

ABSTRACT
Vibrio parahaemolyticus is an important human foodborne pathogen whose transmission is associated with the consumption of contaminated seafood, with a growing number of infections reported over recent years worldwide. A multilocus sequence typing (MLST) database for V. parahaemolyticus was created in 2008, and a large number of clones have been identified, causing severe outbreaks worldwide (sequence type 3 [ST3]), recurrent outbreaks in certain regions (e.g., ST36), or spreading to other regions where they are nonendemic (e.g., ST88 or ST189). The current MLST scheme uses sequences of 7 genes to generate an ST, which results in a powerful tool for inferring the population structure of this pathogen, although with limited resolution, especially compared to pulsed-field gel electrophoresis (PFGE). The application of whole-genome sequencing (WGS) has become routine for trace back investigations, with core genome MLST (cgMLST) analysis as one of the most straightforward ways to explore complex genomic data in an epidemiological context. Therefore, there is a need to generate a new, portable, standardized, and more advanced system that provides higher resolution and discriminatory power among V. parahaemolyticus strains using WGS data. We sequenced 92 V. parahaemolyticus genomes and used the genome of strain RIMD 2210633 as a reference (with a total of 4,832 genes) to determine which genes were suitable for establishing a V. parahaemolyticus cgMLST scheme. This analysis resulted in the identification of 2,254 suitable core genes for use in the cgMLST scheme. To evaluate the performance of this scheme, we performed a cgMLST analysis of 92 newly sequenced genomes, plus an additional 142 strains with genomes available at NCBI. cgMLST analysis was able to distinguish related and unrelated strains, including those with the same ST, clearly showing its enhanced resolution over conventional MLST analysis. It also distinguished outbreak-related from non-outbreak-related strains within the same ST. The sequences obtained from this work were deposited and are available in the public database (http://pubmlst.org/vparahaemolyticus). The application of this cgMLST scheme to the characterization of V. parahaemolyticus strains provided by different laboratories from around the world will reveal the global picture of the epidemiology, spread, and evolution of this pathogen and will become a powerful tool for outbreak investigations, allowing for the unambiguous comparison of strains with global coverage.
KEYWORDS: whole-genome sequencing (WGS), core genome multilocus sequence typing, cgMLST, Vibrio parahaemolyticus, clinical, phylogenetic analysis, phylogeny, single nucleotide polymorphism (SNP)
INTRODUCTION
Vibrio parahaemolyticus is an important human foodborne pathogen whose transmission is associated with the consumption of contaminated seafood (1). Most V. parahaemolyticus strains that are considered pathogenic carry genes encoding thermostable direct hemolysin (tdh) and/or thermostable direct hemolysin-related hemolysin (trh) (2). Usually, these potential pathogenic strains represent a small fraction of all environmental strains (3). In addition to these two virulence genes, pathogenic V. parahaemolyticus strains carry other virulence-related genes, usually located in pathogenicity islands (4–7).
The V. parahaemolyticus “pandemic clonal complex” has been the dominant clone causing diseases around the world (3, 8–14). The emergence and cross-border spreading of strains, mostly belonging to sequence type 3 (ST3), raised public health concerns regarding the possibility of a pandemic spread, an uncharacteristic trait for V. parahaemolyticus. It was believed that this pandemic strain was the only strain that was spreading among distant regions. However, recent findings have shown that this was not the case, and other V. parahaemolyticus strains belonging to diverse clonal complexes have been spreading between Asia and other parts of the world (15–18). The dispersal routes of these strains remain uncertain at the moment, but at least three different mechanisms have been identified as being associated with the introduction of pathogenic V. parahaemolyticus: ballast water, ocean currents, and transport of oysters or other mollusks between regions (11, 15, 16).
A first glance into the population structure and diversity of V. parahaemolyticus populations was accomplished by the establishment of the multilocus sequence typing (MLST) scheme for V. parahaemolyticus (19) and a centralized database (http://pubmlst.org/vparahaemolyticus) in 2008. This MLST database has enabled researchers from around the world to compare isolates. Currently, more than 2,477 strains from diverse regions of the world, belonging to 1,681 STs, are available for analyses. Genetic variants identified as prevalent in the different regions of the world can be mapped to identify potential connections between populations from diverse geographical areas and delineate potential routes of dispersion. Although useful, MLST is based on sequence analysis of 7 chosen housekeeping genes and therefore lacks enough resolution when used in outbreak scenarios to discriminate between related and unrelated strains at the ST level (19).
The prices for performing whole-genome sequencing (WGS) have decreased dramatically during the last 5 years, with genomes costing around $50 to $100 USD. Scientists have been using WGS to reanalyze historical collections of pathogens and outbreak strains, resulting in a new way of performing outbreak investigations. WGS analyses, such as WGS-single nucleotide polymorphism (WGS-SNP) (20–26) and core genome MLST analyses (15, 16, 27–33), have been used extensively for epidemiological trace back investigations of outbreaks. WGS data analyses allow us to better understand both population dynamics and the mechanisms which contribute to increased virulence among foodborne bacterial pathogens.
cgMLST schemes have already been successfully used for the analysis of different epidemiological investigations, such as the two recent V. parahaemolyticus outbreaks in Maryland (pandemic ST3 strains in MD in 2014 and a retrospective analysis of ST8 strains in MD in 2010) (15), the identification of a novel clone of V. parahaemolyticus causing infections in Peru (16), and the description of an emergent V. parahaemolyticus pathogenic strain (ST631) causing illnesses in the North Atlantic coast of the United States (34). All of the cgMLST schemes used in these analyses were custom-made for each strain type and according to a specific epidemiological context where strains were very similar and shared most of the genes with the reference strain (>83%) (12, 15, 16, 34). Therefore, there is a need to generate a portable, standardized, and more advanced system for the analysis of V. parahaemolyticus strains. Using WGS data will introduce a higher level of resolution and discrimination into the study of populations collected from all around the world, which can be analyzed using a universal cgMLST scheme for V. parahaemolyticus.
To establish this universal V. parahaemolyticus cgMLST scheme, we sequenced 92 V. parahaemolyticus genome representatives from the STs prevailing in different areas of the world. We used the genome of strain RIMD 2210633, which contained 4,832 total genes, as a reference, of which 2,254 genes were selected to create the new V. parahaemolyticus cgMLST scheme after analyzing those 92 genomes. Additionally, another 142 genomes available at NCBI were included in the study to evaluate the performance of the new cgMLST scheme. The cgMLST analysis was able to distinguish related and unrelated strains, including those with the same ST, clearly showing its enhanced resolution over conventional MLST analysis. The sequences obtained from this work were deposited and are available online in a public cgMLST V. parahaemolyticus database (http://pubmlst.org/vparahaemolyticus).
RESULTS
Sequencing of representative strains of V. parahaemolyticus for setting up the cgMLST scheme.
Ninety-two V. parahaemolyticus strains, previously used for setting up the MLST scheme for this bacterium (19), were sequenced to reach >25× average coverage using MiSeq (Illumina) (Table 1). Genome sequences with low coverage (<25×) usually result in low sequencing qualities and incorrect assemblies. Forty-eight additional strains previously sequenced by Ion Torrent (5) were resequenced by MiSeq in order to generate better-quality genomes (Table 2) and to validate the cgMLST scheme. In silico multilocus sequence typing (MLST; http://pubmlst.org/vparahaemolyticus) analysis of the de novo assembled contigs confirmed the identity of every V. parahaemolyticus strain (Tables 1 and 2).
TABLE 1.
