

ABSTRACT
In addition to their chemical antimicrobial nature, bile acids are thought to have other functions in the homeostatic control of gastrointestinal immunity. However, those functions have remained largely undefined. In this work, we used ileal explants and mouse models of bile acid administration to investigate the role of bile acids in the regulation of the intestinal antimicrobial response. Mice fed on a diet supplemented with 0.1% chenodeoxycholic acid (CDCA) showed an upregulated expression of Paneth cell α-defensins as well as an increased synthesis of the type-C lectins Reg3b and Reg3g by the ileal epithelium. CDCA acted on several epithelial cell types, by a mechanism independent from farnesoid X receptor (FXR) and not involving STAT3 or β-catenin activation. CDCA feeding did not change the relative abundance of major commensal bacterial groups of the ileum, as shown by 16S analyses. However, administration of CDCA increased the expression of ileal Muc2 and induced a change in the composition of the mucosal immune cell repertoire, decreasing the proportion of Ly6G+ and CD68+ cells, while increasing the relative amount of IgGκ+ B cells. Oral administration of CDCA to mice attenuated infections with the bile-resistant pathogens Salmonella enterica serovar Typhimurium and Citrobacter rodentium, promoting lower systemic colonization and faster bacteria clearance, respectively. Our results demonstrate that bile acid signaling in the ileum triggers an antimicrobial program that can be potentially used as a therapeutic option against intestinal bacterial infections.
KEYWORDS: bile acids, Citrobacter rodentium, intestinal antimicrobial peptides, mucins, mucosal immunity, Salmonella enterica
INTRODUCTION
Bile acids are synthetized by hepatocytes and excreted to the bile in their primary glycine- or taurine-conjugated forms. Approximately 50% of bile is collected and stored in the gallbladder (1), where the concentration of bile acids rises 6- to 10-fold (2, 3). Eventually, the bile is discharged into the duodenum in response to feeding, incorporating concentrated bile acids into the process of digestion and absorption of dietary lipids and liposoluble compounds (4). Most of the intestinal bile acids are later reabsorbed in the ileum by active transport, carried back to the liver by the entero-portal circulation, and incorporated again into the bile. The bile acids not reclaimed by the ileum are metabolized by the intestinal commensal microbiota or excreted in the feces (4). Microbial transformation of primary conjugated bile acids starts in the terminal ileum (5) with amino acid deconjugation, which produces primary, unconjugated bile acids. Those may be subsequently converted into secondary bile acids by other chemical modifications, such as oxidation and dehydroxylation (6). Once deconjugated, bile acids do not require active transport and can be passively absorbed by the intestinal mucosa (5). In humans, the primary, conjugated bile acid pool is composed mostly of cholic acid (CA) and chenodeoxycholic acid (CDCA) conjugated to either glycine or taurine (7). However, in mice, it is composed of approximately 49% β-muricholic acid, 46% CA, and only 2% chenodeoxycholic acid (CDCA), exclusively conjugated to taurine (3).
Due to their detergent nature and membranolytic activity (8), bile acids are microbicidal (9, 10), a feature considered important for limiting the growth of bacteria in the proximal small intestine (10, 11). However, recent evidence suggests that in addition to that, bile acid signaling to the gut epithelium is important for intestinal immunity and homeostasis, as genetic deficiency of the bile acid nuclear receptor FXR (farnesoid X receptor) facilitates intestinal bacterial overgrowth and leads to a compromised intestinal barrier (12). This barrier is a functional entity formed by the continuous single-cell layer of intestinal epithelial cells, antimicrobial peptides and proteins (AMPPs), immunoglobulins produced by mucosal resident B cells (13), the mucus layer that separates the epithelium from the luminal content (14), dispersed mucosal secondary lymphoid tissue, and a full complement of mucosal immune cells (15). All of these elements work together to maintain the delicate homeostatic balance of the gastrointestinal tract. Defects of the epithelial barrier lead to increased intestinal permeability, mucosal inflammation, and physiological dysfunction (15, 16). In the intestines, enterocytes, Paneth cells, and goblet cells of the epithelium produce a large repertoire of AMPPs (17) that has been associated with the control of the intestinal microbiota number and composition (18, 19) and the defense from enteric pathogens (20, 21). The regulation of AMPP synthesis and secretion involves the microbiota, the innate immune system, bacterial pathogens, mucosal immune cells, or cytokines, depending on the specific AMPP (19, 22–24). Secreted AMPPs, together with immunoglobulins, accumulate to their highest antimicrobial concentrations within the intestinal mucus layer (25–28).
