

ABSTRACT
Most Helicobacter pylori strains express the BabA adhesin, which binds to ABO/Leb blood group antigens on gastric mucin and epithelial cells and is found more commonly in strains that cause peptic ulcers or gastric cancer, rather than asymptomatic infection. We and others have previously reported that in mice, gerbils, and rhesus macaques, expression of babA is lost, either by phase variation or by gene conversion, in which the babB paralog recombines into the babA locus. The functional significance of loss of babA expression is unknown. Here we report that in rhesus monkeys, there is independent selective pressure for loss of babA and for overexpression of BabB, which confers a fitness advantage. Surprisingly, loss of babA by phase variation or gene conversion is not dependent on the capacity of BabA protein to bind Leb, which suggests that it may have other, unrecognized functions. These findings have implications for the role of outer membrane protein diversity in persistent H. pylori infection.
KEYWORDS: Helicobacter pylori, adhesin, babA, rhesus
INTRODUCTION
Helicobacter pylori is a human pathobiont that causes a characteristic inflammatory response in the gastric mucosa. In most individuals, this inflammation (gastritis) is asymptomatic, but there is a ∼10% lifetime risk of the development of peptic ulcer disease and a 1 to 3% risk of gastric cancer (1), the third most common cause of cancer deaths worldwide. Understanding how H. pylori sometimes leads to clinical disease versus asymptomatic infection, or perhaps even mutualism (2), is one of the leading challenges in the field. Emerging evidence suggests the importance of host and bacterial genetics (1), and their interaction (3), as well as dietary (4, 5) and other environmental variables, including the gastric microbiota (6). Among the bacterial virulence factors found more commonly in strains isolated from patients with clinical disease, the best studied is the cytotoxin-associated gene pathogenicity island (cagPAI), which encodes a type IV secretion system (T4SS) that is essential for injection of the CagA bacterial oncoprotein (7, 8).
Another important H. pylori virulence factor is the capacity for adherence, which is mediated by adhesins that belong to a diverse family of outer membrane proteins (OMPs) called Hops (9). BabA is a member of the Hop family (HopS) that binds with high affinity to mono (H)- or di (Leb)-fucosylated blood group antigens that can each be modified into blood group A or B (either unmodified is O) and are expressed on gastric epithelial cells and the overlying mucin glycoproteins (10–13). Some studies suggest that patients infected with H. pylori strains expressing BabA are more likely to develop peptic ulcer disease or gastric cancer (14, 15), particularly if the cagPAI is also present. These epidemiological observations have been supported by in vitro and in vivo mechanistic studies demonstrating that BabA-mediated attachment can potentiate CagA translocation by the T4SS (16) and promote autoantibody-mediated gastritis, parietal cell loss, and mucosa-associated lymphoid tissue in Leb transgenic mice (17).
The extensive 5′ and 3′ DNA homology between babA and two other OMP paralogs of unknown function, designated babB (HopT) and babC (HopU), generates the potential for recombination events and allelic variation. Sequence analysis of diverse clinical isolates demonstrates that indeed there appears to be frequent DNA shuffling between babA and babB and, to a lesser extent, babC (18–20). Dinucleotide repeats in the 5′ coding region of babA, babB, and other OMPs could also promote on-off phase variation by slipped-strand mispairing during DNA replication (21, 22), which further contributes to the potential for variable BabA expression. We previously demonstrated in rhesus macaques that expression of BabA and attachment to Leb were lost early during experimental infection, either by babA phase variation or by gene conversion, with replacement of babA with a duplicate copy of babB (23). Similar results were obtained more recently with mice and gerbils (24, 25). DNA sequence analysis of H. pylori strains isolated sequentially from chronically infected humans also demonstrated BabA-mediated loss of Leb binding, though less commonly than in animal models (26). This may reflect the fact that animal studies exploit a mutation burst during acute H. pylori infection (27) that would be detected less frequently in isolates from chronically infected humans.
The functional significance of loss of BabA expression is unknown. We have speculated that H. pylori lives a “life at the margins” (28) in which attachment has benefits—acquisition of iron (29) and probably other nutrients, for example—but at the cost of a close encounter with the host immune system. Other factors likely contribute as well, such as differences in host glycan expression that may make BabA or one of its paralogs relatively more advantageous for persistent infection. Here we begin to address these issues by comparing the responses of rhesus macaques to different isogenic babA and babB mutants. The results suggest that in rhesus monkeys there is independent selective pressure for both loss of BabA expression, which surprisingly was not dependent on the capacity to bind Leb, and overexpression of BabB. These findings have implications for the role of OMP diversity in persistent H. pylori infection.
RESULTS
Colonization of rhesus macaques with wild-type (WT) H. pylori and isogenic babA or babB mutants.
We compared the outcome of H. pylori infection in four groups of rhesus monkeys challenged with WT H. pylori J166 (n = 6) or a babA::cat (n = 5), babB::cat (n = 6), or babA-CL2 (n = 6) mutant, a site-directed mutant in which Cys-to-Ala replacements at residues 189 and 197 result in a BabA protein that is expressed but cannot bind Leb (Table 1). In the deletion mutants, the cat cassette was inserted so as to replace the unique middle region of babA (babA::cat) or babB (babB::cat), creating knockout strains that left the 5′ and 3′ homologous regions intact (Fig. 1A). Initial in vitro experiments confirmed loss of BabA and BabB expression in the respective deletion mutants and loss of Leb attachment in the babA::cat and babA-CL2 mutants (Fig. 1B). Monkeys underwent endoscopy with culture of the gastric antrum and corpus at 2, 8, 14, and 20 weeks postinfection (p.i.). Compared to all other groups, fewer challenged monkeys were successfully colonized with the babB::cat mutant and the percent colonized declined markedly over time (Fig. 2A). The bacterial load was also significantly lower in animals colonized with the babB::cat mutant (Fig. 2B). These results suggest that BabB but not BabA confers a fitness advantage on rhesus monkeys.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Antibiotic resistancea | Source or reference(s) |
|---|---|---|---|
| Strains | |||
| H. pylori J166 | WT clinical isolate | 23, 27 | |
| H. pylori J166 babA::cat | J166 with cat inserted in babA | Cm | This study |
| H. pylori J166 babB::cat | J166 with cat inserted in babB | Cm | This study |
| H. pylori J166 babA-CL2 | J166 with cysteines 189 and 197 replaced with alanines in babA | Str | 13 |
| H. pylori J166 babBB | J166 with babA replaced with second copy of babB | Str | This study |
| E. coli Top10 | Cloning strain | Invitrogen | |
| Plasmids | |||
| pBluescript SK− | Cloning vector | Ap | Agilent |
| pJ272 | J166 babA, including 1,459 bp upstream and 1,173 bp downstream cloned into pBluescript SK−, bp 167–1335 of babA replaced with chloramphenicol resistance cassette | Cm | This study |
| pJ270 | J166 babB, including 1,510 bp upstream and 1,919 bp downstream cloned into pBluescript SK−, bp 167–1227 of babB replaced with chloramphenicol resistance cassette | Cm | This study |
| pJ231 | Bp 1638–312 upstream of J166 babA joined to chloramphenicol resistance cassette and bp 1343–2224 of babA cloned into pBluescript SK− | Cm | This study |
| pJ232 | Bp 1638–312 upstream of J166 babA joined to kanamycin resistance cassette and bp 1–2116 of babB, including 348 bp upstream, cloned into pBluescript SK− | Km | This study |
Cm, chloramphenicol; Str, streptomycin; Ap, ampicillin; Km, kanamycin.
