences of stress. The guinea pig in particular serves as a useful model of early-life stress with excellent early-life translatability that recapitulates findings from nonhuman primates and humans. Further validation of the guinea pig model, and expansion of genetic tools and antibodies directed toward guinea pig-specific proteins, will allow for bridging the findings in rats and mice with a complementary, tractable model system. The use of nonhuman primates confers significant advantages not only in being able to bridge findings in rodent models to humans, but also in that they lead highly social lives allowing for examination of stress and social interactions (eg, buffering effect of a cage partner on social separation discussed earlier in the review.168). Through studying conserved transcriptional responses to stress that have been examined in circulating leukocytes in humans, researchers have made connections between clinically relevant psychological disorders and expression levels of immune and inflammatory factors.133,136 The finding that epigenetic modulation of cytokines differs in individuals with stress-related disorders, such as MDD and PTSD,138 provides an intriguing avenue for animal model research in examining how stress can affect future gene expression. Thus, viewing neuroimmune consequences of stress through a multispecies lens provides a compelling argument for the highly conserved nature of the relationship between cytokines, stress, and multiple forms of psychopathology. Our challenge for the future, therefore, will be to dive deeper into individual differences in stress reactivity, using a combination of highly tractable animal models from different species to better discriminate between those neuroimmune consequences of stress that are constant within and across species, relative to those that differ as a function of the individual (sex, age, recent stress history) or that are species-specific. In doing so, we can exploit the strengths of individual model systems while at the same time circumventing their limitations, with the hope of defining novel therapeutic strategies to ameliorate adverse health consequences of stress.
Acknowledgments
Dr Terrence Deak is currently supported by NIH grant numbers R01AGG43467 and P50AA017823. Dr Michael B. Hennessy is currently supported by R15MH068228. Dr Brandon E. Gibb is currently supported by NIH grant number R01MH098060. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the above-stated funding agencies. The authors have no conflicts of interest to declare.
Selected abbreviations and acronyms
- CNS
central nervous system
- CRP
C-reactive protein
- GC
glucocorticoid
- HPA
hypothalamic-pituitary-adrenal
- IL
interleukin
- LPS
lipopolysaccharide
- MDD
major depressive disorder
- PVN
paraventricular nucleus
- TNF-α
tumor necrosis factor α
Contributor Information
Terrence Deak, Center for Affective Science and Department of Psychology, Binghamton University-State University of New York (SUNY), Binghamton, New York, USA.
Anastacia Kudinova, Center for Affective Science and Department of Psychology, Binghamton University-State University of New York (SUNY), Binghamton, New York, USA.
Dennis F. Lovelock, Center for Affective Science and Department of Psychology, Binghamton University-State University of New York (SUNY), Binghamton, New York, USA.
Brandon E. Gibb, Center for Affective Science and Department of Psychology, Binghamton University-State University of New York (SUNY), Binghamton, New York, USA.
Michael B. Hennessy, Department of Psychology, Wright State University, Dayton, Ohio, USA.
REFERENCES
- 1.Selye H. A syndrome produced by diverse nocuous agents. Nature. 1936;138(3479):32. doi: 10.1176/jnp.10.2.230a. [DOI] [PubMed] [Google Scholar]
- 2.Catanzaro JM., Hueston CM., Deak MM., Deak T. The impact of the P2X7 receptor antagonist A-804598 on neuroimmune and behavioral consequences of stress. Behav Pharmacol. 2014;25(5-6):582–598. doi: 10.1097/FBP.0000000000000072. [DOI] [PubMed] [Google Scholar]
- 3.Vitkovic L., Konsman J., Bockaert J., Dantzer R., Homburger V., Jacque C. Cytokine signals propagate through the brain. Mol Psychiatry. 2000;5(6):604–615. doi: 10.1038/sj.mp.4000813. [DOI] [PubMed] [Google Scholar]
- 4.Holguin A., Frank MG., Biedenkapp JC., et al Characterization of the temporo-spatial effects of chronic bilateral intrahippocampal cannulae on interleukin-1β. J Neurosci Methods. 2007;161(2):265–272. doi: 10.1016/j.jneumeth.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deak T., Bordner K., McElderry N., et al Stress-induced increases in hypothalamic IL-1: a systematic analysis of multiple stressor paradigms. Brain Res Bulletin. 2005;64(6):541–556. doi: 10.1016/j.brainresbull.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Blandino P Jr., Hueston CM., Barnurn CJ., Bishop C., Deak T. The impact of ventral noradrenergic bundle lesions on increased IL-1 in the PVN and hormonal responses to stress in male Sprague Dawley rats. Endocrinology. 2013;154(7):2489–2500. doi: 10.1210/en.2013-1075. [DOI] [PubMed] [Google Scholar]