In this study, using mice as a model, we investigated the role of bile acids on the regulation of ileal AMPP synthesis and their impact on the intestinal antimicrobial environment. We found that dietary supplementation with chenodeoxycholic acid (CDCA, the unconjugated form of the primary low-abundance bile acid taurochenodeoxycholic acid [TCDCA]), stimulated the synthesis of several AMPPs in the terminal ileum, in vivo. CDCA feeding also upregulated the ileal expression of Muc2, caused a significant decrease in the number of mucosal macrophages and neutrophils, and modestly increased the number of mucosal B cells. All of these effects occurred without significant changes in the relative abundance of several major commensal bacterial groups of the ileal microbiota. CDCA feeding enhanced the intestinal antimicrobial environment such as to limit the systemic spread of orally administered Salmonella enterica serovar Typhimurium and accelerate the clearance of enteric Citrobacter rodentium infections. Taken together, our results show that bile acids play important roles in the regulation of the antimicrobial program of the terminal ileum and suggest they act as regulators of critical aspects of the intestinal epithelial barrier and immunity. These findings also uncover a potential therapeutic use of bile acids in the control of enteric bacterial infections.
RESULTS
CDCA induces the synthesis of multiple ileal antimicrobial peptides.
To determine the direct effect of bile acids in the intestinal production of antimicrobial peptides, we used an ex vivo system based in cultured ileal explants (23). Under the experimental conditions used here, these explants are essentially devoid of microbiota due to the use of antibiotics and the aerobic environment. Explants were exposed to a panel of primary conjugated bile acids (taurocholic acid [TCA] and taurochenodeoxycholic acid [TCDCA]), their primary unconjugated derivatives (cholic acid [CA] and chenodeoxycholic acid [CDCA], respectively) or their secondary derivatives (deoxycholic acid [DCA] and lithocholic acid [LCA], respectively) at 5 μM concentrations for 6 h. The ileum was selected as the target tissue because (i) it is involved in the reabsorption of bile acids from the intestinal lumen (4), (ii) bacterial modification of bile acids starts in the ileum, and thus it is exposed to different types of bile acids (5), and (iii) it is the region of the small intestine that harbors the highest abundance of Paneth cells, the professional antimicrobial-producing cells and the sole producers of multiple intestinal α-defensins (Defa) (29). The relative levels of transcripts for several α-defensin genes were analyzed by quantitative PCR (qPCR). As shown in Fig. 1, primary conjugated and unconjugated bile acids induced the expression of Defa genes to various extents. CDCA induced the strongest and more generalized effect. The secondary bile acids DCA and LCA failed to stimulate the expression of Defa genes in cultured ileal explants.
FIG 1.
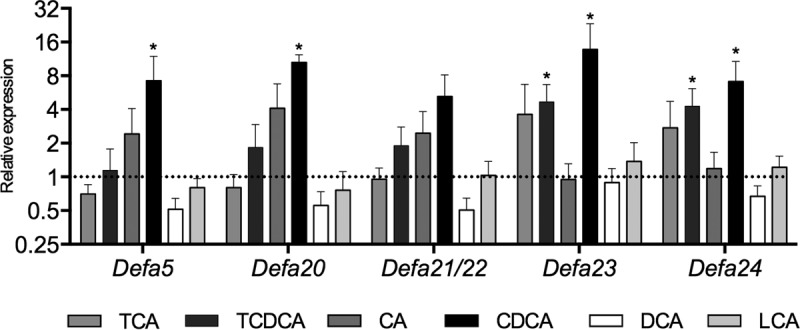
Bile acids induce the expression of AMPPs in ileal explants. Shown are the relative transcript levels of Defa genes in ileal explants treated with various bile acids. TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid. The expression levels in explants treated with vehicle controls are set at 1 and indicated by a dotted line. Data were obtained by qPCR. n = 6 to 8 samples per group. Statistically significant differences are shown by asterisks (*, P < 0.05).