FIG 1.
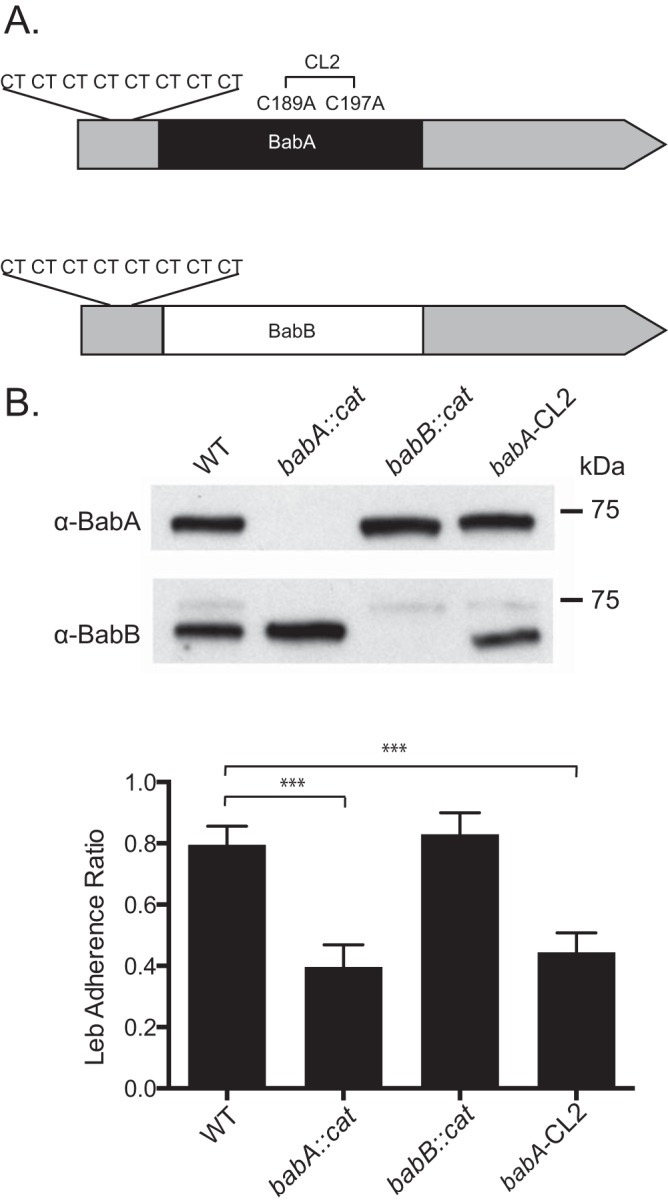
Characterization of WT H. pylori J166 and isogenic mutants. (A) babA and babB differ predominantly in the midregion of the genes, where BabA (black) binds Leb but BabB (white) does not, but are very similar at the 5′ and 3′ ends (gray). Both babA and babB have a series of eight CT repeats at the 5′ end that is in frame. Cys189 and Cys197 in babA form a redox-sensitive disulfide-clasped loop designated CL2, which is essential for Leb binding. Modification of Cys to Ala at residues 189 and 197 (babA-CL2 mutant) eliminates Leb binding. (B) Immunoblotting of WT H. pylori J166, the babA-CL2 mutant, and isogenic strains with deletions of babA (babA::cat mutant) and babB (babB::cat mutant). The faint band obtained with the anti-BabB antibody is cross-reactivity with BabA, and it is not seen in the babA::cat mutant. ELISA (bottom) demonstrates that the WT and the babB::cat mutant attach to Leb but the babA::cat and babA-CL2 mutants do not. ***, P < 0.001.
FIG 2.
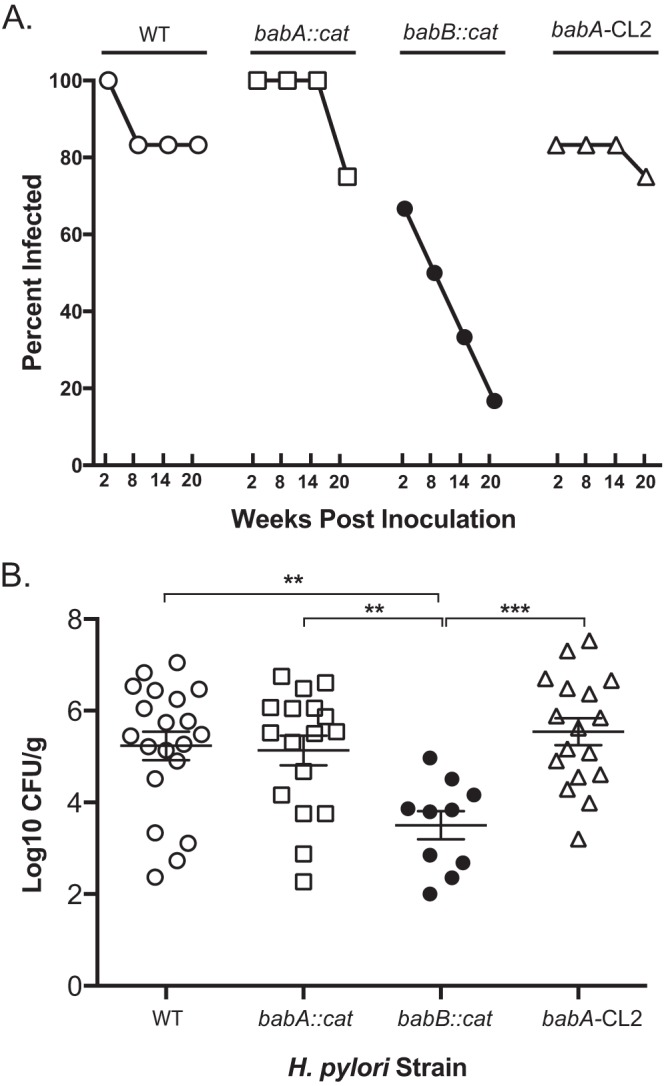
Loss of babB reduces H. pylori colonization in rhesus macaques. (A) The percentage of animals infected over time after a challenge with WT H. pylori (n = 6), the babA::cat mutant (n = 5), the babB::cat mutant (n = 6), or the babA-CL2 mutant (n = 6). For the babA::cat mutant, one animal was unavailable at the 14- and 20-week time points. For the babA-CL2 mutant, two animals were unavailable at the 20-week time point. (B) Level of colonization in each group collapsed over all time points, excluding animals without detectable colonization. Together, the results suggest that babB but not babA confers increased fitness in rhesus macaques. **, P < 0.01; ***, P < 0.001.