- 7.Hueston CM., Deak T. The inflamed axis: the interaction between stress, hormones, and the expression of inflammatory-related genes within key structures comprising the hypothalamic-pituitary-adrenal axis. Physiol Behav. 2014;124:77–91. doi: 10.1016/j.physbeh.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 8.Blandino P Jr., Barnum CJ., Solomon LG., Larish Y., Lankow BS., Deak T. Gene expression changes in the hypothalamus provide evidence for regionally-selective changes in IL-1 and microglial markers after acute stress. Brain Behav Immun. 2009;23(7):958–968. doi: 10.1016/j.bbi.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 9.Deak T., Bellamy C., D'Agostino LG. Exposure to forced swim stress does not alter central production of IL-1. Brain Res. 2003;972(1-2):53–63. doi: 10.1016/s0006-8993(03)02485-5. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen KT., Deak T., Owens SM., et al Exposure to acute stress induces brain interleukin-1β protein in the rat. Neuroscience. 1998;18(6):2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hueston CM., Barnum CJ., Eberle JA., Ferraioli FJ., Buck HM., Deak T. Stress-dependent changes in neuroinflammatory markers observed after common laboratory stressors are not seen following acute social defeat of the Sprague Dawley rat. Physiol Behav. 2011;104(2):187–198. doi: 10.1016/j.physbeh.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Hodes GE., Pfau ML., Leboeuf M., et al Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci US A. 2014;111(45):16136–16141. doi: 10.1073/pnas.1415191111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood SK., Wood CS., Lombard CM., et al Inflammatory factors mediate vulnerability to a social stress-induced depressive-like phenotype in passive coping rats. Biol Psychiatry. 2015;78(1):38–48. doi: 10.1016/j.biopsych.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roque A., Ochoa-Zarzosa A., Torner L. Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav Immun. 2015;55:39–48. doi: 10.1016/j.bbi.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Wohleb ES., Powell ND., Godbout JP., Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33(34):13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reyes TM., Walker JR., DeCino C., Hogenesch JB., Sawchenko PE. Categorically distinct acute stressors elicit dissimilar transcriptional profiles in the paraventricular nucleus of the hypothalamus. J Neurosci. 2003;23(13):5607–5616. doi: 10.1523/JNEUROSCI.23-13-05607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross AK., Woodroofe MN. Chemokines induce migration and changes in actin polymerization in adult rat brain microglia and a human fetal microglial cell line in vitro. J Neurosci Res. 1999;55(1):17–23. doi: 10.1002/(SICI)1097-4547(19990101)55:1<17::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Eugenin EA., Dyer G., Calderon TM., Berman JW. HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)dependent mechanism: possible role in NeuroAIDS. Glia. 2005;49(4):501–510. doi: 10.1002/glia.20137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marella M., Chabry J. Neurons and astrocytes respond to prion infection by inducing microglia recruitment. J Neurosci. 2004;24(3):620–627. doi: 10.1523/JNEUROSCI.4303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carbonell WS., Murase SI., Horwitz AF., Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J Neurosci. 2005;25(30):7040–7047. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ubogu EE., Cossoy MB., Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27(1):48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Schilling M., Strecker JK., Ringelstein EB., Schabitz WR., Kiefer R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res. 2009;1289:79–84. doi: 10.1016/j.brainres.2009.06.054. [DOI] [PubMed] [Google Scholar]
- 23.Milior G., Lecours C., Samson L., et al Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav Immun. 2015;55:114–125. doi: 10.1016/j.bbi.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Bueno B., Madrigal JL., Perez-Nievas BG., Leza JC. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology. 2008;149(4):1969–1978. doi: 10.1210/en.2007-0482. [DOI] [PubMed] [Google Scholar]
- 25.Furuyashiki T., Narumiya S. Stress responses: the contribution of prostaglandin E2 and its receptors. Nat Rev Endocrinol. 2011;7(3):163–175. doi: 10.1038/nrendo.2010.194. [DOI] [PubMed] [Google Scholar]