List of V. parahaemolyticus strains sequenced in this study
| Isolate | CFSAN no. | Yr | Country | Source | STa | Serotype | Accession no. | Coverage (×) |
|---|---|---|---|---|---|---|---|---|
| 428/00 | CFSAN018752 | 1998 | Spain | C | 17 | O4:K11 | LHAU00000000 | 145 |
| 30824 | CFSAN018753 | 1999 | Spain | C | 17 | O4:K11 | LHAV00000000 | 88 |
| 9808/1 | CFSAN018754 | 2004 | Spain | C | 3 | O3:K6 | LHAW00000000 | 131 |
| UCM-V441 | CFSAN018755 | 2002 | Spain | E | 52 | O4:Kunk | LHAX00000000 | 107 |
| UCM-V586 | CFSAN018756 | 2003 | Spain | E | 45 | O8:K22 | LHAY00000000 | 114 |
| 906-97 | CFSAN018757 | 1997 | Peru | C | 3 | O3:K6 | LHAZ00000000 | 127 |
| 357-99 | CFSAN018758 | 1999 | Peru | C | 19 | O3:Kunk | LHBA00000000 | 148 |
| K0976 | CFSAN001174 | 2004 | USA | E | 50 | O6:K18 | LHBB00000000 | 73 |
| K1068 | CFSAN018760 | 2004 | USA | E | 61 | O5:Kunk | LHBC00000000 | 83 |
| K1297 | CFSAN018761 | 2004 | USA | E | 12 | O5:K17 | LHBD00000000 | 102 |
| K1314 | CFSAN018762 | 2004 | USA | E | 12 | O4:K63 | LHBE00000000 | 34 |
| K1202 | CFSAN018763 | 2004 | USA | E | 43 | O4:K63 | LHBF00000000 | 115 |
| K1322 | CFSAN018764 | 2004 | USA | E | 58 | O3:K56 | LHBG00000000 | 108 |
| K1186 | CFSAN018765 | 2004 | USA | E | 58 | O3:K20 | LHBH00000000 | 72 |
| K1296 | CFSAN018766 | 2004 | USA | E | 9 | O10:K68 | LHBI00000000 | 77 |
| K1303 | CFSAN018767 | 2004 | USA | E | 20 | O1:Kunk | LHBJ00000000 | 131 |
| NY3547 | CFSAN001172 | 1998 | USA | E | 98 | O4:K55 | LHQW00000000 | 53 |
| ATCC 17802 | CFSAN022339 | 1951 | Japan | C | 1 | O1:K1 | MQUE00000000 | 92 |
| K1193 | CFSAN022890 | 2004 | USA | E | 15 | O1:K9 | SRR5070562 | 77 |
| K1317 | CFSAN022891 | 2004 | USA | E | 23 | O1:K54 | SRR5070560 | 129 |
| K1302 | CFSAN022892 | 2004 | USA | E | 50 | O1:K25 | SRR5070559 | 50 |
| 48262 | CFSAN022893 | 1990 | USA | C | 43 | O1:K56 | SRR5070561 | 93 |
| HC-01-22 | CFSAN022894 | 2001 | USA | C | 43 | O4:K63 | SRR5070563 | 78 |
| 049-2A3 | CFSAN022895 | 1997 | USA | E | 57 | O4:K29 | SRR5070568 | 65 |
| HC-01-20 | CFSAN022896 | 2001 | USA | E | 199 | O1:Kunk | SRR5070567 | 96 |
| M25-0B | CFSAN022897 | 1993 | USA | E | 22 | O4:Kunk | SRR5070565 | 84 |
| HC-01-06 | CFSAN022898 | 2001 | USA | E | 199 | O1:Kunk | SRR5070566 | 37 |
| 9546257 | CFSAN022899 | 1995 | USA | C | 32 | O4:K8 | SRR5070569 | 144 |
| 98-506-B102 | CFSAN022900 | 1998 | USA | E | 30 | O5:K11 | SRR5070574 | 91 |
| 98-506-B103 | CFSAN022901 | 1998 | USA | E | 30 | O5:K11 | SRR5070571 | 112 |
| 98-513-F51 | CFSAN022902 | 1998 | USA | E | 34 | O4:K9 | SRR5070570 | 95 |
| 98-548-D11 | CFSAN023517 | 1998 | USA | E | 34 | O4:K9 | SRR5070572 | 110 |
| 98-605-A9 | CFSAN023518 | 1998 | USA | E | 30 | O5:K17 | SRR5070573 | 43 |
| 98-605-A10 | CFSAN023519 | 1998 | USA | E | 30 | O5:K17 | SRR5070586 | 99 |
| 99-524-A9 | CFSAN023520 | 1999 | USA | E | 53 | O3:K34 | SRR5070584 | 98 |
| 99-780-C12 | CFSAN023521 | 1999 | USA | E | 29 | O11:Kunk | SRR5070588 | 148 |
| DI-B11 | CFSAN023522 | 1999 | USA | E | 54 | O1:K22 | SRR5070587 | 110 |
| DI-A8 | CFSAN023523 | 2000 | USA | E | 46 | O1:K30 | SRR5070585 | 136 |
| DI-B-6-4 | CFSAN023524 | 2000 | USA | E | 47 | O1:K30 | SRR5070601 | 102 |
| CP-B-5 | CFSAN023525 | 2000 | USA | E | 23 | O1:K54 | SRR5070598 | 132 |
| DI-B-1 | CFSAN023526 | 2000 | USA | E | 23 | O1:K54 | SRR5070600 | 82 |
| DI-A-6-1 | CFSAN023527 | 2000 | USA | E | 24 | O1:K55 | SRR5070597 | 142 |
| DI-E5 | CFSAN023528 | 2000 | USA | E | 60 | O1:K55 | SRR5070599 | 79 |
| DI-B9 | CFSAN023529 | 1999 | USA | E | 25 | O1:K56 | SRR5070649 | 103 |
| DI-H8 | CFSAN023530 | 1999 | USA | E | 26 | O1:K56 | SRR5070650 | 89 |
| DI-C2 | CFSAN023531 | 1999 | USA | E | 35 | O4:K9 | SRR5070648 | 70 |
| DI-C5 | CFSAN023532 | 1999 | USA | E | 35 | O4:K9 | SRR5070651 | 65 |
| U5474 | CFSAN023549 | 1980 | Bangladesh | C | 87 | O3:K6 | SRR5071102 | 93 |
| PMA 1.5 | CFSAN023550 | 2005 | Chile | E | 28 | O3:K6 | SRR5071104 | 24 |
| PMA 2.5 | CFSAN023551 | 2005 | Chile | E | 10 | O4:Kunk | SRR5071129 | 30 |
| PMA 3.5 | CFSAN023552 | 2005 | Chile | E | 16 | O4:Kunk | SRR5071130 | 71 |
| PMA 16.5 | CFSAN023553 | 2005 | Chile | E | 48 | O4:K12 | SRR5071131 | 95 |
| PMA 45.5 | CFSAN023555 | 2005 | Chile | E | 49 | O3:K6 | SRR5071133 | 122 |
| PMA 79 | CFSAN023557 | 2004 | Chile | E | 56 | O2:Kunk | SRR5071135 | 43 |
| PMA 112 | CFSAN023558 | 2004 | Chile | E | 6 | O3:K6 | SRR5071136 | 45 |
| PMA 189 | CFSAN023559 | 2004 | Chile | E | 7 | O3:K6 | SRR5071134 | 136 |
| PMA 337 | CFSAN023560 | 2004 | Chile | E | 11 | O7:Kunk | SRR5071137 | 59 |
| PMA 339 | CFSAN023561 | 2004 | Chile | E | 55 | O4:Kunk | SRR5071139 | 36 |
| PMA 3316 | CFSAN023562 | 2004 | Chile | E | 13 | O3:K6 | SRR5071141 | 73 |
| VpHY145 | CFSAN023563 | 1999 | Thailand | C | 3 | O4:K68 | SRR5071143 | 83 |
| KXV-641 | CFSAN023564 | 1998 | Japan | C | 3 | O1:K25 | SRR5071140 | 52 |
| AN-2189 | CFSAN023565 | 1998 | Bangladesh | C | 3 | O4:K68 | SRR5071142 | 80 |
| AP-11243 | CFSAN023566 | 2000 | Bangladesh | C | 3 | O1:Kunk | SRR5071144 | 59 |
| PMA 109.5 | CFSAN023556 | 2005 | Chile | E | 3 | O3:K6 | SRR5071138 | 33 |
| PMA 37.5 | CFSAN023554 | 2005 | Chile | E | 3 | O3:K6 | SRR5071132 | 37 |
| TX2103 | CFSAN023541 | 1998 | USA | C | 3 | O3:K6 | SRR5071094 | 103 |
| BAC-98-3372 | CFSAN023542 | 1998 | USA | C | 3 | O3:K6 | SRR5071092 | 104 |
| BAC-98-3374 | CFSAN023543 | 1998 | USA | C | 42 | O3:K6 | SRR5071095 | 118 |
| BAC-98-4092 | CFSAN023544 | 1998 | USA | C | 3 | O3:K6 | SRR5071096 | 126 |
| AN-5034 | CFSAN023545 | 1998 | Bangladesh | C | 3 | O4:K68 | SRR5071093 | 85 |
| AO-24491 | CFSAN023546 | 1999 | Bangladesh | C | 3 | O1:K25 | SRR5071106 | 94 |
| VpHY191 | CFSAN023547 | 1999 | Thailand | C | 3 | O1:K25 | SRR5071105 | 108 |
| AN-16000 | CFSAN023548 | 1998 | Bangladesh | C | 3 | O1:Kunk | SRR5071103 | 90 |
| Vp81 | CFSAN023533 | 1996 | India | C | 3 | O3:K6 | SRR5070652 | 96 |
| Vp155 | CFSAN023535 | 1996 | India | C | 3 | O3:K6 | SRR5071101 | 132 |
| Vp96 | CFSAN023536 | 1996 | India | C | 3 | O3:K6 | SRR5071097 | 92 |
| Vp208 | CFSAN023537 | 1997 | India | C | 3 | O3:K6 | SRR5071099 | 123 |
| AN-8373 | CFSAN023538 | 1998 | Bangladesh | C | 3 | O3:K6 | SRR5071098 | 100 |
| Vp2 | CFSAN023540 | 1998 | South Korea | C | 3 | O3:K6 | SRR5071100 | 95 |
| 029-1(b) | CFSAN001611 | 1997 | USA | E | 36 | O4:K12 | JNTW02000000 | 104 |
| 48057 | CFSAN001612 | 1990 | USA | C | 36 | O4:K12 | JNTX02000000 | 118 |
| K1198 | CFSAN001614 | 2004 | USA | E | 59 | O4:K12 | JNTY02000000 | 150 |
| 10292 | CFSAN001617 | 1997 | USA | C | 50 | O6:K18 | JNTZ02000000 | 85 |
| 48291 | CFSAN001618 | 1990 | USA | C | 36 | O12:K12 | JNUA02000000 | 99 |
| F11-3A | CFSAN001619 | 1988 | USA | E | 36 | O4:K12 | JNUB02000000 | 113 |
| NY-3483 | CFSAN001620 | 1998 | USA | C | 36 | O4:K12 | JNUC02000000 | 72 |
| K1203 | CFSAN001173 | 2004 | USA | E | 59 | O4:K12 | JNUD02000000 | 47 |
| 98-513-F52 | CFSAN001160 | 1998 | USA | E | 34 | O4:K9 | JNUE02000000 | 39 |
| 10290 | CFSAN001613 | 1997 | USA | C | 37 | O4:K12 | JNUF02000000 | 51 |
| JJ21-1C | CFSAN001615 | 1990 | USA | E | 38 | O4:Kunk | LHPD00000000 | 64 |
| W9OA | CFSAN001616 | 1982 | USA | E | 59 | O4:K12 | LHPE00000000 | 39 |
| VP43-1A | CFSAN001621 | 1992 | USA | E | 36 | O4:Kunk | LHQV00000000 | 92 |
C, clinical; E, environmental.