H. pylori loses BabA expression by phase variation and by gene conversion during infection of rhesus macaques.
We previously reported that after infection of rhesus macaques with H. pylori strain J166, expression of BabA was lost by one of two mechanisms (23). In some cases, the babA open reading frame (ORF) was disrupted by phase variation, in which slipped-strand mispairing caused the gain or loss of a 5′ dinucleotide CT repeat, with loss of the ORF. In other cases, babA underwent a nonreciprocal gene conversion in which babA was replaced with a duplicate copy of babB, a paralog of unknown function. In either case, expression of BabA and attachment to Leb were lost. Here we first reproduced those results and examined quantitatively the frequency with which each mechanism occurred. H. pylori strain J166 was inoculated into six rhesus macaques. In one animal, three colonies were recovered 2 weeks p.i., after which infection was cleared. PCR and DNA sequencing were performed on DNA extracted from multiple single colonies recovered from the remaining five monkeys to determine if the babA locus contained babA or babB and if there was an ORF following the CT repeat region, which we previously showed was uniformly associated with expression (25). Early during infection, the babA locus contained predominantly babA with an intact ORF, but over time, the babA gene underwent phase variation (Fig. 3A) or was replaced with babB (Fig. 3B). By 20 weeks p.i., loss of BabA expression was nearly complete, with approximately equal frequency of babA phase variation and babB gene conversion.
FIG 3.
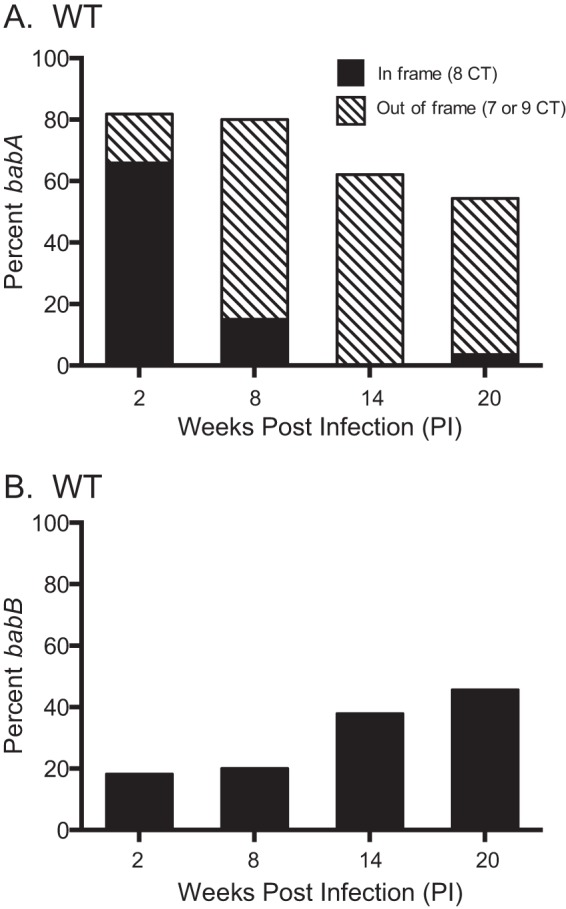
H. pylori loses BabA expression by phase variation and by gene conversion during infection of rhesus macaques. Six monkeys were inoculated with WT H. pylori J166, and output colonies from 2, 8, 14, and 20 weeks p.i. were examined. Amplification and sequencing of the 5′ region of the babA locus from multiple colonies were used to determine if the gene was babA (A) or babB (B) and if the CT repeat region was in frame (eight CT repeats, black bars) or out of frame (seven or nine CT repeats, hatched bars). By 20 weeks p.i., loss of BabA expression by phase variation (hatched bars) and gene conversion (black bars) occurred with approximately equal frequency.
H. pylori infection of rhesus macaques is under independent selective pressure for loss of babA and overexpression of babB.
babA phase variation indicates that BabA expression is under negative selection in rhesus monkeys, though the reason is unknown. babA gene conversion by babB might also reflect selective pressure against BabA expression, but it could be a result of selection for overexpression of BabB. To distinguish these two possibilities, we inserted a chloramphenicol acetyltransferase (CAT) cassette into the unique middle region of babA (babA::cat) or babB (babB::cat), creating knockout strains that left the 5′ and 3′ homologous regions intact so as to permit both phase variation and gene conversion (Fig. 1A). We reasoned that during infection with the babA::cat mutant, gene conversion would result in overexpression of BabB, but neither gene conversion nor phase variation would affect BabA expression because the babA gene was already interrupted. On the other hand, infection with the babB::cat mutant would eliminate any selection for overexpression of BabB, which was deleted, but would leave the potential for babA phase variation intact.
The babA::cat and babB::cat mutants were inoculated into five and six rhesus monkeys, respectively, which were biopsied 2, 8, 14, and 20 weeks p.i. The output strains were characterized as before by DNA sequencing to determine if the babA locus contained babA or babB and if there was a predicted ORF. During infection with the babA::cat mutant, babA was progressively lost over time because of gene conversion but not phase variation, indicating selection for overexpression of BabB (Fig. 4A and B). In all instances of gene conversion, babB was in frame. Infection with the babB::cat mutant showed loss of babA expression by phase variation (Fig. 4C) but no gene conversion (Fig. 4D). These results suggest that during H. pylori infection of rhesus monkeys, there is selection for both loss of babA expression and overexpression of BabB.
FIG 4.

H. pylori infection of rhesus macaques is under independent selective pressure for loss of babA and overexpression of babB. Five or six rhesus macaques were inoculated with the H. pylori J166 babA::cat (A, B) or babB::cat (C, D) mutant, and multiple output colonies from 2, 8, 14, and 20 weeks p.i. were examined. Amplification and sequencing were used to determine if the gene at the babA locus was babA::cat (A) or babB (B) for monkeys inoculated with the babA::cat mutant or if it was babA (C) or babB::cat (D) for monkeys that received the babB::cat mutant. Sequence analysis was also used to determine if the CT repeat region was in frame (eight CT repeats, black bars) or out of frame (seven or nine CT repeats, hatched bars). Loss of BabA expression by gene conversion in the babA::cat mutant (B) and by phase variation in the babB::cat mutant (C) suggests that there is independent selective pressure against expression of BabA and for overexpression of BabB.
Overexpression of BabB enhances H. pylori fitness in rhesus macaques.
The colonization efficiency and bacterial load results (Fig. 2) suggest that BabB increases H. pylori fitness in rhesus monkeys. To address this further, we compared the bacterial loads in monkeys infected predominantly (>50%) with colonies expressing two copies of babB and those colonized with babA in numbers equal to or greater than those of babB. Animals challenged with the babB::cat mutant were excluded, since babB overexpression could not occur. At each time point, H. pylori colonization was greater in monkeys infected predominantly with strains expressing two copies of babB, though the main effect of the strain did not quite achieve statistical significance (analysis of variance, P = 0.08). However, when the results for all animals were collapsed across all time points (Fig. 5A), the bacterial load was approximately 10-fold higher in those colonized predominantly with strains expressing two copies of babB (P < 0.01). To confirm that duplication of babB resulted in increased protein expression, we compared Western blot assays of three output strains with one copy of babB to three colonies with two copies of babB. As controls, we included WT J166, J166 babB::cat, and a J166 strain in which we replaced babA with a second copy of babB, designated J166 babBB. The results demonstrated increased BabB protein in output strains that had undergone gene conversion with duplication of babB (Fig. 5B). In all colonies, babB was present at the babB locus and all babB genes were in frame, whether at the babA or the babB locus. Together with the colonization data in Fig. 2, these results strongly suggest that overexpression of BabB increases H. pylori fitness in rhesus monkeys.