- 26.Blandino P Jr., Barnum CJ., Deak T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1β responses to stress. J Neuroimmunol. 2006;173(1-2):87–95. doi: 10.1016/j.jneuroim.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 27.Nair A., Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol. 2006;171(1-2):72–85. doi: 10.1016/j.jneuroim.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 28.Tynan RJ., Naicker S., Hinwood M., et al Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 2010;24(7):1058–1068. doi: 10.1016/j.bbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Kopp BL., Wick D., Herman JP. Differential effects of homotypicvs. heterotypic chronic stress regimens on microglial activation in the prefrontal cortex. Physiol Behav. 2013;122:246–252. doi: 10.1016/j.physbeh.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bisht K., Sharma KP., Lecours C., et al Dark microglia: a new phenotype predominantly associated with pathological states. Glia. 2016;64(5):826–839. doi: 10.1002/glia.22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frank MG., Baratta MV., Sprunger DB., Watkins LR., Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav immun. 2007;21(1):47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Johnson JD., O'Connor KA., Deak T., Stark M., Watkins LR., Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun. 2002;16(4):461–476. doi: 10.1006/brbi.2001.0638. [DOI] [PubMed] [Google Scholar]
- 33.Frank MG., Barrientos RM., Watkins LR., Maier SF. Aging sensitizes rapidly isolated hippocampal microglia to LPS ex vivo. J Neuroimmunol. 2010;226(1):181–184. doi: 10.1016/j.jneuroim.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arakawa H., Blandino P Jr., Deak T. Central infusion of interleukin-1 receptor antagonist blocks the reduction in social behavior produced by prior stressor exposure. Physiol Behav. 2009;98(1-2):139–146. doi: 10.1016/j.physbeh.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yirmiya R., Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25(2):181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Ohgidani M., Kato TA., Sagata N., et al TNF- from hippocampal microglia induces working memory deficits by acute stress in mice. Brain Behav Immun. 2015;55:17–24. doi: 10.1016/j.bbi.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 37.Walker FR., Yirmiya R. Microglia, physiology and behavior: a brief commentary. Brain Behav Immun. 2016;55:1–5. doi: 10.1016/j.bbi.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Slavich GM., Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140(3):774. doi: 10.1037/a0035302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young JJ., Bruno D., Pomara N. A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Dis. 2014;169:15–20. doi: 10.1016/j.jad.2014.07.032. [DOI] [PubMed] [Google Scholar]
- 40.Clark S., Li X., Tonelli L. Hippocampal brain derived neurotrophic factor levels are mediated by T cells in response to stress. Brain Behav Immun. 2015;49:e20. [Google Scholar]
- 41.Brachman RA., Lehmann ML., Marie D., Herkenham M. Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J Neurosci. 2015;35(4):1530–1538. doi: 10.1523/JNEUROSCI.2278-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wohleb ES., McKirn DB., Shea DT., et al Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 2014;75(12):970–981. doi: 10.1016/j.biopsych.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wohleb ES., McKim DB., Sheridan JF., Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-tobrain communication that influences mood and behavior. Front Neurosci. 2015;8:447. doi: 10.3389/fnins.2014.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watkins LR., Maier SF., Goehler LE. Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci. 1995;57(11):1011–1026. doi: 10.1016/0024-3205(95)02047-m. [DOI] [PubMed] [Google Scholar]
- 45.Menard C., Hodes G., Russo S. Pathogenesis of depression: insights from human and rodent studies. Neuroscience. 2016;321:138–162. doi: 10.1016/j.neuroscience.2015.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hodes GE., Kana V., Menard C., Merad M., Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci. 2015;18(10):1386–1393. doi: 10.1038/nn.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen KT., Deak T., Will MJ., et al Timecourse and corticosterone sensitivity of the brain, pituitary, and serum interleukin-1β protein response to acute stress. Brain Res. 2000;859(2):193–201. doi: 10.1016/s0006-8993(99)02443-9. [DOI] [PubMed] [Google Scholar]
- 48.Johnson JD., Campisi J., Sharkey CM., et al Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005;135(4):1295– 1307. doi: 10.1016/j.neuroscience.2005.06.090. [DOI] [PubMed] [Google Scholar]
- 49.Jander S., Schroeter M., Stoll G. Role of NMDA receptor signaling in the regulation of inflammatory gene expression after focal brain ischemia. J Neuroimmunol. 2000;109(2):181–187. doi: 10.1016/s0165-5728(00)00317-9. [DOI] [PubMed] [Google Scholar]
- 50.Vladychenskaya E., Tyulina O., Urano S., Boldyrev A. Rat lymphocytes express NMDA receptors that take part in regulation of cytokine production. Cell Biochem Funct. 2011;29(7):527–533. doi: 10.1002/cbf.1771. [DOI] [PubMed] [Google Scholar]
- 51.Koo JW., Duman RS. Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett. 2009;456(1):39–43. doi: 10.1016/j.neulet.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lord B., Aluisio L., Shoblock JR., et al Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-422 53432. J Pharmacol Exp Then. 2014;351(3):628–641. doi: 10.1124/jpet.114.218487. [DOI] [PubMed] [Google Scholar]