TABLE 2.
List of V. parahaemolyticus genomes from NCBI used for further testing of the newly created cgMLST
| Isolate | CFSAN no.a | Yr | Country | Sourceb | ST | Serotypec | Accession no. | Reference or source |
|---|---|---|---|---|---|---|---|---|
| From our lab | ||||||||
| MDVP1d | CFSAN007429 | 2012 | USA | C | 631 | unk | JNSM02000000 | This study |
| MDVP8d | CFSAN007430 | 2012 | USA | C | 631 | unk | JNSN02000000 | This study |
| MDVP9d | CFSAN007431 | 2012 | USA | C | 631 | unk | JNSO02000000 | This study |
| MDVP31d | CFSAN007432 | 2013 | USA | C | 631 | unk | JNSP02000000 | This study |
| MDVP35d | CFSAN007433 | 2013 | USA | C | 631 | unk | JNSQ02000000 | This study |
| MDVP41d | CFSAN007434 | 2013 | USA | C | 631 | unk | JNSR02000000 | This study |
| MDVP44d | CFSAN007435 | 2013 | USA | C | 631 | unk | JNSS02000000 | This study |
| MDVP45d | CFSAN007436 | 2013 | USA | C | 631 | unk | JNST02000000 | This study |
| MDVP2d | CFSAN007437 | 2012 | USA | C | 651 | unk | JNSU02000000 | This study |
| MDVP3d | CFSAN007438 | 2012 | USA | C | 652 | unk | JNSV02000000 | This study |
| MDVP4d | CFSAN007439 | 2012 | USA | C | 653 | unk | JNSW02000000 | This study |
| MDVP34d | CFSAN007440 | 2013 | USA | C | 653 | unk | JNSX02000000 | This study |
| MDVP5d | CFSAN007441 | 2012 | USA | C | 113 | unk | JNSY02000000 | This study |
| MDVP7d | CFSAN007442 | 2012 | USA | C | 34 | unk | JNSZ02000000 | This study |
| MDVP11d | CFSAN007443 | 2012 | USA | C | 1116 | unk | JNTA02000000 | This study |
| MDVP6d | CFSAN007444 | 2012 | USA | C | 677 | unk | JNTB02000000 | This study |
| MDVP10d | CFSAN007445 | 2012 | USA | C | 43 | unk | JNTC02000000 | This study |
| MDVP13d | CFSAN007446 | 2012 | USA | C | 678 | unk | JNTD02000000 | This study |
| MDVP14d | CFSAN007447 | 2012 | USA | C | 162 | unk | JNTE02000000 | This study |
| MDVP15d | CFSAN007448 | 2012 | USA | C | 679 | unk | JNTF02000000 | This study |
| MDVP39d | CFSAN007455 | 2013 | USA | E | 896 | unk | JNTL02000000 | This study |
| 090-96-70d | CFSAN001595 | 1996 | Peru | C | 189a | O4:K8 | JFFP02000000 | This study |
| VP16MDd | CFSAN007449 | 2012 | USA | C | 3 | unk | JNTG02000000 | This study |
| VP17MDd | CFSAN007450 | 2012 | USA | C | 3 | unk | JNTH02000000 | This study |
| VP18MDd | CFSAN007451 | 2012 | USA | C | 3 | unk | JNTI02000000 | This study |
| MDVP19d | CFSAN007452 | 2010 | USA | C | 8 | unk | JNTJ02000000 | 15 |
| MDVP20d | CFSAN007453 | 2010 | USA | C | 8 | unk | JNTK02000000 | 15 |
| MDVP22d | CFSAN007454 | 2010 | USA | E | 676 | unk | JNUO02000000 | 15 |
| MDVP25d | CFSAN007456 | 2010 | USA | E | 810 | unk | JNUK02000000 | 15 |
| MDVP26d | CFSAN007457 | 2010 | USA | E | 811 | unk | JNUL02000000 | 15 |
| MDVP27d | CFSAN007458 | 2010 | USA | E | 34 | unk | JNUM02000000 | 15 |
| MDVP28d | CFSAN007459 | 2010 | USA | E | 768 | unk | JNUN02000000 | 15 |
| MDVP21d | CFSAN012491 | 2010 | USA | E | 8 | unk | JNUG02000000 | 15 |
| MDVP23d | CFSAN012492 | 2010 | USA | E | 8 | unk | JNUH02000000 | 15 |
| MDVP24d | CFSAN012493 | 2010 | USA | E | 8 | unk | JNUI02000000 | 15 |
| MDVP29d | CFSAN012494 | 2010 | USA | E | 8 | unk | JNUJ02000000 | 15 |
| 281-09d | CFSAN025052 | 2009 | Peru | C | 120 | O3:K59 | LKQB00000000 | 16 |
| 283-09d | CFSAN025053 | 2009 | Peru | C | 120 | O3:K59 | LKQA00000000 | 16 |
| C220-09d | CFSAN025054 | 2009 | Peru | C | 120 | O3:KUT | LKQC00000000 | 16 |
| C224-09d | CFSAN025055 | 2009 | Peru | C | 120 | O3:K59 | LKQD00000000 | 16 |
| C226-09d | CFSAN025056 | 2009 | Peru | C | 120 | O3:K59 | LKQE00000000 | 16 |
| C244-09d | CFSAN025057 | 2009 | Peru | C | 120 | O3:K59 | LKQF00000000 | 16 |
| C235d | CFSAN025058 | 2009 | Peru | C | 120 | O3:K59 | LKQG00000000 | 16 |
| PIURA 17d | CFSAN025059 | 2009 | Peru | C | 120 | O3:K59 | LKQH00000000 | 16 |
| C237d | CFSAN025060 | 2009 | Peru | C | 120 | O3:K59 | LKQI00000000 | 16 |
| 239-09d | CFSAN025061 | 2009 | Peru | C | 120 | O3:K59 | LKQJ00000000 | 16 |
| 241-09d | CFSAN025062 | 2009 | Peru | C | 120 | O3:K59 | LKQK00000000 | 16 |
| 245-09d | CFSAN025063 | 2009 | Peru | C | 120 | O3:K59 | LKQL00000000 | 16 |
| CO1409d | CFSAN025064 | 2009 | Peru | C | 120 | O3:K59 | LKQM00000000 | 16 |
| CO1609d | CFSAN025065 | 2009 | Peru | C | 120 | O3:K59 | LKQN00000000 | 16 |
| 285-09d | CFSAN025066 | 2009 | Peru | C | 120 | O3:K59 | LKQO00000000 | 16 |
| 287-09d | CFSAN025067 | 2009 | Peru | C | 120 | O3:K59 | LKQP00000000 | 16 |
| 379-09d | CFSAN025068 | 2009 | Peru | C | 120 | O3:K59 | LKQQ00000000 | 16 |
| P306d | CFSAN029653 | 2009 | Peru | E | 120 | O3:K59 | LKQR00000000 | 16 |
| Guillen 151 Perud | CFSAN029654 | 2009 | Peru | E | 120 | O3:K59 | LKQS00000000 | 16 |
| P310d | CFSAN029656 | 2009 | Peru | E | 120 | O3:K59 | LKQT00000000 | 16 |
| From other labs | ||||||||
| 10-4287d | NA | 2003 | Canada | C | 50 | O6:K18 | JYJU00000000 | Unpublished datai |
| BB22OPg | NA | 1995 | Bangladesh | E | 88 | O4:K8 | NC_019955.1, NC_019971.1 | 51 |
| CDC_K4557e | NA | 2006 | USA | C | 799 | O1:K53 | NC_021822.1, NC_021848.1 | 52 |
| FDA_R31e | NA | 2007 | USA | E | 23 | O1:Kunk | NC_021847.1, NC_021821.1 | 52 |
| RIMD 2210633h | NA | 2003 | Japan | C | 3 | O3:K6 | NC_004605.1, NC_004603.1 | 35 |
| FORC_008d,e,g | NA | 2004 | South Korea | E | 984 | unk | NZ_CP009982.1, NZ_CP009983.1 | Unpublished dataj |
| UCM-V493d,e | NA | 2002 | Spain | E | 471 | O2:K28 | CP007004, CP007005 | 53 |
| CHN25g | NA | 2011 | China | E | 395 | unk | NZ_CP010884.1, NZ_CP010883.1 | Unpublished datak |
| FORC_004e | NA | 2014 | South Korea | E | 1628 | unk | NZ_CP009848.1, NZ_CP009847.1 | Unpublished datak |
| FORC_006d,e | NA | 2014 | South Korea | E | 1630 | unk | NZ_CP009765.1, NZ_CP009766.1 | Unpublished datak |
| FORC_014e | NA | 2015 | South Korea | E | 1629 | unk | NZ_CP011407.1, NZ_CP011406.1 | Unpublished datak |
| KVp10d | NA | 2007 | Sweden | E | 1579 | unk | MBTR01 | Unpublished datal |
| R10B2_71d | NA | 1997 | USA | E | 1556 | unk | MCFR01 | Unpublished datam |
| 04-2192d | NA | 2004 | Canada | C | 629 | unk | LQCB01 | Unpublished datan |
| 04-2550d | NA | 2004 | Canada | C | 630 | unk | LRAH01 | Unpublished datan |