FIG 5.
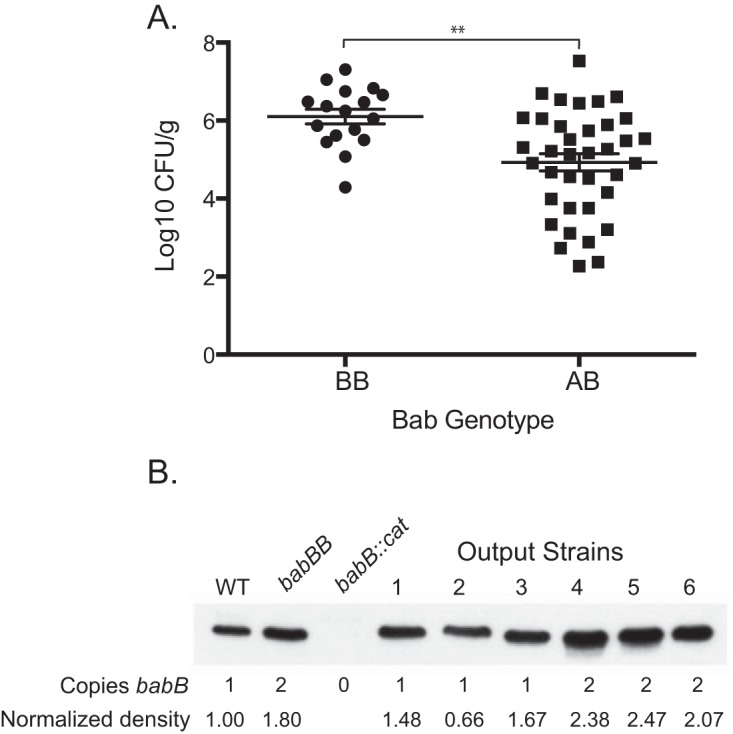
Overexpression of BabB enhances H. pylori fitness in rhesus macaques. (A) The Bab genotype at the babA locus was determined for an average of 10.6 (SD = 3.2) colonies recovered from monkeys challenged with the WT, the babA::cat mutant, or the babA-CL2 mutant and biopsied 2 to 20 weeks p.i. The genotype was defined as BB when >50% of the colonies contained two copies of babB and AB when ≥50% contained one copy each of babA and babB. When the results were collapsed over time, animals colonized predominantly with strains that duplicated babB (BB) showed approximately 10-fold greater bacterial loads (**, P = 0.001). Animals without detectable colonization were excluded. (B) Immunoblotting with BabB antiserum of H. pylori J166 (WT with one copy of babB), the babBB mutant (engineered to have two copies of babB), the babB::cat mutant, and rhesus output strains having one or two copies of babB. Immunoblotting and densitometry normalized to the total protein (see Materials and Methods) demonstrated that strains with duplicated babB express approximately 2× BabB protein.
Loss of BabA expression in rhesus monkeys is not dependent on its capacity to bind Leb.
The only known function of BabA is attachment to fucosylated ABO(H) type 1 or Leb blood group antigens expressed on gastric epithelial cells and the overlying mucin (10–12). We recently demonstrated by high-resolution structural analysis that BabA residues Cys189 and Cys197 form a redox-sensitive disulfide-clasped loop designated CL2, which is essential for binding of the ABO blood group antigen-determining α1.2-fucose residue of Leb (13). Cys-to-Ala replacements at BabA residues 189 and 197 in H. pylori J166 (designated the babA-CL2 mutant) were sufficient to eliminate all Leb binding activity, though the protein was expressed at levels similar to those in the WT (Fig. 1B) and reached the cell surface as a folded protein (13). To determine if Leb binding is required for loss of BabA protein by phase variation or gene conversion, we challenged six rhesus macaques with the babA-CL2 mutant and characterized the babA locus in output strains recovered 2, 8, 14, and 20 weeks p.i. At 2 and 8 weeks p.i., the proportion of colonies expressing babA was greater than that in monkeys challenged with WT J166 (compare Fig. 6 with Fig. 3). However, after 14 weeks p.i., the expression of babA was lost completely by either phase variation or gene conversion with babB, in approximately equal proportions, similar to what was observed in the WT. When the data were collapsed over all time points, babA expression was retained in 30% (61 of 204) of the output colonies of the babA-CL2 mutant but only 19% (41 of 222) and 21% (16 of 78) of the colonies of the WT and the babB::cat mutant, respectively. Chi-square analysis confirmed that significantly more babA expression was retained in monkeys challenged with the babA-CL2 mutant than in monkeys challenged with the WT (P = 0.006). These results suggest that Leb binding contributes to but is not essential for loss of babA expression in rhesus monkeys.
FIG 6.
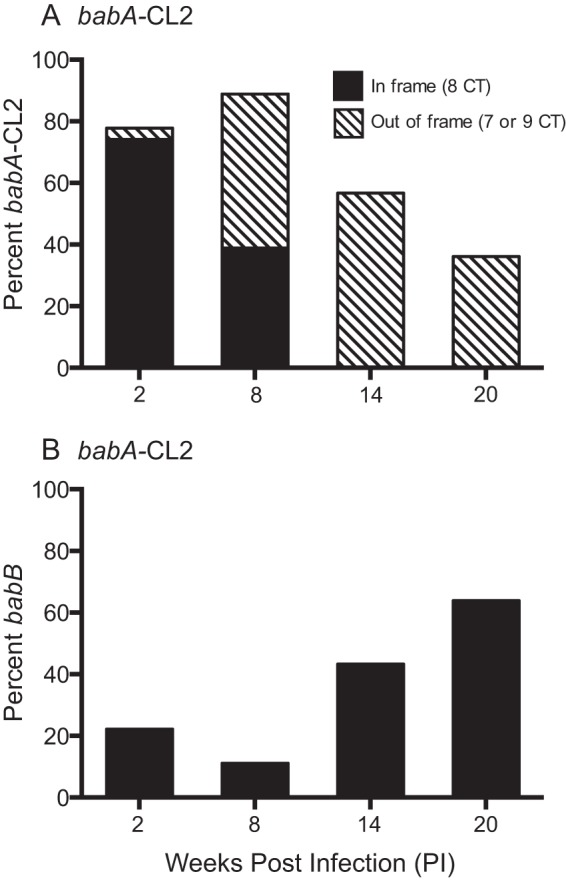
Loss of BabA expression in rhesus monkeys is not dependent on its capacity to bind Leb. Six monkeys were inoculated with the H. pylori babA-CL2 mutant, and output colonies from 2, 8, 14, and 20 weeks p.i. were examined. Amplification and sequencing of the 5′ region of the babA locus from multiple colonies were used to determine if the gene was babA (A) or babB (B) and if the CT repeat region was in frame (eight CT repeats, black bars) or out of frame (seven or nine CT repeats, hatched bars). By 20 weeks p.i., loss of BabA expression by phase variation (hatched bars) and gene conversion (black bars) occurred with approximately equal frequency.