CDCA was selected for further studies because it induced significantly higher transcript levels for 4 of the 5 α-defensin genes tested. Also, we reasoned that since CDCA is a low-abundance bile acid in mice, variations of its concentrations could be more meaningful to the intestinal environment than changes of the same magnitude in the concentration of highly abundant bile acids. Under this assumption, CDCA could be more likely to evoke an adaptive response in vivo. Furthermore, as activation of the MyD88 pathway by the intestinal microbiota is involved in the homeostatic expression of multiple AMPP genes in vivo (19, 23), we evaluated the impact of ileal Toll-like receptor 4 (TLR4) activation (as a way of mimicking signaling from the microbiota) in the context of CDCA treatment. We focused on one member of the Defa gene family (Defa20) and expanded the analyses to other AMPPs that, in contrast to the α-defensins, are also produced by other cell types of the intestinal epithelium. Ileal explants were independently or simultaneously treated with 10 ng/ml lipopolysaccharide (LPS) from Escherichia coli and 5 μM CDCA for 6 h in culture. The results in Fig. 2 show that independent treatments with either CDCA or LPS significantly increase the relative transcript levels for Defa20, Reg3b, and Reg3g, whereas lysozyme (Lyz1), angiogenin 4 (Ang4) and matrix metalloproteinase 7 (Mmp7, coding for the enzyme responsible for processing precursor Paneth cell α-defensins to their mature functional form) were rather insensitive to both CDCA and LPS exposure. Moreover, cotreatment with CDCA and LPS caused a higher increase in Defa20 transcripts, although not that of other AMPPs. These results indicate that the regulatory pathways of ileal AMPP production by bile acids and by microbial activation of TLR4 are independent of each other and suggest that at least for some of the α-defensin genes, those pathways may operate in synergy.
FIG 2.
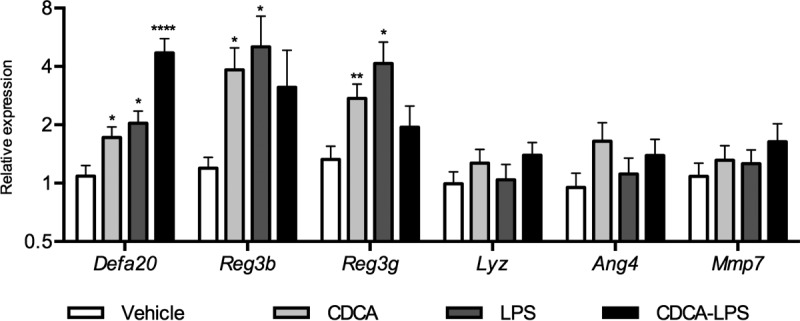
CDCA induces the synthesis of AMPPs in ileal explants independently of TLR4 activation. Shown are the relative transcript levels of AMPP genes in ileal explants treated with 5 μM CDCA, 10 nM E. coli LPS, or a combination of both. Data were obtained by qPCR. n = 12 to 14 samples per group. Statistically significant differences are shown by asterisks (*, P < 0.05; **, P < 0.01; ****, P < 0.0001).
To evaluate whether this effect of CDCA occurs in a physiological context in vivo, C57BL/6 mice were given a diet supplemented with 0.1% CDCA ad libitum for 16 h (from 5:00 p.m. to 9:00 a.m., including the dark period). The animals were euthanized at the end of the feeding period, and ileal AMPP transcripts and protein levels were analyzed by quantitative PCR, Western blotting, and immunofluorescence (Fig. 3). The CDCA diet induced the transcript abundance of Defa20 and other α-defensin genes, as well as Reg3b and Reg3g (Fig. 3A). The protein levels of Reg3b and Reg3g were also increased, as shown by Western blotting (Fig. 3B). Immunofluorescent staining for Reg3b and Reg3g revealed a marked increase in the number of positive cells in the ileum of CDCA-fed animals (Fig. 3C and D), whereas lysozyme did not show any significant difference between the two diets (Fig. 3E). Reg3b staining was absent in the crypts and more strongly localized to the base of the villi, in agreement with others (30). Stimulation of Reg3g synthesis occurs not only in cells of the crypt (presumably Paneth cells) but also in cells localized along the villus. These results demonstrate that CDCA can upregulate antimicrobial peptide synthesis in more than one type of intestinal epithelial cells in vivo.
FIG 3.

CDCA induces the synthesis of ileal AMPPs in vivo. (A) Relative transcript levels of AMPP genes in the ileum of animals fed on a 0.1% CDCA-supplemented diet in comparison to animals fed on a normal diet for 16 h. Data were obtained by qPCR. n = 11 to 13 samples per group. Statistically significant differences are shown by asterisks (*, P < 0.05; **, P < 0.01). (B) Reg3b and Reg3g Western blots of total ileum lysates from two animals each fed with CDCA or on a normal diet (ND). (C to E) Immunofluorescent microscopy of ileal tissues from animals fed CDCA or normal diet stained for Reg3g (C), Reg3b (D), or lysozyme (E). Scale bars are 25 μm.