| 05-3133d | NA | 2005 | Canada | C | 43 | unk | LRAI01 | Unpublished datan |
| 05-4792d | NA | 2005 | Canada | C | 199 | unk | LPUZ01 | Unpublished datan |
| 07-2964d | NA | 2007 | Canada | C | 8 | unk | LRSV01 | Unpublished datan |
| 09-1772d | NA | 2009 | Canada | C | 417 | unk | LRSX01 | Unpublished datan |
| 09-3219d | NA | 2009 | Canada | C | 36 | unk | LRSW01 | Unpublished datan |
| 09-4436d | NA | 2009 | Canada | C | 631 | unk | LRAJ01 | Unpublished datan |
| 09-4661d | NA | 2009 | Canada | C | 417 | unk | LNTR01 | Unpublished datan |
| 09-4662d | NA | 2009 | Canada | C | 417 | unk | LRTH01 | Unpublished datan |
| 09-4665d | NA | 2009 | Canada | C | 417 | unk | LRFL01 | Unpublished datan |
| 09-4666d | NA | 2009 | Canada | C | 417 | unk | LQCC01 | Unpublished datan |
| A0EZ383d | NA | 2000 | Canada | C | 638 | unk | LRSY01 | Unpublished datan |
| A0EZ608d | NA | 2000 | Canada | C | 36 | unk | LRFM01 | Unpublished datan |
| A0EZ664d | NA | 2000 | Canada | C | 50 | unk | LRFN01 | Unpublished datan |
| A0EZ713d | NA | 2000 | Canada | C | 50 | unk | LRFO01 | Unpublished datan |
| A1EZ679d | NA | 2001 | Canada | C | 36 | unk | LRSZ01 | Unpublished datan |
| A1EZ919d | NA | 2001 | Canada | C | 36 | unk | LNTX01 | Unpublished datan |
| A1EZ952d | NA | 2001 | Canada | C | 43 | unk | LRTI01 | Unpublished datan |
| A2EZ523d | NA | 2002 | Canada | C | 36 | unk | LRTA01 | Unpublished datan |
| A2EZ614d | NA | 2002 | Canada | C | 43 | unk | LRFP01 | Unpublished datan |
| A2EZ715d | NA | 2002 | Canada | C | 36 | unk | LRFQ01 | Unpublished datan |
| A2EZ743d | NA | 2002 | Canada | C | 324 | unk | LRFR01 | Unpublished datan |
| A3EZ136d | NA | 2003 | Canada | C | 3 | unk | LRFS01 | Unpublished datan |
| A3EZ634d | NA | 2003 | Canada | C | 50 | unk | LRTB01 | Unpublished datan |
| A3EZ710d | NA | 2003 | Canada | C | 43 | unk | LRTC01 | Unpublished datan |
| A3EZ711d | NA | 2003 | Canada | C | 43 | unk | LRTD01 | Unpublished datan |
| A3EZ770d | NA | 2003 | Canada | C | 50 | unk | LRTE01 | Unpublished datan |
| A3EZ799d | NA | 2003 | Canada | C | 43 | unk | LRTF01 | Unpublished datan |
| A3EZ936d | NA | 2003 | Canada | C | 1060 | unk | LRTG01 | Unpublished datan |
| A4EZ700d | NA | 2004 | Canada | C | 43 | unk | LOBT01 | Unpublished datan |
| A4EZ703d | NA | 2004 | Canada | C | 141 | unk | LODO01 | Unpublished datan |
| A4EZ724d | NA | 2004 | Canada | C | 43 | unk | LOHO01 | Unpublished datan |
| A4EZ927d | NA | 2004 | Canada | C | 3 | unk | LOHN01 | Unpublished datan |
| A4EZ964d | NA | 2004 | Canada | C | 636 | unk | LQGX01 | Unpublished datan |
| A5Z1022d | NA | 2005 | Canada | C | 15 | unk | LRFT01 | Unpublished datan |
| A5Z273d | NA | 2005 | Canada | C | ? | unk | LQCD01 | Unpublished datan |
| A5Z652d | NA | 2005 | Canada | C | 36 | unk | LQCE01 | Unpublished datan |
| A5Z853d | NA | 2005 | Canada | C | 3 | unk | LQCF01 | Unpublished datan |
| A5Z860d | NA | 2005 | Canada | C | 43 | unk | LQCS01 | Unpublished datan |
| A5Z878d | NA | 2005 | Canada | C | 36 | unk | LQCT01 | Unpublished datan |
| A5Z905d | NA | 2005 | Canada | C | 36 | unk | LQCU01 | Unpublished datan |
| A5Z924d | NA | 2005 | Canada | C | 36 | unk | LQCV01 | Unpublished datan |
| C140d | NA | 2008 | Canada | C | 332 | unk | LQCW01 | Unpublished datan |
| C142d | NA | 2008 | Canada | C | 417 | unk | LPVA01 | Unpublished datan |
| C143d | NA | 2008 | Canada | C | 36 | unk | LPVB01 | Unpublished datan |
| C144d | NA | 2008 | Canada | C | 36 | unk | LPVC01 | Unpublished datan |
| C145d | NA | 2008 | Canada | C | 417 | unk | LPVK01 | Unpublished datan |
| C146d | NA | 2008 | Canada | C | 1060 | unk | LPVL01 | Unpublished datan |
| C147d | NA | 2008 | Canada | C | 36 | unk | LPVM01 | Unpublished datan |
| C148d | NA | 2008 | Canada | C | 43 | unk | LPVN01 | Unpublished datan |
| C150d | NA | 2008 | Canada | C | 417 | unk | LPVU01 | Unpublished datan |
| F1419d | NA | 2006 | Canada | C | 43 | unk | LRSU01 | Unpublished datan |
| F30368d | NA | 2006 | Canada | C | 8 | unk | LRFV01 | Unpublished datan |
| F4395d | NA | 2006 | Canada | C | 36 | unk | LRFU01 | Unpublished datan |
| F63267d | NA | 2006 | Canada | C | 3 | unk | LRFW01 | Unpublished datan |
| H11523d | NA | 2006 | Canada | C | 36 | unk | LRFY01 | Unpublished datan |
| H18983d | NA | 2006 | Canada | C | 36 | unk | LRST01 | Unpublished datan |
| H64024d | NA | 2006 | Canada | C | 36 | unk | LRFZ01 | Unpublished datan |
| M59787d | NA | 2006 | Canada | C | 36 | unk | LRJZ01 | Unpublished datan |
| T8994d | NA | 2006 | Canada | C | 36 | unk | LRGA01 | Unpublished datan |
| W501d | NA | 2006 | Canada | C | 635 | unk | LRFX01 | Unpublished datan |
| HS-06-05d | NA | 2014 | Canada | E | 614 | unk | LIRS01 | Unpublished datan |
| ISF-29-3d | NA | 2011 | Canada | E | 1518 | unk | LFYM01 | Unpublished datan |
| ISF-54-12d | NA | 2011 | Canada | E | 1631 | unk | LIRR01 | Unpublished datan |
| S357-21d | NA | 2010 | Canada | E | 102 | unk | LFYN01 | Unpublished datan |
| S372-5d | NA | 2011 | Canada | E | 324 | unk | LIRQ01 | Unpublished datan |
| ISF-94-1d | NA | 2011 | Canada | E | 1632 | unk | LIRT01 | Unpublished datan |
| RM-14-5d | NA | 2014 | Canada | E | 1663 | unk | LFXK01 | Unpublished datan |
| Gxw_7004f | NA | 2007 | China | C | 3 | unk | LPZS01 | Unpublished datao |
| Gxw_9143f | NA | 2009 | China | C | 265 | unk | LPZT01 | Unpublished datap |
| K23d | NA | 2013 | India | E | 1052 | unk | LQGU01 | 54 |
NA, not applicable.
C, clinical; E, environmental.
unk–unknown.
MiSeq sequencing platform.
PacBio sequencing platform.
HiSeq sequencing platform.
454 sequencing platform.
Sanger sequencing platform.
J. Ronholm, N. Petronella, R. Kenwell, and S. Banerjee.
J.-H. Lee, D.-H. Lee, S. Kim, H.-J. Ku, H. Y. Chung, H. Kim, S. Ryu, and S.-H. Choi.
C. Zhu, B. Sun, T. Liu, H. Zheng, and L. Chen.
J. W. Turner, R. N. Paranjpye, B. Collin, L. J. Pinnell, and J. Tallman.
K. C. Liu.
J. Ronholm, N. Petronella, and S. Banerjee.
Y. Huang, H. Wang, Y. Pang, Z. Tang, Y. Zhou, and G. Sun.
Y. Huang, H. Wang, Y. Pang, Z. Tang, Y. Zhou, C. Qu, L. Lan, C. Wei, and C. Wang.
Development of a cgMLST for V. parahaemolyticus.
