

Abstract
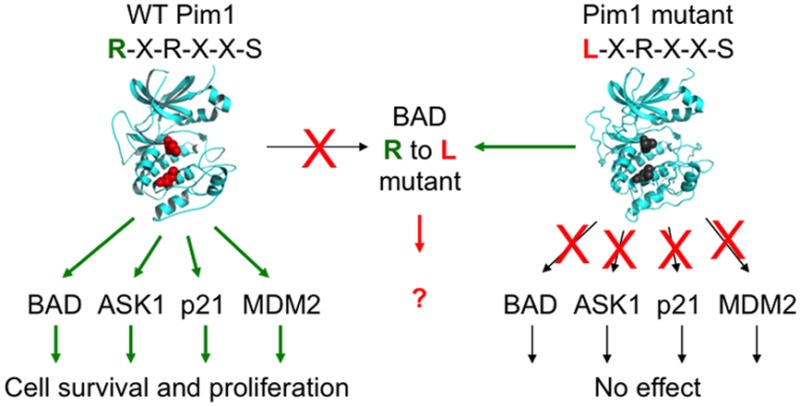
Eukaryotic protein kinases typically phosphorylate substrates in the context of specific sequence motifs, contributing to specificity essential for accurate signal transmission. Protein kinases recognize their target sequences through complementary interactions within the active site cleft. As a step toward the construction of orthogonal kinase signaling systems, we have re-engineered the protein kinase Pim1 to alter its phosphorylation consensus sequence. Residues in the Pim1 catalytic domain interacting directly with a critical arginine residue in the substrate were substituted to produce a kinase mutant that instead accommodates a hydrophobic residue. We then introduced a compensating mutation into a Pim1 substrate, the pro-apoptotic protein BAD, to reconstitute phosphorylation both in vitro and in living cells. Coexpression of the redesigned kinase with its substrate in cells protected them from apoptosis. Such orthogonal kinase–substrate pairs provide tools to probe the functional consequences of specific phosphorylation events in living cells and to design synthetic signaling pathways.
Protein phosphorylation, the most common reversible post-translational modification, is used universally as a regulatory mechanism in cellular signaling networks. Unraveling the molecular mechanisms underlying cellular regulation is dependent on our ability to experimentally control protein phosphorylation in living cells. For example, a phosphorylated residue is generally mutated to alanine to examine the consequence of preventing phosphorylation of a specific substrate protein. In addition, more elaborate synthetic signaling systems, engineered by manipulating interactions among component molecules1−3 or by introduction of artificial phosphorylation sites,4,5 can reveal basic mechanisms of signaling. Despite such impressive control over cellular signaling pathways, our capacity to simply direct phosphorylation to specific sites on proteins in living cells is remarkably limited. Phosphorylation is often simulated by introducing negatively charged Asp or Glu residues into proteins, but such mutations are necessarily constitutive and frequently fail to substitute for true phosphorylation.6 Here, we demonstrate how a protein kinase can be re-engineered by mutating its catalytic domain to phosphorylate a specific substrate in living cells. Thus far, engineering of protein kinase catalytic domains has mainly focused on the nucleotide binding site. Substitution of key residues has produced “analog-sensitive” kinase mutants that can accept modified ATP analogs and inhibitors, facilitating specific inhibition of the kinase and identification of its direct downstream substrates.7,8 By contrast, re-engineering the peptide binding specificity of kinases has been limited to a few cases primarily aimed at validating structural and evolutionary models for kinase–substrate interactions.9−14 Though not yet realized, protein kinase mutants with altered phosphorylation site specificity could potentially offer powerful tools for probing and controlling signaling pathways within living cells. For example, when expressed alone in cells, such mutants are expected to be nonfunctional, as they would not recognize and phosphorylate their endogenous substrates. However, introduction of compensating mutations into a known substrate would restore its ability to be phosphorylated by the mutant kinase. Such an orthogonal kinase–substrate pair would then allow one to directly probe the functional consequences of phosphorylation at a single site. A conceptually similar approach employing orthogonal G protein coupled receptor (GPCR)–ligand pairs (termed receptors activated by solely synthetic ligands, or RASSLs) has been used extensively for functional elucidation of GPCR signaling pathways in vivo.15