- 53.Deak T., Quinn M., Cidlowski JA., Victoria NC., Murphy AZ., Sheridan JF. Neuroimmune mechanisms of stress: sex differences, developmental plasticity, and implications for pharmacotherapy of stress-related disease. Stress. 2015;18(4):367–380. doi: 10.3109/10253890.2015.1053451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson JD., Fleshner M. Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. J LeukocBiol. 2006;79(3):425–434. doi: 10.1189/jlb.0905523. [DOI] [PubMed] [Google Scholar]
- 55.Frank MG., Adhikary S., Sobesky JL., Weber MD., Watkins LR., Maier SF. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav Immun. 2016;51:99–108. doi: 10.1016/j.bbi.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber MD., Frank MG., Tracey KJ., Watkins LR., Maier SF. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci. 2015;35(1):316–324. doi: 10.1523/JNEUROSCI.3561-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailey MT., Dowd SE., Galley JD., Hufnagle AR., Allen RG., Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25(3):397–407. doi: 10.1016/j.bbi.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayer EA., Knight R., Mazmanian SK., Cryan JF., Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34(46):15490–15496. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cryan JF., O'Mahony S. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroeneterol Motil. 2011;23(3):187–192. doi: 10.1111/j.1365-2982.2010.01664.x. [DOI] [PubMed] [Google Scholar]
- 60.Frank MG., Weber MD., Fonken LK., Hershman SA., Watkins LR., Maier SF. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav Immun. 2015;55:215–224. doi: 10.1016/j.bbi.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newton R., Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol. 2007;72(4):799–809. doi: 10.1124/mol.107.038794. [DOI] [PubMed] [Google Scholar]
- 62.Hoffman EP., Reeves E., Damsker J., et al Novel approaches to corticosteroid treatment in Duchenne muscular dystrophy. Phys Med Rehabil Clin NAm. 2012;23(4):821–828. doi: 10.1016/j.pmr.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sorrells SF., Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21(3):259–272. doi: 10.1016/j.bbi.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frank MG., Weber MD., Watkins LR., Maier SF. Stress sounds the alarmin: the role of the danger-associated molecular pattern HMGB1 in stressinduced neuroinflammatory priming. Brain Behav Immun. 2015;48:1–7. doi: 10.1016/j.bbi.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frank MG., Hershman SA., Weber MD., Watkins LR., Maier SF. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology. 2014;40:191–200. doi: 10.1016/j.psyneuen.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frank MG., Watkins LR., Maier SF. Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain Behav Immun. 2013;33:1–6. doi: 10.1016/j.bbi.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whirledge S., Cidlowski JA. A role for glucocorticoids in stress-impaired reproduction: beyond the hypothalamus and pituitary. Endocrinology. 2013;154(12):4450–4468. doi: 10.1210/en.2013-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hueston CM., Deak T. On the time course, generality, and regulation of plasma progesterone release in male rats by stress exposure. Endocrinology. 2014;155(9):3527–3537. doi: 10.1210/en.2014-1060. [DOI] [PubMed] [Google Scholar]
- 69.Arakawa K., Arakawa H., Hueston CM., Deak T. Effects of the estrous cycle and ovarian hormones on central expression of interleukin-1 evoked by stress in female rats. Neuroendocrinology. 2014;100(2-3):162–177. doi: 10.1159/000368606. [DOI] [PubMed] [Google Scholar]
- 70.Reis FG., Marques RH., Starling CM., et al Stress amplifies lung tissue mechanics, inflammation and oxidative stress induced by chronic inflammation. Exp Lung Res. 2012;38(7):344–354. doi: 10.3109/01902148.2012.704484. [DOI] [PubMed] [Google Scholar]
- 71.Shannonhouse JL., Fong LA., Clossen BL., et al Female-biased anorexia and anxiety in the Syrian hamster. Physiol Behav. 2014;133:141–151. doi: 10.1016/j.physbeh.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto N., Sakagami T., Fukuda Y., et al Influence of Helicobacter pylori infection on development of stress-induced gastric mucosal injury. J Gastroenterol. 2000;35(5):332–340. doi: 10.1007/s005350050357. [DOI] [PubMed] [Google Scholar]
- 73.Blatteis CM. Postnatal development of pyrogenic sensitivity in guinea pigs. J Appl Physiol. 1975;39(2):251–257. doi: 10.1152/jappl.1975.39.2.251. [DOI] [PubMed] [Google Scholar]
- 74.Harper LV. Behavior. In: Wagner JE, Manning PJ, eds. The Biology of the Guinea Pig. New York, NY: Academic Press. 1976:31–51. [Google Scholar]
- 75.Konig B. Maternal activity budget during lactation in two species of Caviidae. (Cavia porcellus and Galea musteloides). Z Tierpsychol. 1985;68(3):215–230. [Google Scholar]
- 76.Hennessy MB., Jenkins R. A descriptive analysis of nursing behavior in the guinea pig. (Cavia porcellus). J Comp Psychol. 1994;108(1):23. doi: 10.1037/0735-7036.108.1.23. [DOI] [PubMed] [Google Scholar]
- 77.Hennessy MB., Ritchey RL. Hormonal and behavioral attachment responses in infant guinea pigs. Dev Psychobiol. 1987;20(6):613–625. doi: 10.1002/dev.420200607. [DOI] [PubMed] [Google Scholar]