The initial setup of the cgMLST for V. parahaemolyticus using the genome of strain RIMD 2210633 as the reference genome (4,832 genes total) generated 3,709 potential core gene targets for use in the cgMLST scheme after eliminating duplicated, truncated, and accessory genes. RIMD 2210633 is a prototypic ST3 pandemic strain and was fully sequenced in 2003 using Sanger sequencing technology (35). Only core genes were used for constructing the cgMLST scheme. Of the 3,709 potential core genes identified in the comparison of strain RIMD 2210633 with seven other V. parahaemolyticus strains (BB22OP, CDC_K4557, FDA_R31, UCM-V493, FORC_008, FORC_006, and FORC_004), only 2,254 genes were present in every genome of the additional 92 V. parahaemolyticus strains used to define the final cgMLST scheme (see Table S1 in the supplemental material). These 92 strains represented a diverse set of strains isolated from environmental and clinical sources, as well as from different locations (Table 1).
Implementation of the V. parahaemolyticus cgMLST website.
The cgMLST scheme was implemented into the BIGSdb database hosting the original MLST scheme for V. parahaemolyticus (http://pubmlst.org/vparahaemolyticus). This database allows for testing contigs of new V. parahaemolyticus genomes for the presence and typing of 2,254 genes. Briefly, the BIGSdb genome comparator tool performs a cgMLST analysis, which produces a color-coded cgMLST output (e.g., see Table S2), facilitating comparison among isolates (see Materials and Methods for specific details).
Evaluation of the cgMLST target gene set.
All V. parahaemolyticus genomes generated in this study, as well as a collection of 142 additional V. parahaemolyticus genomes available at NCBI (Table 2), were used to validate this cgMLST scheme (Fig. 1). The average percentage of cgMLST targets called was 99.21%. Only five assembled genomes contained incomplete loci: 97-10290 (two incomplete loci), Guillen_151_Peru (six incomplete loci), P310 (two incomplete loci), C148 (one incomplete locus), and HS-06-05 (seven incomplete loci). The output of this general analysis produced an informative Excel file (Table S2) composed of different sheets, with each one containing different results, as explained in Materials and Methods. cgMLST analysis for the 234 genomes available in the MLST database allowed a fast phylogenetic exploration of V. parahaemolyticus genomes (Fig. 1), clearly differentiating strains belonging to different STs, clustering strains with same STs, and allowing for further discrimination among strains within a specific ST.
FIG 1.
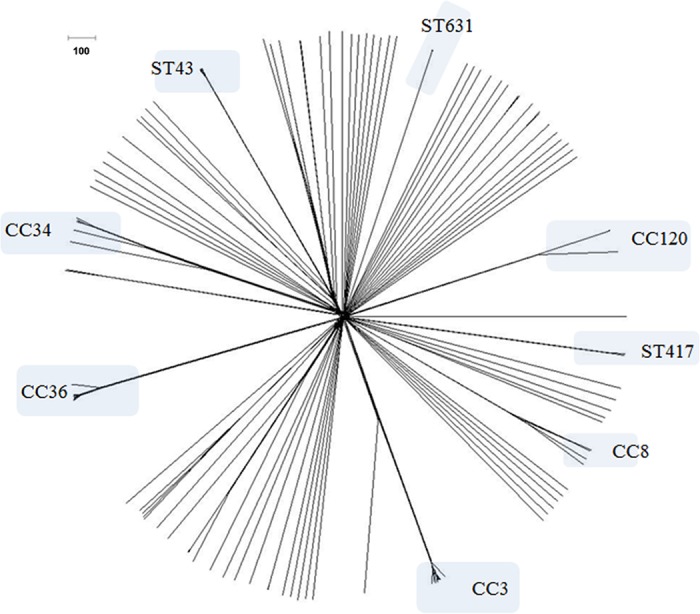
cgMLST analysis of the 234 V. parahaemolyticus genomes available at the V. parahaemolyticus MLST database using the genome comparator tool implemented within the MLST database (NeighborNet phylogenetic network). Visualization of the nexus file exported from the cgMLST analysis report in Splits Tree software (48). The names at the nodes were removed for easy visualization. The original tree with the nodes names is available in Fig. S1 in the supplemental material.
Evaluation of the cgMLST scheme using genomes of strains belonging to four known STs from outbreak-related and non-outbreak-related strains.
The performance of this cgMLST scheme was tested using six different sets of informative V. parahaemolyticus strains whose genomes were available and that clustered together in the global data set (Fig. 1). In addition to the unique pandemic clone of V. parahaemolyticus identified to date (clonal complex 3 [CC3]), other major groups with a relevance on a local or transnational scale were also analyzed: (i) strains belonging to ST36 (CC36) (outbreak related and non-outbreak related) (5, 19, 36) (Fig. 2B), (ii) strains belonging to ST8 (CC8) that were outbreak related, isolated in MD in 2010 (15) (Fig. 2C), (iii) strains belonging to ST120 (CC120) from the same outbreak (Peru, 2009) and that were recently characterized (16) (Fig. 2D), and (iv) strains belonging to ST631, a new emergent clone in the East Coast of the United States (5, 34, 36) (Fig. 2E).
FIG 2.


cgMLST analysis of representative V. parahaemolyticus strains from same outbreaks and/or non-outbreak related displaying the same ST identified in Fig. 1. (A) CC3 outbreak-related (12) and non-outbreak-related (19). (B) CC36-ST36 outbreak-related and non-outbreak-related strains (5, 19, 36). (C) CC8-ST8 outbreak-related and non-outbreak-related strains (15). (D) CC120 outbreak (Peru, 2009 [16]) strains 281-09, 241-09, 379-09, CO1409, CO1609, P310, Guillen_151_Peru, C226-09, C224-09, C235, PIURA_17, C237, and 239-09, were identical by cgMLST (represented by letter a). (E) ST631 strains (5, 34, 36). The scale represents the number of allele differences.
CC3.
The first test of the new cgMLST was performed using strains belonging to the pandemic clone CC3 using a panel of 30 strains (all ST3) epidemiologically unrelated, along with some additional strains collected in the course of a single epidemiological event (typically the same outbreak), including the recently reported strains MDVP16, MDVP7, and MDVP18 that caused a small outbreak in MD in 2014 (12) (Fig. 2A).
The cgMLST analysis of the genomes identified as CC3 by MLST consistently grouped the strains according to their serotype. Strains of serotypes O3:K6, O1:Kunk, O1:K25, and O4:K68 were efficiently discriminated and included in independent clusters. A high level of diversity was found within each cluster, even though these strains were highly related by PFGE profiling and random amplified polymorphic DNA (RAPD). The cgMLST was highly effective in separating strains that were less related to each other (e.g., see O3:K6 group). Noteworthy, cgMLST analysis showed that the first reported outbreaks of pandemic V. parahaemolyticus in the United States in 1998 (NY and TX) were caused by two different strains and differed by at least 14 loci from each other (detailed analysis can be found in Table S3). The strains causing the outbreak in MD in 2014 were grouped together and divergent from the original O3:K6 strains (old ST3 strains) by >30 loci. Strains MDVP17 and MDVP18 were undistinguishable and differed from MDVP16 by 1 locus, confirming that this outbreak in MD in 2014 was caused by a single strain.
CC36.
CC36 includes strains typically causing infections in the Pacific Northwest United States and Canada (2, 19, 37). Figure 2B shows the analysis of strains belonging to CC36 from the United States and Canada isolated over the last 20 years from clinical and environmental sources. The cgMLST analysis clearly separated them into two distinct groups: strains isolated before 2000 (old or classic clone ST36) and strains isolated after 2000 (new clone ST36). The results of the cgMLST analysis can be found in Table S4. For illustration purposes here in this analysis, we focused on the known outbreak strains isolated in MD during the period of 2012 to 2013. In 2012, there was an outbreak on the East Coast of the United States caused by a unique ST36 clone (5, 38). This clone is represented by strain MDVP12 (grouped as strain 2 in the tree). However, as can be observed during the 2013 season, in the remaining MDVP strains, there were at least 3 different strains causing clinical cases during that year.
CC8.
Strains belonging to this CC8 have been described as primarily causing illnesses in Asia (15); however, strains belonging to CC8 caused a small outbreak in MD in 2010 (15). Haendiges et al. (15) showed that these clinical ST8 strains were almost indistinguishable from strains isolated from oysters in MD and that they were different from other ST8 strains that were available at NCBI. Therefore, we chose these strains to test the performance of the newly developed V. parahaemolyticus cgMLST. Figure 2C shows the cgMLST analysis of these ST8 strains from an outbreak in MD in 2010 and their relationship to two other strains isolated in Canada. The qualities of the ST8 sequences available from NCBI were not “up to par” and were not included in this analysis, because they were sequenced at low coverage, and too many contigs were generated in their assembly (>300), indicative of the low quality of the sequences. The cgMLST analysis results (Table S5) clearly indicate that all ST8 strains from the MD outbreak in 2010 clustered together (differing up to 2 loci), revealing that the outbreak was caused by the same strain and differed by >500 loci from the ST8 strains isolated in Canada in 2006 and 2007.