So far, only two cytoplasmic membrane receptors for bile acids have been described. SLC10A2 (apical sodium-dependent bile acid transporter [Asbt]) is responsible for bile acid reabsorption from the intestinal lumen, but its expression is restricted to ileal enterocytes, and it exclusively transports conjugated bile acids (31), thus it is unlikely to mediate the CDCA effect. The other receptor, TGR5 (Gpbar), is a G-protein-coupled receptor present in many cell types, responsive to conjugated and unconjugated CDCA, and highly expressed in the ileum and colon (32). To determine if activation of TGR5 increased AMPP synthesis, ileal explants were treated with the TGR5-specific agonist 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N,5-dimethyl-4-isoxazolecarboxamide. The results in Fig. S1A in the supplemental material show that as previously reported, TGR5 activation induced the expression of the Il1b and Tnf genes (33). However, it failed to induce Defa20, Reg3b, or Reg3g, thus suggesting that TGR5 is not involved in the regulation of the synthesis of these AMPPs by CDCA.
Since bile acids have the ability to bind to and activate the nuclear receptor FXR (11), we probed whether activation of this receptor with the specific agonist GW4064 causes an increase in AMPP gene expression. Different from CDCA, GW4064 treatment failed to significantly upregulate the synthesis of AMPPs in ileal explants, while it still induced the FXR target genes Slc51a (organic solute transporter alpha [Osta]) and Fabp6 (fatty acid binding protein 6) (see Fig. S2 in the supplemental material), suggesting that FXR activation has no effect in the expression of the AMPPs analyzed. In keeping with that, CDCA treatment did not induce the expression of the FXR target genes Slc51a and Fabp6 (see Fig. S3 in the supplemental material). Together, these results strongly suggest that stimulation of AMPP expression by CDCA is not dependent upon activation of FXR and that instead it proceeds through a different pathway.
Bile acid signaling in esophageal cells and hepatocytes has been linked to the interleukin-6 (IL-6)/STAT3 pathway (34, 35). Moreover, STAT3 activation has been associated with Reg3b and Reg3g synthesis in intestinal and lung epithelia, respectively (36, 37): thus, we considered the possibility that CDCA would act over AMPP synthesis by activation of STAT3 either directly or through the induction of IL-6. In fact, activation of STAT3 using IL-6 in rat IEC-6/L1 intestinal epithelial cells and mouse explants correlated with increased production of Reg3b (Fig. S1B). However, IL-6 was undetectable in CDCA-fed animals, and CDCA did not induce phosphorylation of ileal STAT3 in animals, explants, or IEC-6/L1 cells (Fig. S1C). Collectively, these results suggest that a pathway relying on direct or indirect activation of STAT3 by CDCA is not involved in the CDCA-induced synthesis of AMPPs.
Signaling through the Wnt/β-catenin pathway is involved in the control of both Paneth cell development and α-defensin expression (38, 39). Extrapolation of data from cancer studies (40–45) suggested the possibility that a CDCA-Wnt/β-catenin or a CDCA-COX2-Wnt/β-catenin pathway might function in the healthy intestinal epithelium and drive AMPP synthesis. Expression of Ptgs2 (Cox2) in the ileum was in fact upregulated by CDCA feeding (Fig. S1D), but immunohistochemical staining of ileal sections showed no increase of β-catenin nuclear localization (Fig. S1E) or upregulation of expression of its target gene Ccnd1 (cyclin D1) in CDCA-fed animals (Fig. S1D), indicating that activation of β-catenin does not participate in the stimulation of ileal AMPP synthesis by CDCA.
CDCA modifies the intestinal immunological environment.