Altering the target sequence of a protein-modifying enzyme is particularly challenging, as it involves both engineering an enzyme and redesigning a protein interaction interface. We chose to use a structure-based design approach, taking advantage of available X-ray crystal structures of kinase–substrate complexes, as well as insight into the substrate preferences of many protein kinases, collected using peptide library screening methods.14,16−23 We focused our initial efforts on the serine-threonine kinase Pim1, which promotes cell growth and survival and has been implicated as an oncogenic driver in leukemia, breast cancer, and prostate cancer.24 Pim1 is thought to contribute to cell survival in part through phosphorylation of the pro-apoptotic protein Bcl2-related antagonist of cell death (BAD), causing the protein to be inactivated through sequestration by 14–3–3 proteins.25,26 The primary Pim1 phosphorylation site on BAD (Ser112), as well as most other known substrates of the kinase, conforms to its preferred R-x-R-x-x-S/T consensus sequence that has been established through analysis of peptide substrates.25,27,28 X-ray crystallography of Pim1 in complex with a consensus peptide substrate27 revealed four residues (Thr134, Asp170, Asp234, and Asp239) in the peptide-binding cleft in direct contact with the guanidino headgroup of the Arg residue located at the −5 position in the substrate (Figure 1a). Likewise, in the X-ray crystal structure of the MARK2, a related protein kinase selective for aliphatic hydrophobic residues at the −5 position, the corresponding Leu residue of a bound pseudosubstrate inhibitor interacted with a structurally analogous site.29 We therefore focused our efforts to re-engineer Pim1 specificity on mutation of the four residues within this pocket. In previous studies, we have found through peptide library screening that many kinases related to Pim1 select hydrophobic, rather than basic, residues at the −5 position.14,23,30 Sequence alignment of these kinases with Pim1 revealed a correlation between selectivity at the −5 position and the identity of residues found at positions analogous to the Pim1 –5 interaction pocket (Figure 1b), suggesting specific residues to introduce into the pocket to change the substrate specificity of Pim1.
Figure 1.
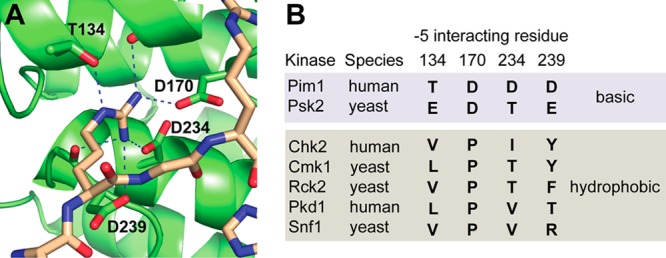
Residues in the −5 interaction pocket of Pim1 and related kinases. (A) The crystal structure of Pim1 in complex with a peptide substrate, showing the four residues (Thr134, Asp170, Asp234, and Asp239) that make direct contact with an Arg residue at the −5 position. (B) Residues found at the analogous positions in related kinases that select basic (top, shaded blue) or hydrophobic (bottom, shaded gray) residues at the −5 position on the basis of published peptide array analyses.14,23,30
To determine whether we could indeed re-engineer Pim1 to phosphorylate a new consensus sequence, we generated a series of single, double, triple, and quadruple mutants in which residues found at the analogous positions in other kinases were introduced into the −5 interaction pocket of Pim1. These mutants involved either direct grafting of residues found in a single kinase onto Pim1 or construction of a consensus kinase including residues found frequently at each of the four positions in hydrophobic-selecting kinases. In total, 12 mutant kinases were expressed, purified, and subjected to peptide library screening14 to assess changes in substrate specificity (Figure 2a; full peptide array analysis is shown in Figure S1). We found that mutation of the three Asp residues in the −5 pocket was sufficient to convert Pim1 from selecting primarily basic to hydrophobic residues at the −5 position but that the quadruple mutants appeared to be the most selective in general. To quantitatively assess substrate specificity and overall level of activity, we assayed each quadruple mutant using a pair of matched peptide substrates differing only in the −5 residue (Arg or Leu, Figure 2b). While wild-type (WT) Pim1 preferred the Arg peptide over the Leu peptide by more than 200-fold, Pim1 quadruple mutants preferred Leu over Arg by between 4- and 13-fold. All Pim1 mutants had reduced catalytic efficiency in comparison to WT Pim1 using their respective preferred substrate. However, one mutant, Pim1-T134L/D170P/D234 V/D239T (Pim1-LPVT), had a level of activity comparable to WT (40% of the WT level by this criterion). In addition, Pim1-LPVT was the most selective mutant we identified, preferring Leu to Arg by 13-fold, indicating a nearly 3000-fold change in relative peptide substrate specificity, the highest yet reported for a protein kinase.9,11,31
Figure 2.