Antral gastritis is not associated with BabA or BabB expression.
Antral biopsy specimens were scored for gastritis according to the Sydney system. Combined scores from 8 and 14 weeks p.i. showed no significant differences among the four experimental groups (Kruskal-Wallis test, P = 0.51).
DISCUSSION
Signature features characteristic of H. pylori adhesins are their polymorphism and capacity for genetic diversification. For example, although most strains express BabA, some do not (18, 30), and in others, it is silent owing to a truncated signal peptide, a frameshift in a dinucleotide repeat at the 5′ end of the gene, or a single base pair mutation that results in a stop codon (12). Even among strains that express BabA, they can be classified as “specialists” or “generalists,” depending on whether they bind only glycans in blood group O individuals or instead bind blood group A, B, and O antigens. Recent structural analysis has demonstrated that the basis for this functional diversity lies in two “diversity loops” within the carbohydrate binding site that represent areas of strong positive selection among babA sequences (13). Similarly, SabA shows polymorphism among clinical isolates in binding affinity to sialyl Lewis x (sLex) (31) and can be modulated by phase variation (32), gene conversion (33), and even variation in the length of a poly-T tract in the promoter region that serves as a rheostat-like mechanism to alter gene expression (34).
The functional significance of diversity among H. pylori adhesins is poorly understood, though sometimes epidemiologic observations or animal experiments offer clues. For example, H. pylori isolates from populations that express all ABO blood types are typically generalists, while those from South American Amerindians, who are predominantly blood group O, are more often specialists (10). Transition from expression of BabA to SabA may result from inflammation-induced downregulation of Leb and upregulation of sLex (35, 36), but attachment may be a mixed blessing for H. pylori, which we have called the “attachment dilemma” (28). On the one hand, intimate attachment to the gastric epithelium may provide nutrients by disrupting cellular tight junctions and injecting the CagA oncoprotein into host cells, as well as avoiding the unacceptably low pH in the gastric lumen. Yet this comes at the cost of an encounter with reactive oxygen species and other inflammatory mediators, as well as the risk of being shed into the lumen as the gastric epithelium is repopulated every few days.
We and others have observed loss of BabA expression during experimental infections of mice, gerbils, and monkeys (23-25, 37). While most strains recovered from humans express BabA, analysis of isolates recovered sequentially from the same individual over time demonstrates decreased Leb binding in nearly 25% of the strains (26). Here we sought to better understand the selection for loss of BabA expression in the rhesus macaque model by using WT and carefully constructed isogenic strains. In macaques infected with WT H. pylori J166, BabA expression is lost as early as 2 weeks p.i. By 20 weeks p.i., loss of BabA expression is complete and is a result of both phase variation and babB duplication with approximately equal frequency (Fig. 3). Experiments in which a CAT cassette replaces the middle region of either babA or babB suggest that there is independent selective pressure for loss of BabA and overexpression of BabB (Fig. 4).
What might these selective pressures be? Since H. pylori strains that express or overexpress BabB show increased colonization efficiency and bacterial loads (Fig. 2 and 5), it is reasonable to suggest that, like BabA, BabB is a lectin that mediates attachment to the gastric epithelium of the macaque and perhaps some humans. This is also consistent with our previous observation that H. pylori strains naturally infecting rhesus macaques contain no babA but two copies of babB (23). The glycan receptor for BabB might be constitutive or perhaps induced by inflammation, much like the sialyl-dimeric-Lewis x glycosphingolipid that binds H. pylori SabA (35). Selection for overexpression of BabB seems to be specific, because in the absence of functional BabB, gene conversion does not occur with other members of the hop family that also have 5′ and 3′ homology to BabA (albeit less), such as hopN. But this explanation is incomplete because BabA expression is also lost by phase variation in the absence of gene conversion by babB (Fig. 4). Surprisingly, this occurs even in a site-directed mutant that expresses BabA on the bacterial surface but does not attach to Leb (Fig. 6). BabA (but not BabB) might be immunogenic and lost by immune evasion. However, neither immunoproteomic studies (38, 39) nor serologic responses in macaques (23) have identified BabA or BabB as an immunodominant antigen and its expression appears to have no effect on gastritis. Furthermore, BabA expression is lost in recombination-activating gene-deficient mice that do not express functional B or T cells (46). Therefore, while the basis for selection against BabA expression is unknown, it is not likely classical immune evasion and it does not require the capacity to attach to Leb. Further studies of the role of H. pylori OMP diversity and plasticity during infection in animal models will be important to better understand H. pylori persistence in humans.
MATERIALS AND METHODS
Ethics statement.
This work was performed at the California National Primate Research Center and the University of California, Davis, in accordance with NIH guidelines, the Animal Welfare Act, and U.S. federal law. All experiments were carried out under protocol 18788 approved by the University of California, Davis, Institutional Animal Care and Use Committee, which is accredited by the Association of Assessment and Accreditation of Laboratory Animal Care. All animals were housed under these guidelines in an accredited research facility fully staffed with trained personnel.
Animals and experimental design.
Colony-bred, specific-pathogen-free male and female rhesus macaques between the ages of 2 and 7 years that were free of H. pylori infection were derived as previously described (40). Four experimental groups were challenged by gavage with 109 CFU/2 ml of H. pylori J166 WT (n = 6), the babA::cat mutant (n = 5), the babB::cat mutant (n = 6), or the babA-CL2 mutant (n = 6) as previously described (41). The genome of H. pylori J166, which has been fully sequenced (GenBank accession no. NZ_CP007603), contains single copies of babA (GenBank accession no. 428593) and babB (GenBank accession no. AY 428591) but does not contain babC.
Endoscopy, gastric biopsy, and H. pylori quantitative culture.
At 2, 8, 14, and 20 weeks p.i., six biopsy specimens each were obtained from the gastric antrum and corpus by endoscopy performed under ketamine anesthesia (10 mg/kg administered intramuscularly). Four biopsy specimens each were collected from the antrum and corpus in brucella broth for H. pylori quantitative culture by serial dilution and plating as described previously (41). The additional four biopsy specimens (two from the antrum, two from the corpus) were collected in 10% formalin for histology.
Bacterial strains and culture.
H. pylori strains (Table 1) were cultured on brucella agar (BBL/Becton Dickinson, Sparks, MD) supplemented with 5% heat-inactivated newborn calf serum (Invitrogen, Carlsbad, CA) and TVPA antibiotics (5 μg/ml trimethoprim, 10 μg/ml vancomycin, 2.5 U/ml polymyxin B, and 2.5 μg/ml amphotericin B), unless otherwise indicated. Cultures were incubated at 37°C under microaerophilic conditions generated by a 5% CO2 incubator or by a fixed 5% O2 concentration (Anoxomat; Advanced Instruments, Norwood, MA). WT H. pylori J166 was initially recovered from a human patient with a duodenal ulcer and has been shown to efficiently colonize rhesus macaques (36, 41–43). It expresses a T4SS encoded on the cagPAI and attaches to Leb blood group antigens by expression of BabA, though both functions are lost during passage in mice and rhesus monkeys (23, 25, 42). The complete genomes of WT H. pylori J166 and its rhesus-passaged variant were recently sequenced (27).