To characterize the impact of CDCA feeding on the immunological intestinal environment, we studied its effects over the ileal mucosa. Hematoxylin and eosin (H&E)-stained sections from the ileum of animals fed normal diet and CDCA were examined for signs of an inflammatory response. Histological examination showed a good preservation of mucosal architecture and did not reveal any apparent epithelial damage or signs of ileal inflammation as a result of CDCA administration (Fig. 4A). An analysis of major types of immune cell populations in the ileal mucosa (Fig. 4B and C) revealed lower relative numbers of mucosal monocytes/macrophages (CD68+) and neutrophils (Ly6G+), whereas no significant change in the frequency of CD4+ cells was observed. The proportion of IgGκ+ B cells was increased, although it did not reach statistical significance. Collectively, the results show that this regimen of CDCA feeding does not trigger ileal inflammation and strongly suggest that an inflammatory response is not the cause underlying the induction of antimicrobial peptide synthesis. The observed changes in the proportion of immune cells are in fact suggestive of a shift toward an attenuated capacity of the ileal mucosa to mount an inflammatory response.
FIG 4.

CDCA does not induce inflammation but alters the relative abundance of major immune cell types of the ileal mucosa. Animals were fed on a CDCA-supplemented diet or a normal diet (ND) for 16 h. (A) H&E staining of ileal cross sections. Scale bars are 100 μm. (B) FACS dot plots of CD68+, Ly6G+, CD4+, and IgGκ+ cells from the ileal mucosa; the results shown are from one representative animal. (C) Percentage of CD68+, Ly6G+, CD4+ and IgGκ+ cells from the ileal mucosa (CDCA, n = 5 animals; normal diet, n = 4 animals). Statistically significant differences are shown by asterisks (*, P < 0.05; **, P < 0.01).
Histological examination did not reveal crypt hyperplasia or gross changes in appearance and abundance of Paneth cells upon CDCA feeding (Fig. 4A), suggesting that increase in Paneth cell number is not the cause of elevated levels of antimicrobial peptides. However, goblet cells, another important producer of antimicrobial peptides such as Reg3b, Reg3g, and angiogenin 4, were more prominent upon side-by-side staining with alcian blue (Fig. 5A and B). Moreover, the transcript levels of Muc2, the gene coding for the major intestinal goblet cells' mucin, were moderately but significantly increased in CDCA-fed animals (Fig. 5C), suggesting that in addition to antimicrobial peptide synthesis, CDCA feeding stimulates mucin production in the ileum.
FIG 5.

Alcian blue staining of ileal sections from mice fed on a normal diet (ND) (A) or a CDCA-supplemented diet (B). Scale bars are 100 μm. (C) Relative transcript levels of ileal Muc2 in animals fed with the CDCA-supplemented diet in comparison to animals fed a normal diet. Data were obtained by qPCR. n = 8 to 9 samples per group (*, P < 0.05).
Given that intestinal commensals are involved in multiple aspects of intestinal mucosal immunity, including immune cell differentiation, proliferation, AMPP synthesis, and mucin production (23, 46, 47), we investigated if the short-term CDCA feeding regimen used here caused gross alterations in the relative abundance of major bacterial groups normally found in the mouse ileal microbiota. 16S qPCR analyses showed a slight but statistically significant increase in the relative abundance of Bacteroidetes on the ileum of CDCA-fed animals (see Fig. S4 in the supplemental material). However, no significant changes were observed for the phylum Firmicutes, the family Enterobacteriaceae, Lactobacillus spp., Bacteroides spp., or segmented filamentous bacteria (SFB) (Fig. S4). These results suggest that the ileal microbiota composition was generally stable in response to CDCA feeding and that major shifts in the composition of the normal microbiota are not associated with the observed effects of CDCA over the ileal epithelium.
CDCA feeding limits the systemic spread of Salmonella Typhimurium and accelerates the clearance of Citrobacter rodentium infection.
To determine the antimicrobial potential of the changes induced by CDCA against intestinal pathogens, we used two well-established models of enteric bacterial infections. Orally administered Salmonella Typhimurium is able to traverse the intestinal epithelial barrier and spread systemically, establishing a lethal typhoid-like disease in susceptible mouse strains such as C57BL/6 (48). Citrobacter rodentium is the mouse model of human enteropathogenic and enterohemorrhagic E. coli (EPEC and EHEC, respectively). In contrast to Salmonella, Citrobacter remains confined to the colon extracellularly attached to the colonic mucosa and in C57BL/6 mice establishes a self-limiting infection that typically peaks around day 10 and clears by 3 to 4 weeks postinfection (49). Both Salmonella and Citrobacter are highly resistant to bile acids and are capable of growing very efficiently in bile (10, 50) (see Fig. S5 in the supplemental material).