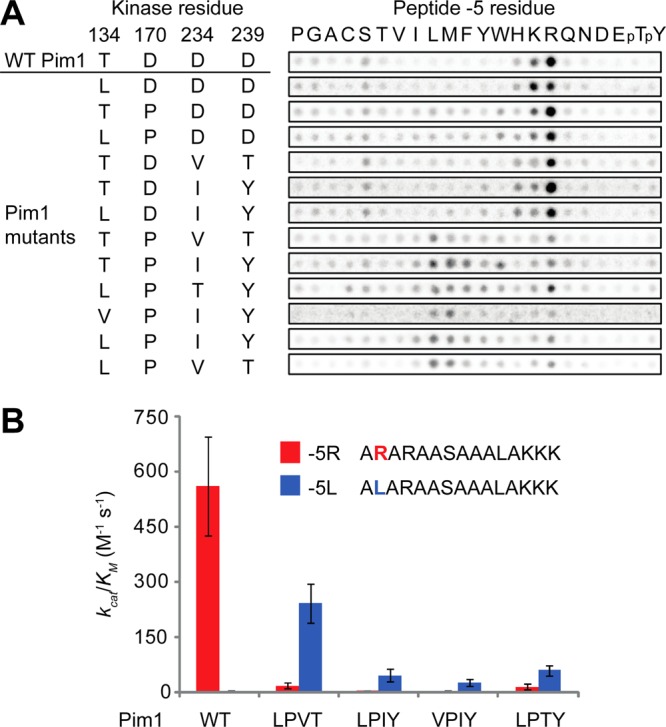
Re-engineering of the phosphorylation site specificity of Pim1. (A) Peptide array analysis of Pim1 mutants showing specificity at the −5 position. Spot intensities reflect the extent of phosphorylation using radiolabeled ATP. pT, phosphothreonine; pY, phosphotyrosine. (B) Quantitative phosphorylation parameters for Pim1 and quadruple mutants on peptide substrates assessed by radiolabel kinase assay. LPVT, Pim1-T134L/D170P/D234 V/D239T; LPIY, Pim1-T134L/D170P/D234I/D239Y; VPIY, Pim1-T134 V/D170P/D234I/D239Y; LPTY, Pim1-T134L/D170P/D234T/D239Y. Error bars indicate standard deviation (n = 3).
We next examined whether Pim1 mutation affected phosphorylation of the protein substrate BAD. As reported,25,26 WT Pim1 robustly phosphorylated recombinant BAD at Ser112 in vitro, while none of the Pim1 quadruple mutants detectably phosphorylated BAD at that site (Figure 3a). By contrast, a BAD-R107L mutant, in which the −5 Arg residue was replaced with Leu, was not a substrate of WT Pim1. However, Pim1-LPVT and, to a lesser extent, the other Pim1 mutants did phosphorylate BAD-R107L, albeit at a much lower rate compared to WT Pim1 phosphorylation of WT BAD. Introduction of an additional point mutation at the +1 position in BAD (Y113G) to more closely match the sequence preference of Pim1 as determined from peptide library screening27 (see Figure S1) greatly restored phosphorylation by Pim1-LPVT without providing substantial phosphorylation by WT Pim1 (Figure 3b). These results indicated that by introducing compensating mutations into BAD, we could reconstitute its phosphorylation by mutant Pim1.