Construction of babA::cat, babB::cat, and babBB isogenic strains.
The babA and babB genes are highly conserved in their 5′ and 3′ regions (Fig. 1). Knockouts were constructed that retained these regions of homology, with the variable region between them replaced with a chloramphenicol resistance (CAT) cassette in the reverse orientation. Upstream, downstream, and CAT fragments were PCR amplified with primers described in Table 2. Following purification and restriction digestion, the amplicons were ligated to pBluescript (Agilent Technologies, Wilmington, DE) and transformed into Escherichia coli One Shot Top10 competent cells (Life Technologies, Grand Island, NY). The resulting plasmids were used to deliver the CAT cassette to J166 by allelic exchange. Disruption of babA or babB was verified by PCR and loss of expression in a Western blot assay. Leb binding activity was determined by enzyme-linked immunosorbent assay (ELISA). DNA sequence analysis was performed to verify the length of the dinucleotide repeat near the 5′ end of the gene. A control J166 strain in which babA was replaced with a second copy of babB (designated babBB) was engineered with the construction of two plasmids. Regions bp 1638 to 312 upstream of babA and bp 1343 to 2224 within babA were PCR amplified, joined with a CAT cassette in the reverse orientation, and ligated into pBluescript SK− to create pJ231. J166 was naturally transformed with this plasmid, resulting in the promoter and variable region of babA being replaced with CAT. This strain was then transformed with a second plasmid (pJ232) that contained the region upstream of the babA promoter joined to a kanamycin resistance cassette, followed by the promoter and the entire babB gene. This resulted in the replacement of babA with an identical second copy of babB, including its promoter. The primers used to prepare these constructs are listed in Table 2.
TABLE 2.
Primers used for PCR, cloning, and sequencing
| Primer purpose and name | Sequence (5′ to 3′)a |
|---|---|
| Construction of babA::cat | |
| HP0898:930U24_NotI | AAC GCGGCCGC TTT AAG CCA CAA AAC CTC TAA AGA |
| HP0896:148L25_HincII | AAC GTCGAC GAT TGT TCA GCT TTT CAT AAT TGT C |
| CAT_F_SacI | AAC GAGCTC GCG GAC AAC GAG TAA AAG AG |
| CAT_R_HincII | AAC GTCGAC GCA GGA CGC ACT ACT CTC G |
| HP0896:1234U23_SacI | AAC GAGCTC TAT GTG GAA CAA ACC ATA ACG AA |
| JHP0830:224L22_XhoI | AAT CTCGAG TCA GCA TTC ACT TCC TTT TTG A |
| Construction of babB::cat | |
| JHP1167:383U18_NotI | AAC GCGGCCGC CTC ACC ACG CAG AGG AAG |
| HP0896:148L25_PstI | AAC CTGCAG GAT TGT TCA GCT TTT CAT AAT TGT C |
| CAT_F_SacI | AAC GAGCTC GCG GAC AAC GAG TAA AAG AG |
| CAT_R_PstI | AAC CTGCAG GCA GGA CGC ACT ACT CTC G |
| HP0896:1234U23_SacI | AAC GAGCTC TAT GTG GAA CAA ACC ATA ACG AA |
| HP1241:772L20_XhoI | AAT CTCGAG TGG AAC TCG CTC GCA TAA TC |
| Construction of pJ231 | |
| HP0898:751U24_NotI | AAC GCGGCCGC ATC CAA TAC AAA AGA GCG GTG AGC |
| BabAR19_SacI | AAC GAGCTC TTA TCG CTT GCT TGA TGC AAG CTC |
| CAT_F_HincII | AAC GTCGAC GCG GAC AAC GAG TAA AAG AG |
| CAT_R_SacI | AAC GAGCTC GCA GGA CGC ACT ACT CTC G |
| BabAF20_HincII | AAC GTCGAC CAA CAA ACC ATA ACG AAT TTA ACC AAC AG |
| HP0896:2097L26_XhoI | AAC CTCGAG AAG CGA ACA CGT AAT TCA AAT ACA CG |
| Construction of pJ232 | |
| HP0898:751U24_NotI | AAC GCGGCCGC ATC CAA TAC AAA AGA GCG GTG AGC |
| BabAR19_HincII | AAC GTCGAC TTA TCG CTT GCT TGA TGC AAG CTC |
| Kan_F_SacI | AAC GAGCTC GGT ACC CGG GTG AC |
| Kan_R_HincII | AAC GTCGAC TCT AGA GGA TCC CC |
| BabBF9_SacI | AAC GAGCTC GAT TTT AGC GGT GAT TTC TTG AGC G |
| HP0896:2097L26_XhoI | AAC CTCGAG AAG CGA ACA CGT AAT TCA AAT ACA CG |
| Amplification and sequencing of babA locus | |
| BabAF14 | GCA TCA AGC AAG CGA TAA CTT TAC TAA |
| P160R | ATA CCC TGG CTC GTT GTT GAA |
| P178R | CAT GTC CTG GCT CAT AAT ACG AA |
| CAT_reverse | CCC TGC ACA TAT AGT ATG ACG GTA A |
| BabAF19 | ATG CTA TAA TAC TCC AAA TAC ATT CCA A |
| Amplification and sequencing of babB locus | |
| BabBF3 | CTT GAG CGT TAT TAG AAA TCT AGT GG |
| P178R | CAT GTC CTG GCT CAT AAT ACG AA |
| CAT_reverse | CCC TGC ACA TAT AGT ATG ACG GTA A |
Restriction sites are underlined.
Immunoblotting.
Expression of BabA and BabB was detected by immunoblotting. Bacterial cells were grown on plates overnight, suspended in Tris-EDTA buffer, and lysed by sonication. Following electrophoresis in a 7.5% polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA), proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and blocked overnight in TTBS (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) containing 5% nonfat dry milk. Blots were incubated with polyclonal antiserum (AK277 [BabA] or AK276 [BabB]) (44) at a 1:10,000 dilution for 1 h and then washed in TTBS. A horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (GE Healthcare, Buckinghamshire, United Kingdom) was diluted to 1:20,000 and applied for 1 h. Following washing in TTBS, the bound protein was visualized with Pierce ECL2 Western blotting substrate (Thermo Fisher Scientific, Waltham, MA). To quantitate BabB expression, a TGX Stain-Free gel (Bio-Rad Laboratories) was used to allow normalization to the total protein loaded. Stain-Free gels contain a trihalo compound that is covalently bound to tryptophan residues upon activation with UV light. Following transfer to PVDF, proteins were visualized with a Gel-Doc XR+ Imaging System (Bio-Rad Laboratories). Immunoblotting with anti-BabB antibody was carried out as already described, and the resulting film image was scanned into the Gel-Doc XR+ imager by using a silver stain protocol. ImageQuant 5.2.1 software (Bio-Rad Laboratories) was used to superimpose the images and normalize the BabB immunoblot assay to the total protein loaded into each lane.