- 78.Jackel M., Trillmich F. Olfactory individual recognition of mothers by young guinea-pigs. (Cavia porcellus). Ethology. 2003;109(3):197–208. [Google Scholar]
- 79.Porter RH., Berryman JC., Fullerton C. Exploration and attachment behaviour in infant guinea pigs. Behaviour. 1973;45(3):312–322. doi: 10.1163/156853974x00705. [DOI] [PubMed] [Google Scholar]
- 80.Heirn C., Binder EB. Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol. 2012;233(1):102–111. doi: 10.1016/j.expneurol.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 81.Mandelli L., Petrelli C., Serretti A. The role of specific early trauma in adult depression: a meta-analysis of published literature. Childhood trauma and adult depression. Eur Psychiatry. 2015;30(6):665–680. doi: 10.1016/j.eurpsy.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 82.Hennessy MB. Enduring maternal influences in a precocial rodent. Dev Psychobiol. 2003;42(3):225–236. doi: 10.1002/dev.10095. [DOI] [PubMed] [Google Scholar]
- 83.Hennessy MB., Moorman L. Factors influencing Cortisol and behavioral responses to maternal separation in guinea pigs. Behav Neurosci. 1989;103(2):378. doi: 10.1037//0735-7044.103.2.378. [DOI] [PubMed] [Google Scholar]
- 84.Hennessy MB., Tamborski A., Schiml P., Lucot J. The influence of maternal separation on plasma concentrations of ACTH, epinephrine, and norepinephrine in guinea pig pups. Physiol Behav. 1989;45(6):1147–1152. doi: 10.1016/0031-9384(89)90101-7. [DOI] [PubMed] [Google Scholar]
- 85.Harvey AT., Moore H., Lucot JB., Hennessy MB. Monoamine activity in anterior hypothalamus of guinea pig pups separated from their mothers. Behav Neurosci. 1994;108(1):171. doi: 10.1037//0735-7044.108.1.171. [DOI] [PubMed] [Google Scholar]
- 86.Tamborski A., Lucot JB., Hennessy MB. Central dopamine turnover in guinea pig pups during separation from their mothers in a novel environment. Behav Neurosci. 1990;104(4):607. doi: 10.1037//0735-7044.104.4.607. [DOI] [PubMed] [Google Scholar]
- 87.Mineka S., Suomi SJ. Social separation in monkeys. Psychol Bull. 1978;85(6):1376. [PubMed] [Google Scholar]
- 88.Spitz RA., Wolf KM. Anaclitic depression; an inquiry into the genesis of psychiatric conditions in early childhood. Psychoanal Study Child. 1946;2:313–342. [PubMed] [Google Scholar]
- 89.Hennessy MB., Long SJ., Nigh CK., Williams MT., Nolan DJ. Effects of peripherally administered corticotropin-releasing factor (CRF) and a CRF antagonist: Does peripheral CRF activity mediate behavior of guinea pig pups during isolation? Behav Neurosci. 1995;109(6):1137. doi: 10.1037//0735-7044.109.6.1137. [DOI] [PubMed] [Google Scholar]
- 90.Hennessy MB., Deak T., Schiml-Webb PA., Wilson SE., Greenlee TM., McCall E. Responses of guinea pig pups during isolation in a novel environment may represent stress-induced sickness behaviors. Physiol Behav. 2004;81(1):5–13. doi: 10.1016/j.physbeh.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 91.Kaufman CI., Rosenblum LA. The reaction to separation in infant monkeys: anaclitic depression and conservation-withdrawal. Psychosom Med. 1967;29(6):648–675. doi: 10.1097/00006842-196711000-00010. [DOI] [PubMed] [Google Scholar]
- 92.Hart BL. Biological basis of the behavior of sick animals. Neurosci BiobehavRev. 1988;12(2):123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 93.Hennessy MB., Deak T., Schiml-Webb PA., Carlisle CW., O'Brien E. Maternal separation produces, and a second separation enhances, core temperature and passive behavioral responses in guinea pig pups. Physiol Behav. 2010;100(4):305–310. doi: 10.1016/j.physbeh.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hennessy MB., Deak T., Schiml-Webb PA., Barnum CJ. Immune influences on behavior and endocrine activity in early-experience and maternal separation paradigms. In: Czerbska MT, ed. Psychoneuroendocrinology Research Trends. Hauppauge, NY: Nova Science Publishers;2007:293–319. [Google Scholar]
- 95.Schiml-Webb PA., Deak T., Greenlee TM., Maken D., Hennessy MB. Alpha-melanocyte stimulating hormone reduces putative stress-induced sickness behaviors in isolated guinea pig pups. Behav Brain Res. 2006;168(2):326–330. doi: 10.1016/j.bbr.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 96.Hennessy MB., Schiml-Webb PA., Miller EE., Maken DS., Bullinger KL., Deak T. Anti-inflammatory agents attenuate the passive responses of guinea pig pups: evidence for stress-induced sickness behavior during maternal separation. Psychoneuroendocrinology. 2007;32(5):508–515. doi: 10.1016/j.psyneuen.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perkeybile AM., Schiml-Webb PA., O'Brien E., Deak T., Hennessy MB. Antiinflammatory influences on behavioral, but not Cortisol, responses during maternal separation. Psychoneuroendocrinology. 2009;34(7):1101–1108. doi: 10.1016/j.psyneuen.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hennessy MB., Stafford NP., Yusko-Osborne B., Schiml PA., Xanthos ED., Deak T. Naproxen attenuates sensitization of depressive-like behavior and fever during maternal separation. Physiol Behav. 2015;139:34–40. doi: 10.1016/j.physbeh.2014.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raedler TJ. Inflammatory mechanisms in major depressive disorder. Curr Opin Psychiatry. 2011;24(6):519–525. doi: 10.1097/YCO.0b013e32834b9db6. [DOI] [PubMed] [Google Scholar]