Figure 3.

Compensating mutation of the Pim1 substrate BAD producing a functional orthogonal kinase–substrate pair. (A) Phosphorylation of purified recombinant BAD (equal amounts of WT or R107L) in vitro with Pim1 (WT and quadruple mutants) was assessed by immunoblotting with a phospho-specific antibody against the primary Pim1 phosphorylation site (Ser112). (B) Phosphorylation of recombinant BAD (2 μM of WT or the indicated mutant) by WT Pim1 or Pim1-LPVT (100 nM) was assessed by radiolabel incorporation from [γ-33P]ATP. (C) Phosphorylation of BAD coexpressed with Pim1 in cultured cells. HEK293 cells were transiently transfected with the indicated Pim1 and BAD constructs, and expression levels and BAD phosphorylation were determined by immunoblotting. Bar graphs show mean ± SD (n = 3). For the chart on the left, statistical significance for the indicated differences was calculated by unpaired Student’s t test. For the chart on the right, statistical comparisons were made using a one-way ANOVA, followed by a Fisher’s LSD posthoc test comparing conditions to GFP control. For both charts, *, p < 0.05; **, p < 0.01; ***, p < 0.0001; ns, not significant. (D) Mutant Pim1 specifically rescues cells expressing BAD-R017L from apoptosis. Viability of COS7 cells stably transduced with a doxycycline-inducible Pim1-GFP construct (WT, LPVT mutant or empty vector [EV] control) following transfection with the indicated BAD expression constructs, induction of Pim1 expression, and serum starvation for 24 h was assessed by trypan blue staining. Error bars indicate SEM (n = 4 for EV, n = 7 for all others). Statistical comparisons for each group were performed as for the right chart in panel C, comparing each condition to EV.
To determine whether this redesigned kinase-substrate interaction was functional, we first examined phosphorylation of BAD by Pim1 in cultured cells, focusing on the Pim1-LPVT mutant, which was most active in vitro (Figure 3c). As previously reported for other cell types,25,26 coexpression of WT Pim1 with WT BAD in HEK293 cells increased the level of Ser112 phosphorylation as assessed by reactivity with a phospho-specific antibody. As anticipated, Pim1-LPVT did not phosphorylate WT BAD at Ser112 in cultured cells. Pim1-LPVT induced phosphorylation of BAD-R107L in cells to approximately 50% of the level of phosphorylation of WT BAD by WT Pim1, in keeping with its reduced phosphorylation efficiency in vitro. Though mutation of Tyr113 abolished reactivity with the phospho-specific antibody, we could assess phosphorylation of BAD-R107L/Y113G in cells by electrophoretic mobility shift. Both BAD-R107L and R107L/Y113G underwent a Pim1-LPVT induced mobility shift that was completely eliminated by S112A mutation (Figure 3c and data not shown). This analysis indicated that Pim1-LPVT phosphorylated BAD-R107L/Y113G to high stoichiometry in cells, though some phosphorylation by WT Pim1 was also evident. These results indicate that our redesigned kinase–substrate system can be used to direct site-specific phosphorylation in living cells.