In vitro Leb binding assay.
H. pylori expression of functional BabA protein was assayed by in vitro attachment to Leb with an ELISA as previously described (23). Briefly, digoxigenin (Roche Applied Biosciences)-labeled H. pylori cells were applied to Leb-coated wells in a 96-well polystyrene plate. Unbound bacteria were removed by washing, and bound bacteria were detected with antidigoxigenin Fab fragments conjugated to horseradish peroxidase (Roche Applied Biosciences) and then incubated with 2,2′-azino-di(3-ethyl-benzthiazoline-6-sulfonate). The color change was measured by subtraction of the absorbance at 490 nm from that at 405 nm. Attachment ratio values were reported as the average readings of two Leb-positive wells divided by the average readings of two Leb-negative wells.
PCR and DNA sequencing of the babA and babB loci.
DNA was extracted from an average of 10.6 (standard deviation [SD] = 3.2) single colonies per animal at each time point. A fragment of the babA locus was amplified from each colony by PCR with primers BabAF14 and P160R, P178R, or CAT_reverse (Table 2), which amplifies the 5′ end of babA, babB, or babA::cat, respectively (25). The PCR product was sequenced with primer BabAF19 by dye terminator chemistry to determine the CT repeat length. Eight CT repeats encode a babA ORF; seven or nine repeats result in a stop codon at position 49 or 79, respectively (25). This DNA-based assessment of BabA expression in a sample of colonies correlates strongly (Pearson R2 = 0.57, P < 0.0001) (46) with a radioimmunoassay (10–13) that detects Leb binding in a population (sweep) of bacteria from the mouse stomach. In addition, three single colonies per animal from the latest infected time point were sequenced at the babB locus. Primers BabBF3 and either P178R (WT) or CAT_reverse (babB::cat mutant) were used for amplification, and the corresponding reverse primer was used for sequencing. Similar to babA, eight CT repeats encode a babB ORF, while seven or nine repeats result in a stop codon as described above.
Histology.
Gastric biopsy specimens from the antrum of macaques infected at 8 and 14 weeks p.i. were paraffin imbedded and sectioned. Slides stained with hematoxylin and eosin were analyzed for inflammation (mononuclear cells) and activity (polymorphonuclear leukocytes) by a primate pathologist without knowledge of the experimental condition. Scores were generated as a composite of inflammation and activity according to the modified Sydney system (45).
ACKNOWLEDGMENTS
We thank Mary E. Kable and other members of the Solnick laboratory for suggestions and technical support.
This work was supported by NIH grants AI070803 and AI081037 to J.V.S. The funding agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Wroblewski LE, Peek RM Jr, Wilson KT. 2010. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaser MJ, Falkow S. 2009. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol 7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, Sicinschi LA, Shaffer CL, Romero-Gallo J, de Sablet T, Harder RH, Bravo LE, Peek RM Jr, Wilson KT, Cover TL, Williams SM, Correa P. 2014. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A 111:1455–1460. doi: 10.1073/pnas.1318093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaddy JA, Radin JN, Loh JT, Zhang F, Washington MK, Peek RM Jr, Algood HM, Cover TL. 2013. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect Immun 81:2258–2267. doi: 10.1128/IAI.01271-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, Romero-Gallo J, Suarez G, Loh J, Slaughter JC, Tan S, Morgan DR, Wilson KT, Bravo LE, Correa P, Cover TL, Amieva MR, Peek RM Jr. 2013. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest 123:479–492. doi: 10.1172/JCI64373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, Wang TC, Fox JG. 2011. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 140:210–220. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 8.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, Yamada G, Azuma T, Hatakeyama M. 2008. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A 105:1003–1008. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. 2000. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun 68:4155–4168. doi: 10.1128/IAI.68.7.4155-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikstrom S, Sjostrom R, Linden S, Backstrom A, Lundberg C, Arnqvist A, Mahdavi J, Nilsson UJ, Velapatino B, Gilman RH, Gerhard M, Alarcon T, Lopez-Brea M, Nakazawa T, Fox JG, Correa P, Dominguez-Bello MG, Perez-Perez GI, Blaser MJ, Normark S, Carlstedt I, Oscarson S, Teneberg S, Berg DE, Borén T. 2004. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- 11.Borén T, Falk P, Roth KA, Larson G, Normark S. 1993. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 12.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. 1998. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 13.Moonens K, Gideonsson P, Subedi S, Bugaytsova J, Romao E, Mendez M, Norden J, Fallah M, Rakhimova L, Shevtsova A, Lahmann M, Castaldo G, Brannstrom K, Coppens F, Lo AW, Ny T, Solnick JV, Vandenbussche G, Oscarson S, Hammarstrom L, Arnqvist A, Berg DE, Muyldermans S, Borén T, Remaut H. 2016. Structural insights into polymorphic ABO glycan binding by Helicobacter pylori. Cell Host Microbe 19:55–66. doi: 10.1016/j.chom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. 1999. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A 96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaoka Y, Souchek J, Odenbreit S, Haas R, Arnqvist A, Borén T, Kodama T, Osato MS, Gutierrez O, Kim JG, Graham DY. 2002. Discrimination between cases of duodenal ulcer and gastritis on the basis of putative virulence factors of Helicobacter pylori. J Clin Microbiol 40:2244–2246. doi: 10.1128/JCM.40.6.2244-2246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishijima N, Suzuki M, Ashida H, Ichikawa Y, Kanegae Y, Saito I, Borén T, Haas R, Sasakawa C, Mimuro H. 2011. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J Biol Chem 286:25256–25264. doi: 10.1074/jbc.M111.233601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guruge JL, Falk PG, Lorenz RG, Dans M, Wirth HP, Blaser MJ, Berg DE, Gordon JI. 1998. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci U S A 95:3925–3930. doi: 10.1073/pnas.95.7.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colbeck JC, Hansen LM, Fong JM, Solnick JV. 2006. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect Immun 74:4375–4378. doi: 10.1128/IAI.00485-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saberi S, Schmidt A, Eybpoosh S, Esmaili M, Talebkhan Y, Mohajerani N, Oghalaie A, Eshagh Hosseini M, Mohagheghi MA, Bugaytova J, Borén T, Mohammadi M. 2016. Helicobacter pylori strains from duodenal ulcer patients exhibit mixed babA/B genotypes with low levels of BabA adhesin and Lewis b binding. Dig Dis Sci 61:2868–2877. doi: 10.1007/s10620-016-4217-z. [DOI] [PubMed] [Google Scholar]