Animals given the CDCA-supplemented diet were infected with either Salmonella or Citrobacter and examined for systemic colonization at day 3 postinfection (Salmonella) or monitored for fecal bacterial shedding for a period of 28 days (Citrobacter). In both cases, CDCA feeding was maintained for the duration of the experiments. The results in Fig. 6A and B show that Salmonella burden in the liver and spleen was significantly lower in CDCA-fed animals, possibly as a consequence of more efficient intestinal bacterial killing resulting in lower numbers of Salmonella cells translocated through the epithelium. However, overall survival of infected animals in this model was not affected by CDCA treatment (see Fig. S6 in the supplemental material).
FIG 6.

CDCA feeding attenuates the bacterial burden of enteric infections. (A and B) Bacterial counts (CFU per milligram of tissue) in the liver (A) and spleen (B) of mice fed with CDCA-supplemented diet or on a normal diet (ND) and orally infected with Salmonella Typhimurium SL1344. Counts were taken at day 3 postinfection. n = 12 to 15 mice/group. (C) Bacteria counts in the feces (CFU per milligram of feces) of mice fed the CDCA-supplemented or normal diet and orally infected with Citrobacter rodentium DS100. Counts were taken up to day 28 postinfection. n = 10 to 11 mice/group (*, P < 0.05).
In contrast, infections with Citrobacter progressed similarly during the first days of infection, as evaluated by bacterial fecal shedding. However, by day 12, Citrobacter counts in the CDCA-fed group started to decrease faster than in the group fed the normal diet (Fig. 6C). By day 20 postinfection, bacterial counts were significantly different and only 3 out of 11 CDCA-fed animals had detectable Citrobacter in their feces, whereas in the normal-diet-fed group, 8 out of 10 mice were still colonized. By day 28, all CDCA-fed animals had cleared the infection, whereas most in the control group remained infected. Collectively, our results show that oral CDCA administration decreases the bacterial burden of experimental Salmonella and Citrobacter infections in mouse models of typhoid fever and gastroenteritis.
DISCUSSION
In recent years, it has become clear that in addition to their digestive function, bile acids are involved in multiple other functions, including the control of whole-body immunity and metabolism (11, 51). Using an experimental mouse model of dietary administration, we demonstrate here that CDCA, a bile acid whose conjugated form constitutes only 2% of the total bile acids that reach the gastrointestinal tract of mice, acts as a positive regulator of the intestinal antimicrobial environment in vivo. CDCA impacted the ileal expression of genes from two major families of intestinal AMPPs, namely, Paneth cell α-defensins and C-type lectins, while not influencing the synthesis of others, such as lysozyme, also produced by Paneth cells.
It is apparent that in vivo, CDCA evokes an antimicrobial program in more than one type of ileal epithelial cells. While intestinal α-defensins are exclusively produced by Paneth cells, the C-type lectins Reg3b and Reg3g are made by several cell types, including Paneth cells, goblet cells, and enterocytes (19, 30, 52). In keeping with that, immunofluorescence microscopy showed enhanced positive staining for Reg3b and Reg3g in the villi, outside the Paneth cell niche on the crypts. The notion of a multicell effect of CDCA is further supported by the increase of goblet cells' Muc2 expression, although it is unclear how CDCA signals to all of these cell types.
Our data show that activation of TGR5 with a specific agonist failed to induce ileal AMPP expression, suggesting that TGR5 is not part of the acting pathway. Given these results and the fact that unconjugated bile acids such as CDCA do not require active transport to enter the intestinal epithelium (5), passive uptake of CDCA seems the most likely entry mechanism. Once inside the cells, bile acids can interact with the nuclear receptor FXR, which has been involved in the immunity of the gut (12). However, CDCA induced AMPP genes but failed to induce the FXR target genes Slc51a and Fabp6 in intestinal explants (although taurine-conjugated CDCA did). Moreover, FXR activation with a specific agonist failed to induce AMPP expression (although it induced Slc51a and Fabp6), suggesting that bile acid control of ileal AMPP expression is not mediated by FXR. CDCA also failed to activate STAT3 and β-catenin, the central components of two signaling pathways known to regulate intestinal AMPP expression (36–39). The mechanism by which CDCA induces the synthesis of ileal AMPPs remains to be determined.