We next examined the effect of BAD phosphorylation on Pim1-mediated cell survival, using combinations of WT and mutant kinase and substrate (Figure 3d). BAD overexpression is reported to sensitize multiple cell types to apoptotic stimuli in a manner that can be inhibited by coexpression of kinases that phosphorylate BAD.32 We found that expression of WT BAD, the nonphosphorylatable mutant BAD-S112A, or the −5 substituted BAD-R107L and BAD-R107L/Y113G mutants each induced similar levels of cell death when overexpressed in COS7 cells subjected to serum withdrawal (Figure 3d and data not shown). As expected, cell death induced by WT BAD expression was largely reversed by coexpression with WT Pim1. In keeping with its inability to phosphorylate BAD-S112A or BAD-R107L, WT Pim1 was incapable of protecting cells from expression of these BAD mutants. Pim1-LPVT, by contrast, was unable to block cell death induced by WT BAD, yet rescued cells from BAD-R107L expression, correlating with its ability to substantially phosphorylate mutant BAD in cells. Unexpectedly, Pim1-LPVT did not block cell death induced by BAD-R107L/Y113G expression (data not shown). Pim1 inactivation of BAD may depend on Tyr113, which could be required for example for optimal binding of phosphorylated BAD to 14–3–3 proteins. Nonetheless, the ability of Pim1-LPVT to inhibit cell death specifically induced by BAD-R107L indicates that a redesigned Pim1-BAD kinase–substrate pair can be biologically functional in living cells.
In summary, we have successfully directed a functional phosphorylation event to mutant protein by means of a re-engineered protein kinase. Such an approach may find utility in elucidating the cellular consequences of individual phosphorylation events. We also note that the ability of a compensating mutation in the substrate to restore phosphorylation by the mutant provides unambiguous confirmation that a kinase-substrate interaction is direct in intact cells. Theoretically, this approach is applicable to any kinase with well-defined substrate specificity. In the example described here, we have changed the target sequence of the kinase to match that of other known kinases, which could potentially lead to “off-target” phosphorylation when the mutant kinase is expressed in cells. The use of genetic selection methods, which have been successfully applied to proteases and other enzymes,33 could be used in the future to confer non-native selectivity onto kinases to achieve more complete orthogonality in synthetic kinase signaling systems.
Methods
Vector Construction and Mutagenesis
The mammalian expression vector for N-terminally GFP-tagged Pim1 was generated by PCR amplification of the full length human coding sequence and insertion into the EcoRI and XbaI sites of the vector pEGFP-C2 (Clontech). The inducible GFP-Pim1 expression vector was generated by first shuttling the entire GFP-Pim1 coding sequence into the Gateway entry vector pDONR221 by BP recombination, followed by LR recombination into the lentiviral vector pINDUCER-20. The bacterial expression to produce GST-BAD-His6 was produced by PCR amplification of the mouse BAD coding sequence with a 3′ primer encoding a His6 tag sequence at the C-terminus and inserting into pGEX-4T2. Pim1 and BAD point mutants were generated via successive rounds of site-directed mutagenesis using the QuikChange procedure (Stratagene) with primers listed in the Supporting Information. All mutant constructs were verified by sequencing the entire open reading frame.
In Vitro Kinase Assays
The His6-tagged Pim1 catalytic domain27 was expressed in E. coli. Detailed procedures for protein expression and purification, peptide synthesis, peptide library screening, and peptide kinase assay conditions are described in the Supporting Information. BAD kinase reactions contained 5 or 20 nM kinase and 2 μM purified BAD in kinase assay buffer. Reactions were started by the addition of ATP (to 100 μM, with or without 0.25 μCi/μL [γ-33P]ATP). Cold reactions were quenched after 5 min incubation at 30 °C by adding 4× SDS-PAGE loading buffer and heating to 95 °C for 5 min. Samples were fractionated by SDS-PAGE followed by transfer to the PVDF membrane. Blots were probed with either the α-BAD phosphoSer112 antibody (Cell Signaling Technology #9291) or mouse α-penta-His antibody (Qiagen #34660) followed by the appropriate horseradish peroxidase-linked secondary antibody, and developed by enhanced chemiluminescence. Hot reactions were quenched in the same way after 20 min incubation at 30 °C and fractionated by SDS-PAGE. Gels were dried and radiolabel incorporation detected by phosphor imaging.