- 100.Brown GW., Harris T., Copeland JR. Depression and loss. Br J Psychiatry. 1977;130(1):1–18. doi: 10.1192/bjp.130.1.1. [DOI] [PubMed] [Google Scholar]
- 101.Gold PW., Goodwin FK., Chrousos GP. Clinical and biochemical manifestations of depression. N Engl J Med. 1988;319(6):348–353. doi: 10.1056/NEJM198808113190606. [DOI] [PubMed] [Google Scholar]
- 102.Heim C., Newport DJ., Mletzko T., Miller AH., Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33(6):693–710. doi: 10.1016/j.psyneuen.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 103.McCrory E., De Brito SA., Viding E. The impact of childhood maltreatment: a review of neurobiological and genetic factors. Front Psychiatry. 2011;2(48):1–14. doi: 10.3389/fpsyt.2011.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bertone-Johnson ER., Whitcomb BW., Missmer SA., Karlson EW., RichEdwards JW. Inflammation and early-life abuse in women. Ami J Prev Med. 2012;43(6):611–620. doi: 10.1016/j.amepre.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carpenter LL., Gawuga CE., Tyrka AR., Lee JK., Anderson GM., Price LH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology. 2010;35(13):2617–2623. doi: 10.1038/npp.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gouin JP., Glaser R., Malarkey WB., Beversdorf D., Kiecolt-Glaser JK. Childhood abuse and inflammatory responses to daily stressors. Ann Behav Med. 2012;44(2):287–292. doi: 10.1007/s12160-012-9386-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miller GE., Chen E. Harsh family climate in early life presages the emergence of a proinflammatory phenotype in adolescence. Psychol Sci. 2010;21(6):848–856. doi: 10.1177/0956797610370161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Slopen N., Kubzansky LD., McLaughlin KA., Koenen KC. Childhood adversity and inflammatory processes in youth: a prospective study. Psychoneuroendocrinology. 2013;38(2):188–200. doi: 10.1016/j.psyneuen.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeugmann S., Buehrsch N., Bajbouj M., Heuser I., Anghelescu I., Quante A. Childhood maltreatment and adult proinflammatory status in patients with major depression. Psychiatr Danub. 2013;25(3):227–235. [PubMed] [Google Scholar]
- 110.Miller GE., Chen E., Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol Bull. 2011;137(6):959. doi: 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ganguly P., Brenhouse HC. Broken or maladaptive? Altered trajectories in neuroinflammation and behavior after early life adversity. Dev Cogn Neurosci. 2015;11:18–30. doi: 10.1016/j.dcn.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reader B., Jarrett B., McKim D., Wohleb E., Godbout JP., Sheridan JF. Peripheral and central effects of repeated social defeat stress: monocyte trafficking, microglial activation, and anxiety. Neuroscience. 2015;289:429–442. doi: 10.1016/j.neuroscience.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cole SW., Conti G., Arevalo JM., Ruggiero AM., Heckman JJ., Suomi SJ. Transcriptional modulation of the developing immune system by early life social adversity. Proc Natl Acad Sci U S A. 2012;109(50):20578–20583. doi: 10.1073/pnas.1218253109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qin D., Chu X., Feng X., et al The first observation of seasonal affective disorder symptoms in Rhesus macaque. Behav Brain Res. 2015;292:463–469. doi: 10.1016/j.bbr.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 115.Hennessy MB., McCowan B., Jiang J., Capitanio JP. Depressive-like behavioral response of adult male rhesus monkeys during routine animal husbandry procedure. Front Behavio Neurosci. 2014;8(309) doi: 10.3389/fnbeh.2014.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol. 2004;500(1):399–411. doi: 10.1016/j.ejphar.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 117.Hennessy MB., Chun K., Capitanio JP. Depressive-like behavior, its sensitization, social buffering and altered cytokine responses in rhesus macaques moved from outdoor social groups to indoor housing. Soc Neurosci. 2016;12(1):65–75. doi: 10.1080/17470919.2016.1145595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.De Kloet ER., Joels M., Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6(6):463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 119.Hammen C. Stress and depression. Ann Rev Clin Psych. 2005;1:293–319. doi: 10.1146/annurev.clinpsy.1.102803.143938. [DOI] [PubMed] [Google Scholar]
- 120.Kessler RC., Davis CG., Kendler KS. Childhood adversity and adult psychiatric disorder in the US National Comorbidity Survey. Psychol Med. 1997;27(05):1101–1119. doi: 10.1017/s0033291797005588. [DOI] [PubMed] [Google Scholar]
- 121.Miller AH., Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16(1):22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Iwata M., Ota KT., Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 2013;31:105–114. doi: 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Anisman H., Merali Z., Hayley S. Sensitization associated with stressors and cytokine treatments. Brain Behav Immun. 2003;17(2):86–93. doi: 10.1016/s0889-1591(02)00100-9. [DOI] [PubMed] [Google Scholar]