CC120.
Strains belonging to this CC120 and that were ST120 suddenly emerged in Peru during the course of a cross-country epidemic event in 2009 causing infections in different cities throughout the country (16). Figure 2D shows the cgMLST analysis with a set of 20 strains belonging to ST120 previously characterized by another cgMLST (custom reference based), causing an outbreak of gastroenteritis in Peru in 2009 (16). The results from the cgMLST analysis (Table S6) identified 11 of the 20 strains as undistinguishable, and the remaining 9 strains differed by 1 to 3 loci, indicating the high clonality of these strains and that they were indeed part of the same outbreak.
ST631.
Strains belonging to ST631, which were also previously characterized by another custom-made cgMLST (34), were tested with the V. parahaemolyticus cgMLST. These strains belong to a new emergent V. parahaemolyticus clone causing the second highest number of V. parahaemolyticus illnesses in the East Coast of the United States. The cgMLST analysis results (Table S7) identified a highly clonal structure within this group (with two strains, MDVP8 and MDVP9, being undistinguishable) differing by between 1 and 10 loci, which contrasted with the differences found compared to ST631 strains isolated in Canada (>22 loci) (see Table S7).
DISCUSSION
This study describes the implementation and evaluation of a cgMLST scheme for V. parahaemolyticus using a geographically diverse panel of V. parahaemolyticus strains with global coverage. A database from this study was created and is freely available online (http://pubmlst.org/vparahaemolyticus). The cgMLST scheme consisted of 2,254 target genes and was validated using 142 additional V. parahaemolyticus strains from diverse sources and geographical locations. The new database is a valuable and reliable tool for the unambiguous comparison of data generated from laboratories around the world.
The resequenced 140 genomes provided by this study to the NCBI database encompass a diverse repertoire of strains of historical importance. These genomes were instrumental in the creation of this universal cgMLST scheme for V. parahaemolyticus and represent a diverse set that can be used for other research endeavors, such as virulence typing, PCR detection of specific lineages, evolution, and spreading of different V. parahaemolyticus strains around the world (39, 40). This database will allow for testing contigs of new V. parahaemolyticus genomes for the presence and typing of 2,254 genes. The steady incorporation of new genomes into this database will improve surveillance of this important foodborne pathogen worldwide and provide early detection of new variants being introduced into locations where they are not usually found, as was shown for ST189 (17), ST3 in MD 2014 (12), ST8 in MD 2010 (15), and ST120 in Peru 2009 (16), among others.
The suggested analysis starts with running a default cgMLST analysis with all of the V. parahaemolyticus genomes available in the database, and the new V. parahaemolyticus genomes being tested can be localized in the NeighborNet tree (Fig. 1). This type of analysis allows for a fast phylogenetic examination of V. parahaemolyticus genomes. Then, a more detailed analysis can be produced that includes only the relevant strains contained in the initial tree that clustered with the V. parahaemolyticus genomes tested (Fig. 2). Also, two types of output of the analysis can be performed: a fast analysis output, in which only the allelic information is used, and a more detailed (although slower) output, where not only are the alleles differences, but also an alignment containing the sequences for all the variable genes (loci), are provided. The more detailed output (which is generated in order to be able to generate phylogenetic trees outside the website) can be used to perform additional tests, such as SNP-based phylogeny reconstruction using sequenced-based algorithms, such as maximum likelihood (41), time of evolution (42), or to find a specific sequence signature for an specific lineage or clone.
The evaluation of this universal V. parahaemolyticus cgMLST was performed using five sets of strains known to be part of the same outbreak or unrelated but having the same ST. As expected, cgMLST was extremely efficient in partitioning even among the highly clonal ST3 (pandemic strains), dividing the strains causing an outbreak in the United States in 1998 in two different locations (NY and TX) into two different groups (Fig. 2A). This result is in line with findings from other ongoing studies also identifying these two strains (TX and NY, 1998) having a different origin (our unpublished data). Furthermore, it partitioned the pool of strains in concordance with their serotypes, with all the O3:K6 strains clustering loosely together, while strains from each other serotype were grouped consistently according to serotype. This analysis also showed that the ST3 strains from the outbreak in MD in 2014 (12) were almost identical strains (only 1 SNP difference in one strain among the 2,254 genes analyzed) and very different from the other ST3 strains analyzed. This conclusion was not possible to arrive at previously due to the inherent problems with the sequence quality and analysis performed in the earlier publication (12).
A similar result was achieved with the other sets of strains employed for each individual analysis. Strains belonging to CC36 from the United States and Canada were separated by the V. parahaemolyticus cgMLST analysis into two distinct groups, as observed preliminarily elsewhere (5, 36), with strains isolated before 2000 (classic ST36 clone) and after 2000 (new ST36 clone). It also showed that ST36 strains causing an outbreak in 2013 in MD belonged at least to 3 different lineages. This example clearly shows the performance of the cgMLST for fast clustering and differentiation of strains during an outbreak. The overall MLST discriminatory power expressed by the formula of Simpson's index of diversity (D) for the genomes analyzed was 0.947, which shows that MLST is quite discriminatory but is not enough to discriminate within strains of the same ST. Overall, however, the D of cgMLST was 0.9921, showing a significantly higher discriminatory power than MLST.
This cgMLST analysis has several advantages compared to SNP-based methodologies: it is rapid, reproducible, there is no need for high-performance computers or bioinformatic skills, it allows easy visualization and location on the genome of the loci that differ between or among strains analyzed, the results can be easily transferred between different laboratories, and the information for each genome from all around the world will be stored in the database for future use. In contrast, a limitation of the cgMLST approach is that the analysis is reduced to only coding regions. Of the 4,832 open reading frames (ORFs) used as references (present in RIMD strain), only 50% are shared by the highly diverse V. parahaemolyticus strains used in this study, representing only a fraction of the genome. Therefore, if more detailed or enhanced resolution is needed, whole-genome MLST (wgMLST) using an uploaded annotated reference of a related strain (supported within the website) or a genome-wide SNP analysis is recommended.
V. parahaemolyticus is a natural inhabitant of a wide range of marine habitats, with a life cycle encompassing different stages as free-living organism in seawater, as a component of the microbiota of a vast range of marine organisms, but also as a pathogen in the human gut (43). As a result of this complex lifestyle, this organism is extremely diverse in terms of genomic variation, with a large genomic repertory which enables it to adapt and survive in different habitats under the constant variations in the environmental conditions typical of coastal areas. In addition to mutation, homologous recombination and horizontal gene transfer have been found to represent major contributions to genomic variation in V. parahaemolyticus populations in the need for a rapid adaptation to new habitats under changing environmental conditions (17, 19, 44, 45). These particular features make the phylogenetic analysis of V. parahaemolyticus especially challenging where the identification of the different sources contributing to genetic variation of genomes is needed. For all these reasons, the cgMLST scheme described here represents a notable advance in the genomic analysis of complex organisms, such as V. parahaemolyticus, providing a permanent platform to store available genomes, streamlining the analytical process with the selection of the core genes shared by all the genome and a rapid identification of the variation within each gene, without the need to deal with complex and time-consuming bioinformatics tools, and enabling an urgent response within a context of epidemiological investigation.
In conclusion, we have created a standardized cgMLST scheme that allows for fast typing of V. parahaemolyticus from WGS data in a publicly available database. This cgMLST scheme was tested with a diverse set of strains belonging to the same or unrelated outbreaks and was able to differentiate them accordingly, therefore showing a great potential for use in outbreak investigations. Application of this cgMLST scheme to V. parahaemolyticus strains collected by different laboratories around the world will help define the global picture of the epidemiology, spread, and evolution of this pathogen. All of this information will be critical in its application to outbreak investigations, providing a unique repository of genomes that can be used for unambiguous comparisons of data generated worldwide. Finally, since V. parahaemolyticus is a bacterium highly intertwined with environmental changes, it is our goal to develop a tool that would be able to integrate the results obtained from the cgMLST scheme analysis of the entire database, as it continues to grow, into a geographical visualization that together with environmental variables (e.g., salinity and temperature) would help to determine worldwide dispersal rates of this pathogen and help in modifying risk assessments for this bacterium in different regions.
MATERIALS AND METHODS
Bacterial strains and media.
The V. parahaemolyticus strains sequenced in this study are listed, along with their assigned CFSAN numbers, in Table 1. Strains were selected based on their origin, ST, and date of isolation, with representatives of all the major clinical clones of V. parahaemolyticus prevailing in the different regions of the world. All isolates were retrieved from storage (−80°C freezer), transferred to Luria-Bertani (LB) medium with 3% NaCl, and incubated at 37°C with shaking at 250 rpm. Strains were confirmed in the original studies as belonging to V. parahaemolyticus and subsequently confirmed in this study by in silico MLST and in silico presence of a V. parahaemolyticus-specific gene (Vp-toxR-AB029907) in the genome.
DNA extraction and quantification.
Genomic DNA from each strain was isolated from overnight cultures using the DNeasy blood and tissue kit (Qiagen, Valencia, CA). The concentration was determined using a Qubit double-stranded DNA high-sensitivity (HS) assay kit and a Qubit 2.0 fluorometer (Thermo Scientific, Waltham, MA), according to each manufacturer's instructions.
Whole-genome sequencing, contig assembly, and annotation.