Analysis of BAD Phosphorylation in Cultured Cells
HEK293 cells in six-well plates were cotransfected with pEGFP-Pim1 (0.25 μg for WT Pim1 or 0.5 μg for mutant Pim1 per well, adjusted to achieve equal levels of protein expression), pEBG-BAD (Cell Signaling Technology, 3.5 μg WT or mutant per well), and empty control (to bring the total DNA to 4 μg per well) plasmids using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions. After 24 h, cells were exchanged into reduced serum medium (0.1% FBS) and cultured for an additional 18 h. Wortmannin (200 nM final concentration) was added 30 min prior to lysis. Cell lysates were prepared by washing once briefly with PBS, extracting into lysis buffer (20 mM Tris at pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM DTT, 1 mM PMSF, 10 μg/mL leupeptin, 2 μg/mL pepstatin A, and 10 μg/mL aprotinin) for 10 min at 4 °C, and centrifuging (10 min at 13 500g). Samples were separated by SDS-PAGE and analyzed by immunoblotting with α-BAD pSer112 antibody, mouse α-GST antibody (Cell Signaling Technology #2624), and mouse α-GFP antibody (Clontech #632381) followed by the appropriate secondary antibodies and analyzed on a LI-COR Odyssey CLX infrared imaging system. Signal intensities were quantified using ImageStudio software.
Cell Death Assays
COS7 cell lines harboring doxycycline-inducible GFP or GFP-Pim1 (WT or LPVT mutant) cDNA were generated by infection with lentiviruses derived from the pINDUCER20 vector and selection with puromycin. For cell death assays, 3 × 105 cells per well were seeded in six-well plates, and the following day, cells were transfected with pcDNA3-HA-BAD34 (6 μg WT or mutant per well) and empty pcDNA3 (to a final amount of 12 μg DNA per well) using Lipofectamine 2000, according to the standard protocol as per the manufacturer’s instructions. At 24 h post-transfection, cells were serum starved (0.1% FBS in growth medium) and treated with doxycycline (500 ng/mL) for 24 h. Cells were harvested by trypsinization, and the proportion of dead cells was determined by staining with 0.4% trypan blue followed by manual counting on a hemocytometer.
Acknowledgments
We thank D. Stern and D. Wu for advice and T. Boggon for comments on the manuscript. This work was supported by National Institutes of Health Grants R01 GM102262 and R01 GM104047 to B.E.T. Support for C.C. and C.J.M. was provided by training grants from the National Institutes of Health (T32 CA009085 and T32 GM007324, respectively).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00089.
Supplementary methods and supporting Figure S1 (PDF)
Author Present Address
‡ Department of Pharmacology, Faculty of Medicine, Chiang Mai University, 110 Intawaroros Road, Amphur Muang, Chiang Mai 50200, Thailand
Author Contributions
† These authors contributed equally to this report.
The authors declare no competing financial interest.
Supplementary Material
References
- Lim W. A. (2010) Designing customized cell signalling circuits. Nat. Rev. Mol. Cell Biol. 11, 393–403. 10.1038/nrm2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert E. M.; Schepartz A. (2012) Rewiring kinase specificity with a synthetic adaptor protein. J. Am. Chem. Soc. 134, 3976–3978. 10.1021/ja211089v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui N.; Findlay G. M.; Gish G. D.; Hsiung M. S.; Huang J.; Tucholska M.; Taylor L.; Smith L.; Boldridge W. C.; Koide A.; Pawson T.; Koide S. (2014) Directed network wiring identifies a key protein interaction in embryonic stem cell differentiation. Mol. Cell 54, 1034–1041. 10.1016/j.molcel.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal T.; Hibi M.; Karin M. (1994) Altering the specificity of signal transduction cascades: positive regulation of c-Jun transcriptional activity by protein kinase A. EMBO J. 13, 6006–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh B. J.; Rutigliano R. J.; Deb A.; Bar-Sagi D.; Lim W. A. (2007) Rewiring cellular morphology pathways with synthetic guanine nucleotide exchange factors. Nature 447, 596–600. 10.1038/nature05851. [DOI] [PubMed] [Google Scholar]