The relative abundance of several important groups of the normal ileal microbiota was not affected by CDCA feeding, with the exception of a subtle but statistically significant increase of Bacteroidetes. The phylum Bacteroidetes includes a large number of bacterial species, and changes in its abundance in the gut upon bile acid (cholic acid) feeding have been previously reported (53 [although in that instance, the Bacteroidetes population was almost wiped out]). Whether the higher abundance of Bacteroidetes observed by us is directly responsible for the stimulation of AMPP synthesis is unclear. Decreased ileal Bacteroidetes abundance after prednisolone treatment has been seen in association with lower Reg3b and Reg3g expression, opening the possibility of a causal connection (although changes in Defa20 were not observed) (54). On the other hand, it has also been shown that streptomycin treatments, which decrease the abundance of Bacteroidetes (55), also reduce the synthesis of ileal AMPPs (23), which suggest that no such causal connection exists.
CDCA induced changes in the relative numbers of several types of mucosal immune cells, resulting in an attenuated effector cell profile (fewer neutrophils and macrophages) and a potentially enhanced immunoglobulin secretory capacity of the ileal mucosa. To our knowledge, these effects of a bile acid over intestinal immune cell populations have never been reported, and it is intriguing that those changes are detectable after the short time of CDCA administration used here. This is suggestive of a highly dynamic homeostatic turnover of immune cells on the ileal mucosa in response to changes in the lumen chemical composition. The mechanisms behind these variations are not known, but two plausible nonexclusive explanations are that CDCA signaling influences immune cell differentiation and/or their homing to the intestinal mucosa.
Intestinal AMPPs, immunoglobulins, and a healthy mucus layer are all important factors in the defense against enteric infections (20, 56–61). CDCA administration, which increased AMPP production, the mucosal B cell fraction, and Muc2 expression, also accelerated the clearance of Citrobacter rodentium infection and limited the systemic spread of Salmonella Typhimurium. Extraintestinal dissemination of orally acquired Salmonella occurs by traversing the gut epithelium, primarily at the distal ileum (62). Translocated bacteria are then picked up by mucosal macrophages, transported to mesenteric lymph nodes, and from there spread systemically through the circulation (63). Host defense against Salmonella infection involves Reg3b, Reg3g, and Paneth cell α-defensins (20, 58, 59, 64), which are all induced in the ileum by CDCA. It is fair to assume that the combination of higher intestinal AMPP concentrations and a relatively lower number of mucosal macrophages would create a restrictive scenario for the systemic spread of Salmonella. However, CDCA did not have an impact on survival, as it did not fully prevent systemic colonization. In this model of infection (C57BL/6 mice and wild-type Salmonella Typhimurium), once systemic colonization is established, the pathogenesis rapidly proceeds to full lethality.
Citrobacter rodentium on the other hand, is an attaching-effacing extracellular pathogen mostly restricted to the colon (49). IL-22-induced Reg3g is involved in protection from Citrobacter infection (65), and Paneth cell α-defensins kill it (66). In addition, its clearance is mediated by B cells and immunoglobulins (56, 61). In this work, we did not study the effects of CDCA on the colonic immunological environment; however, it is possible that ileal immunoglobulin secretion is increased. That, together with the fact that Paneth cell α-defensins from the small intestine are found intact and functional up to the distal large bowel (67) may explain the beneficial effect of CDCA in the clearance of Citrobacter infections. The finding that CDCA interferes with two bile-resistant intestinal pathogens with such different life styles is striking and provides a rationale to explore the use of targeted bile acids for the treatment of enteric infections.
The fact that CDCA stimulates the synthesis of ileal AMPPs, mucins, and possibly immunoglobulins and at the same time seems to restrain cellular effector arms of the innate mucosal immunity is intriguing and suggests that CDCA coordinates a homeostatic antimicrobial mechanism that relies on the secretory response to control microbial overgrowth in the terminal ileum. Such a mechanism could be very important to limit the nutrient-induced bacterial growth that occurs after meals (68), while preventing an unnecessary inflammatory burst in response to a homeostatic activity such as feeding. It could be expected to be active also in humans and would offer the opportunity to be manipulated and exploited as a therapeutic tool against intestinal infections. It should be noted that different from mice, CDCA constitutes approximately 50% of the primary bile acid pool of humans, and it may fall to another bile acid species to exert the same effects over the ileal antimicrobial environment. Therefore, CDCA's function as a regulator of AMPP synthesis and its potential to counteract enteric bacterial infections in humans need to be experimentally determined.
MATERIALS AND METHODS
Animals.