- 124.Danese A., Pariante CM., Caspi A., Taylor A., Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A. 2007;104(4):1319–1324. doi: 10.1073/pnas.0610362104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Danese A., Moffitt TE., Pariante CM., Ambler A., Poulton R., Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry. 2008;65(4):409–415. doi: 10.1001/archpsyc.65.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kudinova AY., Deak T., Hueston CM., et al Cross-species evidence for the role of interleukin-33 in depression risk. J Abnorm Psychol. 2016;125(4):482–494. doi: 10.1037/abn0000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen E., Miller GE., Kobor MS., Cole SW. Maternal warmth buffers the effects of low early-life socioeconomic status on pro-inflammatory signaling in adulthood. Mol Psychiatry. 2011;16(7):729–737. doi: 10.1038/mp.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cole SW. Social regulation of human gene expression. Curr Dir Psychol Sci. 2009;18(3):132–137. doi: 10.1111/j.1467-8721.2009.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cole SW., Hawkley LC., Arevalo JM., Sung CY., Rose RM., Cacioppo JT. Social regulation of gene expression in human leukocytes. Genome Biol. 2007;8(9):R189. doi: 10.1186/gb-2007-8-9-r189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Miller GE., Chen E., Sze J., et al A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kB signaling. Biol Psychiatry. 2008;64(4):266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O'Donovan A., Sun B., Cole S., et al Transcriptional control of monocyte gene expression in post-traumatic stress disorder. Dis Markers. 2011;30(23):123–132. doi: 10.3233/DMA-2011-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Powell ND., Sloan EK., Bailey MT., et al Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via p-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A. 2013;110(41):16574–16579. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arion D., Unger T., Lewis DA., Levitt P., Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62(7):711–721. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim S., Hwang Y., Webster M., Lee D. Differential activation of immune/ inflammatory response-related co-expression modules in the hippocampus across the major psychiatric disorders. Mol Psychiatry. 2016;21(3):376–385. doi: 10.1038/mp.2015.79. [DOI] [PubMed] [Google Scholar]
- 135.Noto C., Ota VK., Santoro ML., et al Depression, cytokine, and cytokine by treatment interactions modulate gene expression in antipsychotic naive first episode psychosis. Mol Neurobiol. 2016;53(8):5701–5709. doi: 10.1007/s12035-015-9489-3. [DOI] [PubMed] [Google Scholar]
- 136.Powell TR., McGuffin P., D'Souza UM., et al Putative transcriptomic biomarkers in the inflammatory cytokine pathway differentiate major depressive disorder patients from control subjects and bipolar disorder patients. PloS One. 2014;9(3):1–9. doi: 10.1371/journal.pone.0091076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Arloth J., Bogdan R., Weber P., et al Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron. 2015;86(5):1189–1202. doi: 10.1016/j.neuron.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Uddin M., Aiello AE., Wildman DE., et al Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2010;107(20):9470–9475. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Myint AM., Kim YK. Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses. 2003;61(5):519–525. doi: 10.1016/s0306-9877(03)00207-x. [DOI] [PubMed] [Google Scholar]
- 140.Bam M., Yang X., Zhou J., et al Evidence for epigenetic regulation of pro-inflammatory cytokines, interleukin-1 2 and interferon gamma, in peripheral blood mononuclear cells from PTSD patients. J Neuroimmune Pharmacol. 2016;11(1):168–181. doi: 10.1007/s11481-015-9643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Smith AK., Conneely KN., Kilaru V., et al Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156(6):700–708. doi: 10.1002/ajmg.b.31212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Harrison NA., Brydon L., Walker C., Gray MA., Steptoe A., Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry. 2009;66(5):407–414. doi: 10.1016/j.biopsych.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Slavich GM., Way BM., Eisenberger Nl., Taylor SE. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci U S A. 2010;107(33):14817–14822. doi: 10.1073/pnas.1009164107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Berking M., Wupperman P. Emotion regulation and mental health: recent findings, current challenges, and future directions. Curr Opin Psychiatry. 2012;25(2):128–134. doi: 10.1097/YCO.0b013e3283503669. [DOI] [PubMed] [Google Scholar]
- 145.Snaith P. Anhedonia: a neglected symptom of psychopathology. Psychol Med. 1993;23(04):957–966. doi: 10.1017/s0033291700026428. [DOI] [PubMed] [Google Scholar]