- Park H. S.; Hohn M. J.; Umehara T.; Guo L. T.; Osborne E. M.; Benner J.; Noren C. J.; Rinehart J.; Soll D. (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333, 1151–1154. 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop A. C.; Ubersax J. A.; Petsch D. T.; Matheos D. P.; Gray N. S.; Blethrow J.; Shimizu E.; Tsien J. Z.; Schultz P. G.; Rose M. D.; Wood J. L.; Morgan D. O.; Shokat K. M. (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401. 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Allen J. J.; Li M.; Brinkworth C. S.; Paulson J. L.; Wang D.; Hubner A.; Chou W. H.; Davis R. J.; Burlingame A. L.; Messing R. O.; Katayama C. D.; Hedrick S. M.; Shokat K. M. (2007) A semisynthetic epitope for kinase substrates. Nat. Methods 4, 511–516. 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama N.; Miller W. T. (1999) Identification of residues involved in v-Src substrate recognition by site-directed mutagenesis. FEBS Lett. 456, 403–408. 10.1016/S0014-5793(99)00992-8. [DOI] [PubMed] [Google Scholar]
- Amoui M.; Miller W. T. (2000) The substrate specificity of the catalytic domain of Abl plays an important role in directing phosphorylation of the adaptor protein Crk. Cell. Signalling 12, 637–643. 10.1016/S0898-6568(00)00107-8. [DOI] [PubMed] [Google Scholar]
- Konkol L.; Hirai T. J.; Adams J. A. (2000) Substrate specificity of the oncoprotein v-Fps: site-specific mutagenesis of the putative P+1 pocket. Biochemistry 39, 255–262. 10.1021/bi992096j. [DOI] [PubMed] [Google Scholar]
- Chen C.; Ha B. H.; Thevenin A. F.; Lou H. J.; Zhang R.; Yip K. Y.; Peterson J. R.; Gerstein M.; Kim P. M.; Filippakopoulos P.; Knapp S.; Boggon T. J.; Turk B. E. (2014) Identification of a major determinant for serine-threonine kinase phosphoacceptor specificity. Mol. Cell 53, 140–147. 10.1016/j.molcel.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard C. J., Hanson-Smith V., Kennedy K. J., Miller C. J., Lou H. J., Johnson A. D., Turk B. E., and Holt L. J. (2014) Ancestral resurrection reveals evolutionary mechanisms of kinase plasticity, eLife, 3, DOI: 10.7554/eLife.04126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J.; Kim P. M.; Lam H. Y.; Piccirillo S.; Zhou X.; Jeschke G. R.; Sheridan D. L.; Parker S. A.; Desai V.; Jwa M.; Cameroni E.; Niu H.; Good M.; Remenyi A.; Ma J. L.; Sheu Y. J.; Sassi H. E.; Sopko R.; Chan C. S.; De Virgilio C.; Hollingsworth N. M.; Lim W. A.; Stern D. F.; Stillman B.; Andrews B. J.; Gerstein M. B.; Snyder M.; Turk B. E. (2010) Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signaling 3, ra12. 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin B. R.; Hsiao E. C.; Claeysen S.; Dumuis A.; Srinivasan S.; Forsayeth J. R.; Guettier J. M.; Chang W. C.; Pei Y.; McCarthy K. D.; Nissenson R. A.; Wess J.; Bockaert J.; Roth B. L. (2008) Engineering GPCR signaling pathways with RASSLs. Nat. Methods 5, 673–678. 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z.; Blechner S.; Hoagland N.; Hoekstra M. F.; Piwnica-Worms H.; Cantley L. C. (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 4, 973–982. 10.1016/S0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Songyang Z.; Lu K. P.; Kwon Y. T.; Tsai L. H.; Filhol O.; Cochet C.; Brickey D. A.; Soderling T. R.; Bartleson C.; Graves D. J.; DeMaggio A. J.; Hoekstra M. F.; Blenis J.; Hunter T.; Cantley L. C. (1996) A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 16, 6486–6493. 10.1128/MCB.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata T.; Yaffe M. B.; Leparc G. G.; Piro E. T.; Maegawa H.; Kashiwagi A.; Kikkawa R.; Cantley L. C. (2000) Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J. Biol. Chem. 275, 36108–36115. 