- 20.Sheu SM, Sheu BS, Chiang WC, Kao CY, Wu HM, Yang HB, Wu JJ. 2012. H. pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol 12:89. doi: 10.1186/1471-2180-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salaün L, Linz B, Suerbaum S, Saunders NJ. 2004. The diversity within an expanded and redefined repertoire of phase-variable genes in Helicobacter pylori. Microbiology 150:817–830. doi: 10.1099/mic.0.26993-0. [DOI] [PubMed] [Google Scholar]
- 22.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 23.Solnick JV, Hansen LM, Salama N, Boonjakuakul JK, Syvanen M. 2004. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci U S A 101:2106–2111. doi: 10.1073/pnas.0308573100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohno T, Vallstrom A, Rugge M, Ota H, Graham DY, Arnqvist A, Yamaoka Y. 2011. Effects of blood group antigen-binding adhesin expression during Helicobacter pylori infection of Mongolian gerbils. J Infect Dis 203:726–735. doi: 10.1093/infdis/jiq090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Styer CM, Hansen LM, Cooke CL, Gundersen AM, Choi SS, Berg DE, Benghezal M, Marshall BJ, Peek RM Jr, Borén T, Solnick JV. 2010. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect Immun 78:1593–1600. doi: 10.1128/IAI.01297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nell S, Kennemann L, Schwarz S, Josenhans C, Suerbaum S. 2014. Dynamics of Lewis b binding and sequence variation of the babA adhesin gene during chronic Helicobacter pylori infection in humans. mBio 5:e02282-14. doi: 10.1128/mBio.02281-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linz B, Windsor HM, McGraw JJ, Hansen LM, Gajewski JP, Tomsho LP, Hake CM, Solnick JV, Schuster SC, Marshall BJ. 2014. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nat Commun 5:4165–4172. doi: 10.1038/ncomms5165. [DOI] [PubMed] [Google Scholar]
- 28.Moore ME, Borén T, Solnick JV. 2011. Life at the margins: modulation of attachment proteins in Helicobacter pylori. Gut Microbes 2:42–46. doi: 10.4161/gmic.2.1.14626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan S, Noto JM, Romero-Gallo J, Peek RM Jr, Amieva MR. 2011. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog 7:e1002050. doi: 10.1371/journal.ppat.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim A, Servetas SL, Kang J, Kim J, Jang S, Cha HJ, Lee WJ, Kim J, Romero-Gallo J, Peek RM Jr, Merrell DS, Cha JH. 2015. Helicobacter pylori bab paralog distribution and association with cagA, vacA, and homA/B genotypes in American and South Korean clinical isolates. PLoS One 10:e0137078. doi: 10.1371/journal.pone.0137078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aspholm M, Olfat FO, Norden J, Sonden B, Lundberg C, Sjostrom R, Altraja S, Odenbreit S, Haas R, Wadstrom T, Engstrand L, Semino-Mora C, Liu H, Dubois A, Teneberg S, Arnqvist A, Borén T. 2006. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog 2:e110. doi: 10.1371/journal.ppat.0020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodwin AC, Weinberger DM, Ford CB, Nelson JC, Snider JD, Hall JD, Paules CI, Peek RM Jr, Forsyth MH. 2008. Expression of the Helicobacter pylori adhesin SabA is controlled via phase variation and the ArsRS signal transduction system. Microbiology 154:2231–2240. doi: 10.1099/mic.0.2007/016055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talarico S, Whitefield SE, Fero J, Haas R, Salama NR. 2012. Regulation of Helicobacter pylori adherence by gene conversion. Mol Microbiol 84:1050–1061. doi: 10.1111/j.1365-2958.2012.08073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Åberg A, Gideonsson P, Vallstrom A, Olofsson A, Ohman C, Rakhimova L, Borén T, Engstrand L, Brannstrom K, Arnqvist A. 2014. A repetitive DNA element regulates expression of the Helicobacter pylori sialic acid binding adhesin by a rheostat-like mechanism. PLoS Pathog 10:e1004234. doi: 10.1371/journal.ppat.1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Borén T. 2002. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooke CL, An HJ, Kim J, Canfield DR, Torres J, Lebrilla CB, Solnick JV. 2009. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterology 137:1061–1071, 1071.e1-8. doi: 10.1053/j.gastro.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Fero JB, Mendez M, Carpenter BM, Servetas SL, Rahman A, Goldman MD, Borén T, Salama NR, Merrell DS, Dubois A. 2015. Analysis of a single Helicobacter pylori strain over a 10-year period in a primate model. Int J Med Microbiol 305:392–403. doi: 10.1016/j.ijmm.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haas G, Karaali G, Ebermayer K, Metzger WG, Lamer S, Zimny-Arndt U, Diescher S, Goebel UB, Vogt K, Roznowski AB, Wiedenmann BJ, Meyer TF, Aebischer T, Jungblut PR. 2002. Immunoproteomics of Helicobacter pylori infection and relation to gastric disease. Proteomics 2:313–324. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Kimmel B, Bosserhoff A, Frank R, Gross R, Goebel W, Beier D. 2000. Identification of immunodominant antigens from Helicobacter pylori and evaluation of their reactivities with sera from patients with different gastroduodenal pathologies. Infect Immun 68:915–920. doi: 10.1128/IAI.68.2.915-920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solnick JV, Canfield DR, Yang S, Parsonnet J. 1999. Rhesus monkey (Macaca mulatta) model of Helicobacter pylori: noninvasive detection and derivation of specific pathogen free monkeys. Lab Anim Sci 49:197–201. [PubMed] [Google Scholar]
- 41.Solnick JV, Hansen LM, Canfield DR, Parsonnet J. 2001. Determination of the infectious dose of Helicobacter pylori during primary and secondary infection in rhesus monkeys (Macaca mulatta). Infect Immun 69:6887–6892. doi: 10.1128/IAI.69.11.6887-6892.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek RM Jr, Cover TL, Solnick JV. 2013. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 9:e1003189. doi: 10.1371/journal.ppat.1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubois A, Berg DE, Incecik ET, Fiala N, Heman-Ackah LM, Del Valle J, Yang M, Wirth HP, Perez-Perez GI, Blaser MJ. 1999. Host specificity of Helicobacter pylori strains and host responses in experimentally challenged nonhuman primates. Gastroenterology 116:90–96. doi: 10.1016/S0016-5085(99)70232-5. [DOI] [PubMed] [Google Scholar]
- 44.Odenbreit S, Kavermann H, Puls J, Haas R. 2002. CagA tyrosine phosphorylation and interleukin-8 induction by Helicobacter pylori are independent from alpAB, HopZ and bab group outer membrane proteins. Int J Med Microbiol 292:257–266. doi: 10.1078/1438-4221-00205. [DOI] [PubMed] [Google Scholar]
- 45.Dixon MF, Genta RM, Yardley JH, Correa P. 1996. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 20:1161–1181. [DOI] [PubMed] [Google Scholar]
- 46.Kable ME, Hansen LM, Styer CM, Deck SL, Rakhimova O, Shevtisova A, Eaton KA, Martin ME, Gideonsson P, Borén T, Solnick JV. 2017. Host determinants of expression of the Helicobacter pylori BabA adhesin. Sci Rep 7:46499. doi: 10.1038/srep46499. [DOI] [PMC free article] [PubMed] [Google Scholar]