Strains were sequenced (Table 1 and some in Table 2) using an Illumina MiSeq sequencer (Illumina, CA) with 2 × 250-bp paired-end chemistry, according to the manufacturer's instructions, with >25× average coverage. The genome libraries were constructed using the Nextera XT DNA sample prep kit (Illumina). Genomic sequence contigs were de novo assembled using default settings within CLC Genomics Workbench version 8.5.1 (Qiagen), with a minimum contig size threshold of 500 bp in length. The draft genomes were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP [http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html]) (46).
In silico MLST phylogenetic analysis.
The initial analysis and identification of the strains were performed using an in silico V. parahaemolyticus MLST, based on information available at the V. parahaemolyticus MLST website (http://pubmlst.org/vparahaemolyticus/) and using Ridom SeqSphere+ software version 3.1.0 (Ridom, Münster, Germany). Seven loci (dnaE, gyrB, recA, dtdS, pntA, pyrC, and tnaA), previously described for V. parahaemolyticus (19), were used for MLST analysis. The same V. parahaemolyticus MLST database was also used to assign numbers for alleles and sequence types (STs).
cgMLST target gene definition.
The cgMLST scheme for V. parahaemolyticus was created using Ridom SeqSphere software version 3.1.0, with the genome of strain RIMD 2210633 as a reference (Ridom, Münster, Germany). The cgMLST scheme was composed using the cgMLST target definer tool, using the default settings within the software. The reference genome contains 4,832 genes in total (35). The only seven closed V. parahaemolyticus genomes available at NCBI were used to establish a list of core and accessory genome genes (strains BB22OP, CDC_K4557, FDA_R31, UCM-V493, FORC_008, FORC_006, and FORC_004). Core genes, genes shared by all the strains queried, and accessory genes that were only present in some, but not all, of the queried genomes were identified. Genes that were present in more than one copy in any of the eight genomes were removed from the analysis. A genome-wide gene-by-gene cgMLST comparison was performed with every genome queried against the reference.
Establishment of the cgMLST for V. parahaemolyticus website.
The V. parahaemolyticus MLST website (http://pubmlst.org/vparahaemolyticus/) is run using the BIGSdb platform (47) designed for gene-by-gene analysis of whole-genome assemblies. Establishing the cgMLST scheme was a matter of defining the core gene loci within the database and grouping these into a scheme. The first allele (allele 1) for each locus was defined from the RIMD 2210633 strain and added to the database in order to seed it. New variants of each locus were found using the BIGSdb manual Web-based scan tools and automated offline allele definer. This identified new variants by performing a BLAST query of the genome assembly against a database of known alleles. New alleles were assigned automatically if they had an identity of ≥98% with an existing allele over an alignment length of ≥98% of the allele and contained an initial start codon, a final stop codon, and were in frame with no internal stop codons. New alleles that did not match the description above were manually curated. Allele designations and positions for each locus in each genome assembly were recorded within the database.
Genealogical reconstructions using the cgMLST scheme.
Gene-by-gene analysis was performed using the BIGSdb Genome Comparator tool (47). This analysis produced an output showing allelic variation at each locus, further categorized into loci that are (i) varied among all strains, (ii) same among all strains, and (iii) incomplete in some isolates; also included in the output are (iv) unique strains, (v) a distance matrix, and (vi) the parameters used for comparison. The distance matrix generated by the analysis is based on allelic differences across the cgMLST loci, with every locus with a different allele counted as a single difference in pairwise comparisons of isolates. The genealogies were reconstructed from this distance matrix using the NeighborNet algorithm (48) implemented in SplitsTree4 (49) and were either integrated into the PubMLST website or the desktop package was used.
Evaluation of the cgMLST target gene set.
A collection of 142 additional V. parahaemolyticus genomes available at NCBI (Table 2) was used to validate the cgMLST scheme. Some of these genomes were sequenced de novo, because cgMLST performed best with high-quality sequences, which were those with >25× coverage and without indels due to homopolymers or sequencing errors that might arise from some sequencing techniques, such as 454 and Ion Torrent (Table 2). These strains have been isolated from various sources (environmental and clinical) around the world and constitute a diverse set of V. parahaemolyticus strains. Some of them belonged to the same outbreak, and others belonged to the same ST but were not epidemiologically related. All isolates have been previously evaluated by MLST (http://pubmlst.org/vparahaemolyticus). The index of discrimination or discriminatory power (D) of cgMLST and MLST was calculated using the Simpson's index of diversity, as described previously (50).
Accession number(s).
The draft genome sequences for all 129 V. parahaemolyticus strains used in our analyses are available in GenBank under the accession numbers listed in Tables 1 (92 strains) and 2 (37 strains).
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the FDA Foods Program Intramural Funds. Development of the PubMLST site is supported by the Wellcome Trust. J.M.-U. was funded through an NERC project (NE/P004121/1).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.00227-17.
REFERENCES
- 1.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. 1999. Food-related illness and death in the United States. Emerg Infect Dis 5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner JW, Paranjpye RN, Landis ED, Biryukov SV, Gonzalez-Escalona N, Nilsson WB, Strom MS. 2013. Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS One 8:e55726. doi: 10.1371/journal.pone.0055726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DePaola A, Kaysner CA, Bowers J, Cook DW. 1998. 2000. Environmental investigations of Vibrio parahaemolyticus in oysters after outbreaks in Washington, Texas, and New York (1997 and 1998). Appl Environ Microbiol 66:4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noriea NF III, Johnson CN, Griffitt KJ, Grimes DJ. 2010. Distribution of type III secretion systems in Vibrio parahaemolyticus from the northern Gulf of Mexico. J Appl Microbiol 109:953–962. doi: 10.1111/j.1365-2672.2010.04722.x. [DOI] [PubMed] [Google Scholar]
- 5.Haendiges J, Timme R, Allard MW, Myers RA, Brown EW, Gonzalez-Escalona N. 2015. Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012–2013) and comparisons to a locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front Microbiol 6:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park KS, Iida T, Yamaichi Y, Oyagi T, Yamamoto K, Honda T. 2000. Genetic characterization of DNA region containing the trh and ure genes of Vibrio parahaemolyticus. Infect Immun 68:5742–5748. doi: 10.1128/IAI.68.10.5742-5748.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyd EF, Cohen AL, Naughton LM, Ussery DW, Binnewies TT, Stine OC, Parent MA. 2008. Molecular analysis of the emergence of pandemic Vibrio parahaemolyticus. BMC Microbiol 8:110. doi: 10.1186/1471-2180-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto C, Okuda J, Ishibashi M, Iwanaga M, Garg P, Rammamurthy T, Wong HC, DePaola A, Kim YB, Albert MJ, Nishibuchi M. 2000. Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J Clin Microbiol 38:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okuda J, Ishibashi M, Hayakawa E, Nishino T, Takeda Y, Mukhopadhyay AK, Garg S, Bhattacharya SK, Nair GB, Nishibuchi M. 1997. Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J Clin Microbiol 35:3150–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury NR, Chakraborty S, Ramamurthy T, Nishibuchi M, Yamasaki S, Takeda Y, Nair GB. 2000. Molecular evidence of clonal Vibrio parahaemolyticus pandemic strains. Emerg Infect Dis 6:631–636. doi: 10.3201/eid0606.000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.González-Escalona N, Cachicas V, Acevedo C, Rioseco ML, Vergara JA, Cabello F, Romero J, Espejo RT. 2005. Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg Infect Dis 11:129–131. doi: 10.3201/eid1101.040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haendiges J, Rock M, Myers RA, Brown EW, Evans P, Gonzalez-Escalona N. 2014. Pandemic Vibrio parahaemolyticus, Maryland, USA, 2012. Emerg Infect Dis 20:718–720. doi: 10.3201/eid2004.130818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Urtaza J, Simental L, Velasco D, DePaola A, Ishibashi M, Nakaguchi Y, Nishibuchi M, Carrera-Flores D, Rey-Alvarez C, Pousa A. 2005. Pandemic Vibrio parahaemolyticus O3:K6, Europe. Emerg Infect Dis 11:1319–1320. doi: 10.3201/eid1108.050322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansaruzzaman M, Lucas M, Deen JL, Bhuiyan NA, Wang XY, Safa A, Sultana M, Chowdhury A, Nair GB, Sack DA, von Seidlein L, Puri MK, Ali M, Chaignat CL, Clemens JD, Barreto A. 2005. Pandemic serovars (O3:K6 and O4:K68) of Vibrio parahaemolyticus associated with diarrhea in Mozambique: spread of the pandemic into the African continent. J Clin Microbiol 43:2559–2562. doi: 10.1128/JCM.43.6.2559-2562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haendiges J, Jones J, Myers RA, Mitchell CS, Butler E, Toro M, Gonzalez-Escalona N. 2016. A nonautochthonous U.S. strain of Vibrio parahaemolyticus isolated from Chesapeake Bay oysters caused the outbreak in Maryland in 2010. Appl Environ Microbiol 82:3208–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.González-Escalona N, Gavilan RG, Toro M, Zamudio ML, Martinez-Urtaza J. 2016. Outbreak of Vibrio parahaemolyticus sequence type 120, Peru, 2009. Emerg Infect Dis 22:1235–1237. doi: 10.3201/eid2207.151896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.González-Escalona N, Gavilan RG, Brown EW, Martinez-Urtaza J. 2015. Transoceanic spreading of pathogenic strains of Vibrio parahaemolyticus with distinctive genetic signatures in the recA gene. PLoS One 10:e0117485. doi: 10.1371/journal.pone.0117485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Urtaza J, Baker-Austin C, Jones JL, Newton AE, Gonzalez-Aviles GD, DePaola A. 2013. Spread of Pacific Northwest Vibrio parahaemolyticus strain. N Engl J Med 369:1573–1574. doi: 10.1056/NEJMc1305535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.González-Escalona N, Martinez-Urtaza J, Romero J, Espejo RT, Jaykus LA, DePaola A. 2008. Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J Bacteriol 190:2831–2840. doi: 10.1128/JB.01808-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allard MW, Luo Y, Strain E, Li C, Keys CE, Son I, Stones R, Musser SM, Brown EW. 2012. High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC Genomics 13:32. doi: 10.1186/1471-2164-13-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bakker HC, Switt AI, Cummings CA, Hoelzer K, Degoricija L, Rodriguez-Rivera LD, Wright EM, Fang R, Davis M, Root T, Schoonmaker-Bopp D, Musser KA, Villamil E, Waechter H, Kornstein L, Furtado MR, Wiedmann M. 2011. A whole-genome single nucleotide polymorphism-based approach to trace and identify outbreaks linked to a common Salmonella enterica subsp. enterica serovar Montevideo pulsed-field gel electrophoresis type. Appl Environ Microbiol 77:8648–8655. doi: 10.1128/AEM.06538-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chin C-S, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, Paxinos EE, Yamaichi Y, Calderwood SB, Mekalanos JJ, Schadt EE, Waldor MK. 2011. The origin of the Haitian cholera outbreak strain. N Engl J Med 364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Moller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 365:709–717. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allard MW, Luo Y, Strain E, Pettengill J, Timme R, Wang C, Li C, Keys CE, Zheng J, Stones R, Wilson MR, Musser SM, Brown EW. 2013. On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS One 8:e55254. doi: 10.1371/journal.pone.0055254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.González-Escalona N, Timme R, Raphael BH, Zink D, Sharma SK. 2014. Whole-genome single-nucleotide polymorphism analysis for discrimination of Clostridium botulinum group I strains. Appl Environ Microbiol 80:2125–2132. doi: 10.1128/AEM.03934-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann M, Luo Y, Monday SR, Gonzalez-Escalona N, Ottesen AR, Muruvanda T, Wang C, Kastanis G, Keys C, Janies D, Senturk IF, Catalyurek UV, Wang H, Hammack TS, Wolfgang WJ, Schoonmaker-Bopp D, Chu A, Myers R, Haendiges J, Evans PS, Meng J, Strain EA, Allard MW, Brown EW. 2016. Tracing origins of the Salmonella Bareilly strain causing a food-borne outbreak in the United States. J Infect Dis 213:502–508. doi: 10.1093/infdis/jiv297. [DOI] [PubMed] [Google Scholar]
- 27.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H. 2011. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovanen SM, Kivisto RI, Rossi M, Schott T, Karkkainen UM, Tuuminen T, Uksila J, Rautelin H, Hanninen ML. 2014. Multilocus sequence typing (MLST) and whole-genome MLST of Campylobacter jejuni isolates from human infections in three districts during a seasonal peak in Finland. J Clin Microbiol 52:4147–4154. doi: 10.1128/JCM.01959-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolley KA, Maiden MC. 2014. Using multilocus sequence typing to study bacterial variation: prospects in the genomic era. Future Microbiol 9:623–630. doi: 10.2217/fmb.14.24. [DOI] [PubMed] [Google Scholar]
- 30.Schmid D, Allerberger F, Huhulescu S, Pietzka A, Amar C, Kleta S, Prager R, Preussel K, Aichinger E, Mellmann A. 2014. Whole genome sequencing as a tool to investigate a cluster of seven cases of listeriosis in Austria and Germany, 2011–2013. Clin Microbiol Infect 20:431–436. doi: 10.1111/1469-0691.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohl TA, Diel R, Harmsen D, Rothganger J, Walter KM, Merker M, Weniger T, Niemann S. 2014. Whole-genome-based Mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach. J Clin Microbiol 52:2479–2486. doi: 10.1128/JCM.00567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.González-Escalona N, Toro M, Rump LV, Cao G, Nagaraja TG, Meng J. 2016. Virulence gene profiles and clonal relationships of Escherichia coli O26:H11 isolates from feedlot cattle as determined by whole-genome sequencing. Appl Environ Microbiol 82:3900–3912. doi: 10.1128/AEM.00498-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Gonzalez-Escalona N, Hammack TS, Allard MW, Strain EA, Brown EW. 2016. Core genome multilocus sequence typing for identification of globally distributed clonal groups and differentiation of outbreak strains of Listeria monocytogenes. Appl Environ Microbiol 82:6258–6272. doi: 10.1128/AEM.01532-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu F, Gonzalez-Escalona N, Haendiges J, Myers RA, Ferguson J, Stiles T, Hickey E, Moore M, Hickey JM, Schillaci C, Mank L, DeRosia-Banick K, Matluk N, Robbins A, Sebra RP, Cooper VS, Jones SH, Whistler CA. 2016. Vibrio parahaemolyticus sequence type 631, an emerging foodborne pathogen in North America. J Clin Microbiol 55:645–648. doi: 10.1128/JCM.02162-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. 2003. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 36.Xu F, Ilyas S, Hall JA, Jones SH, Cooper VS, Whistler CA. 2015. Genetic characterization of clinical and environmental Vibrio parahaemolyticus from the Northeast USA reveals emerging resident and non-indigenous pathogen lineages. Front Microbiol 6:272. doi: 10.3389/fmicb.2015.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee SK, Kearney AK, Nadon CA, Peterson CL, Tyler K, Bakouche L, Clark CG, Hoang L, Gilmour MW, Farber JM. 2014. Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J Clin Microbiol 52:1081–1088. doi: 10.1128/JCM.03047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newton AE, Garrett N, Stroika SG, Halpin JL, Turnsek M, Mody RK. 2014. Notes from the field: increase in Vibrio parahaemolyticus infections associated with consumption of Atlantic Coast shellfish—2013. MMWR Morb Mortal Wkly Rep 63:335–336. [PMC free article] [PubMed] [Google Scholar]
- 39.Baker-Austin C, Trinanes J, Gonzalez-Escalona N, Martinez-Urtaza J. 2016. Non-cholera vibrios: the microbial barometer of climate change. Trends Microbiol 25:76–84. doi: 10.1016/j.tim.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Urtaza J, Trinanes J, Gonzalez-Escalona N, Baker-Austin C. 2016. Is El Niño a long-distance corridor for waterborne disease? Nat Microbiol 1:16018. doi: 10.1038/nmicrobiol.2016.18. [DOI] [PubMed] [Google Scholar]
- 41.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph SW, Colwell RR, Kaper JB. 1982. Vibrio parahaemolyticus and related halophilic vibrios. Crit Rev Microbiol 10:77–124. doi: 10.3109/10408418209113506. [DOI] [PubMed] [Google Scholar]
- 44.Gavilan RG, Zamudio ML, Martinez-Urtaza J. 2013. Molecular epidemiology and genetic variation of pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl Trop Dis 7:e2210. doi: 10.1371/journal.pntd.0002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui Y, Yang X, Didelot X, Guo C, Li D, Yan Y, Zhang Y, Yuan Y, Yang H, Wang J, Wang J, Song Y, Zhou D, Falush D, Yang R. 2015. Epidemic clones, oceanic gene pools, and Eco-LD in the free living marine pathogen Vibrio parahaemolyticus. Mol Biol Evol 32:1396–1410. doi: 10.1093/molbev/msv009. [DOI] [PubMed] [Google Scholar]
- 46.Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, Kiryutin B, O'Neill K, Resch W, Resenchuk S, Schafer S, Tolstoy I, Tatusova T. 2009. The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res 37:D216–D223. doi: 10.1093/nar/gkn734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bryant D, Moulton V. 2004. Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21:255–265. doi: 10.1093/molbev/msh018. [DOI] [PubMed] [Google Scholar]
- 49.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 50.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol 26:2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jensen RV, Depasquale SM, Harbolick EA, Hong T, Kernell AL, Kruchko DH, Modise T, Smith CE, McCarter LL, Stevens AM. 2013. Complete genome sequence of prepandemic Vibrio parahaemolyticus BB22OP. Genome Announc 1(1):e00002-12. doi: 10.1128/genomeA.00002-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lüdeke CH, Kong N, Weimer BC, Fischer M, Jones JL. 2015. Complete genome sequences of a clinical isolate and an environmental isolate of Vibrio parahaemolyticus. Genome Announc 3(2):e00216-15. doi: 10.1128/genomeA.00216-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalburge SS, Polson SW, Boyd CK, Katz L, Turnsek M, Tarr CL, Martinez-Urtaza J, Boyd EF. 2014. Complete genome sequence of Vibrio parahaemolyticus environmental strain UCM-V493. Genome Announc 2(2):e00159-14. doi: 10.1128/genomeA.00159-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prabhakaran DM, Chowdhury G, Pazhani GP, Ramamurthy T, Thomas S. 2016. Draft genome sequence of an environmental trh+ Vibrio parahaemolyticus K23 strain isolated from Kerala, India. Genome Announc 4(2):e00282-16. doi: 10.1128/genomeA.00282-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.