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- Manke I. A.; Nguyen A.; Lim D.; Stewart M. Q.; Elia A. E.; Yaffe M. B. (2005) MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17, 37–48. 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Alexander J.; Lim D.; Joughin B. A.; Hegemann B.; Hutchins J. R.; Ehrenberger T.; Ivins F.; Sessa F.; Hudecz O.; Nigg E. A.; Fry A. M.; Musacchio A.; Stukenberg P. T.; Mechtler K.; Peters J. M.; Smerdon S. J.; Yaffe M. B. (2011) Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci. Signaling 4, ra42. 10.1126/scisignal.2001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K.; Zhu G.; Liu Y.; Hallam J.; Chen L.; Herrero J.; Shaw S. (2004) Kinase peptide specificity: improved determination and relevance to protein phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 101, 13744–13749. 10.1073/pnas.0401881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina N. V.; Liu Y.; Hao J. J.; Karasuyama H.; Shaw S. (2009) LOK is a major ERM kinase in resting lymphocytes and regulates cytoskeletal rearrangement through ERM phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 106, 4707–4712. 10.1073/pnas.0805963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti J. E.; Jarrell E. T.; Chang J. D.; Abbott D. W.; Storz P.; Toker A.; Cantley L. C.; Turk B. E. (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29. 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- Nawijn M. C.; Alendar A.; Berns A. (2011) For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 11, 23–34. 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- Aho T. L.; Sandholm J.; Peltola K. J.; Mankonen H. P.; Lilly M.; Koskinen P. J. (2004) Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 571, 43–49. 10.1016/j.febslet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Macdonald A.; Campbell D. G.; Toth R.; McLauchlan H.; Hastie C. J.; Arthur J. S. (2006) Pim kinases phosphorylate multiple sites on Bad and promote 14–3-3 binding and dissociation from Bcl-XL. BMC Cell Biol. 7, 1. 10.1186/1471-2121-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock A. N.; Debreczeni J.; Amos A. L.; Knapp S.; Turk B. E. (2005) Structure and substrate specificity of the Pim-1 kinase. J. Biol. Chem. 280, 41675–41682. 10.1074/jbc.M510711200. [DOI] [PubMed] [Google Scholar]
- Palaty C. K.; Clark-Lewis I.; Leung D.; Pelech S. L. (1997) Phosphorylation site substrate specificity determinants for the Pim-1 protooncogene-encoded protein kinase. Biochem. Cell Biol. 75, 153–162. 10.1139/o97-026. [DOI] [PubMed] [Google Scholar]
- Nesic D.; Miller M. C.; Quinkert Z. T.; Stein M.; Chait B. T.; Stebbins C. E. (2010) Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat. Struct. Mol. Biol. 17, 130–132. 10.1038/nsmb.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike A. C.; Rellos P.; Niesen F. H.; Turnbull A.; Oliver A. W.; Parker S. A.; Turk B. E.; Pearl L. H.; Knapp S. (2008) Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 27, 704–714. 10.1038/emboj.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till J. H.; Chan P. M.; Miller W. T. (1999) Engineering the substrate specificity of the Abl tyrosine kinase. J. Biol. Chem. 274, 4995–5003. 10.1074/jbc.274.8.4995. [DOI] [PubMed] [Google Scholar]
- Danial N. N. (2008) BAD: undertaker by night, candyman by day. Oncogene 27 (Suppl 1), S53–S70. 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- Varadarajan N.; Rodriguez S.; Hwang B. Y.; Georgiou G.; Iverson B. L. (2008) Highly active and selective endopeptidases with programmed substrate specificities. Nat. Chem. Biol. 4, 290–294. 10.1038/nchembio.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. R.; Dudek H.; Tao X.; Masters S.; Fu H.; Gotoh Y.; Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241. 10.1016/S0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.