- 146.Felger J., Li Z., Haroon E., et al Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2016;21(10):1358–1365. doi: 10.1038/mp.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Eisenberger Nl., Berkman ET., Inagaki TK., Rameson LT., Mashal NM., Irwin MR. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748–754. doi: 10.1016/j.biopsych.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Capuron L., Pagnoni G., Drake DF., et al Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. 2012;69(10):1044–1053. doi: 10.1001/archgenpsychiatry.2011.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen HY., Cheng IC., Pan YJ., et al Cognitive-behavioral therapy for sleep disturbance decreases inflammatory cytokines and oxidative stress in hemodialysis patients. Kidney Int. 2011;80(4):415–422. doi: 10.1038/ki.2011.151. [DOI] [PubMed] [Google Scholar]
- 150.Irwin MR., Olmstead R., Breen EC., et al Cognitive behavioral therapy and tai chi reverse cellular and genomic markers of inflammation in late-life insomnia: a randomized controlled trial. Biol Psychiatry. 2015;78(10):721–729. doi: 10.1016/j.biopsych.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kiecolt-Glaser JK., Christian L., Preston H., et al Stress, inflammation, and yoga practice. Psychosom Med. 2010;72(2):113. doi: 10.1097/PSY.0b013e3181cb9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rosenkranz MA., Davidson RJ., MacCoon DG., Sheridan JF., Kalin NH., Lutz A. A comparison of mindfulness-based stress reduction and an active control in modulation of neurogenic inflammation. Brain Behav Immun. 2013;27(1):174–184. doi: 10.1016/j.bbi.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Keller WR., Kum LM., Wehring HJ., Koola MM., Buchanan RW., Kelly DL. A review of anti-inflammatory agents for symptoms of schizophrenia. J Psychopharmacol. 2013;27(4):337–342. doi: 10.1177/0269881112467089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Köhler O., Benros ME., Nordentoft M., et al Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry. 2014;71(12):1381–1391. doi: 10.1001/jamapsychiatry.2014.1611. [DOI] [PubMed] [Google Scholar]
- 155.Koola MM., Varghese SP., Fawcett JA. High-dose prazosin for the treatment of post-traumatic stress disorder. Ther Adv Psychopharmacol. 2014;4(1):43–47. doi: 10.1177/2045125313500982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Muller N., Schwarz M., Dehning S., et al The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a doubleblind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680–684. doi: 10.1038/sj.mp.4001805. [DOI] [PubMed] [Google Scholar]
- 157.Muller N., Riedel M., Schwarz M., Engel R. Clinical effects of COX-2 inhibitors on cognition in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2005;255(2):149–151. doi: 10.1007/s00406-004-0548-4. [DOI] [PubMed] [Google Scholar]
- 158.Sommer IE., De Witte L., Begemann M., Kahn RS. Nonsteroidal antiinflammatory drugs in schizophrenia: ready for practice or a good start? A meta-analysis. J Clin Psychiatry. 2011;73(4):414–419. doi: 10.4088/JCP.10r06823. [DOI] [PubMed] [Google Scholar]
- 159.Nitta M., Kishimoto T., Muller N., et al Adjunctive use of nonsteroidal anti-inflammatory drugs for schizophrenia: a meta-analytic investigation of randomized controlled trials. Schizophr Bull. 2013;39(6):1230–1241. doi: 10.1093/schbul/sbt070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Miller AH., Raison CL. Are anti-inflammatory therapies viable treatments for psychiatric disorders?: where the rubber meets the road. JAMA Psychiatry. 2015;72(6):527–528. doi: 10.1001/jamapsychiatry.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Raison CL., Rutherford RE., Woolwine BJ., et al A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Verma R., Balhara YPS., Gupta CS. Gender differences in stress response: role of developmental and biological determinants. Ind Psychiatriy J. 2011;20(1):4–10. doi: 10.4103/0972-6748.98407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kajantie E., Phillips DL. The effects of sex and hormonal status on the physiological response to acute psychosocial stress. Psychoneuroendocrinology. 2006;31(2):151–178. doi: 10.1016/j.psyneuen.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 164.Kessler RC., Berglund P., Dernier O., et al The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 165.Kessler RC., Bromet EJ. The epidemiology of depression across cultures. Annu Rev Public Health. 2013;34:119–138. doi: 10.1146/annurev-publhealth-031912-114409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gleicher N., Barad DH. Gender as risk factor for autoimmune diseases. J Autoimmun. 2007;28(1):1–6. doi: 10.1016/j.jaut.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 167.Lundberg U. Stress hormones in health and illness: the roles of work and gender. Psychoneuroendocrinology. 2005;30(10):1017–1021. doi: 10.1016/j.psyneuen.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 168.Rooks CR., Ibeanu I., Shah A., et al Young women post-MI have higher plasma concentrations of interleukin-6 before and after stress testing. Brain Behav Immun. 2016;51:92–98. doi: 10.1016/j.bbi.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]