Eight- to 12-week-old male C57BL/6 mice were purchased from Charles River Laboratories and maintained on standard diet (D12102C) or normal diet supplemented with 0.1% CDCA (D12020705). Both diets were formulated and manufactured by Research Diets, Inc., NJ. Tissue samples of the terminal ileum were taken approximately 2 cm from the ileo-cecal junction for RNA/DNA isolation, protein analyses, and microscopy. Animal protocols were approved by the Animal Care Committee of the Université de Sherbrooke.
Culture of cells and ileal explants.
Cells of the stable Cdx2-transfected rat intestinal epithelial cell line IEC-6/L1 (69) were a kind gift from F. Boudreau (Université de Sherbrooke). They were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 4.5 g/liter d-glucose, 5% fetal bovine serum, and 0.1 U/ml of insulin. The cells were treated with 4 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to express Cdx2, which under these conditions induces their limited differentiation (70). After 3 days, the cells were treated with 10 ng/ml IL-6 (BioShop, Ontario, Canada) for 6 h and analyzed for synthesis of PAP1 (rat Reg3b [NCBI accession no. P25031.1]). Ileal explants were prepared as described previously (23). Briefly, the distal region of the ileum was resected and flushed with sterile phosphate-buffered saline (PBS). The tissue was opened longitudinally and washed repeatedly with sterile PBS to eliminate the remaining intestinal content and immediately placed in DMEM–10% charcoal-treated bovine serum–100 μg/ml streptomycin–100 U/ml penicillin. The tissue was divided into 3- to 4-mm-long sections under sterile conditions and incubated at 37°C in a humidified 5% CO2 atmosphere for 1 h. Following this, the medium was replaced with fresh medium and incubated for an additional hour. Explants were then placed on the same medium containing 5 μM bile acids (taurocholic acid [TCA], taurochenodeoxycholic acid [TCDCA], cholic acid [CA], chenodeoxycholic acid [CDCA], deoxycholic acid [DCA], and lithocholic acid [LCA]: Sigma catalogue no. T4009, T6260, C1129, C9377, D4297, and L6250, respectively), E. coli LPS, or vehicle (dimethyl sulfoxide [DMSO] or water, as required) and incubated for 6 h at 37°C in a humidified 5% CO2 atmosphere. Treatment of explants with the TGR5 and FXR agonists 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N,5-dimethyl-4-isoxazolecarboxamide (AbCam; ab142091) and GW4064 (Sigma; G5172), respectively, was done in a similar way.
Analysis of expression.
Ileal tissue sections were collected in RNAlater (Qiagen); explants were directly processed for RNA purification. RNA was isolated using the RNeasy kit (Qiagen), and cDNA was prepared from 1 μg of RNA using the Quantitech reverse transcription kit (Qiagen). Quantitative PCRs were done in an Eppendorf Realplex 2 apparatus using the DyNamo SYBR green PCR kit from ThermoFisher. The qPCR primers and conditions for the Defa genes are as in reference 23; all other primers are given in Table 1. Relative expression was calculated with respect to controls fed normal diet or explants treated with vehicle, using the threshold cycle (ΔΔCT) method corrected for primer efficiencies according to Pfaffl et al. (71). Western blots were performed using total ileum lysates prepared from snap-frozen tissue. No luminal flush washes were done in order to quantitate AMPPs in both the tissue and the luminal content, including the mucus layer in which the highest concentrations of AMPPs typically accumulate (26, 28). SDS-PAGE gels were loaded with 20 μg of protein/well, determined in triplicate using a bicinchoninic acid (BCA) assay (ThermoFisher). The antibodies used were against Reg3g (Aviva System Biology; OAAB03017 [1:200]), Reg3b (R&D Systems; AF5110 [1:200]), PAP1 (R&D Systems; AF1996 [1:200]), and horseradish peroxidase (HRP)-conjugated β-tubulin (AbCam; ab21058 [1:10,000]).
TABLE 1.
Genes analyzed in this study and sequences of the qPCR primer sets
| Target gene or bacteria | NCBI accession no. and/or product | Primer set sequencea |
|---|---|---|
| Genes | ||
| Reg3b | NM_011036.1 (regenerating islet-derived 3 beta) | GGCTTCATTCTTGTCCTCCA |
| TCCACCTCCATTGGGTTCT | ||
| Reg3g | NM_011260.2 (regenerating islet-derived 3 gamma) | AAGCTTCCTTCCTGTCCTCC |
| TCCACCTCTGTTGGGTTCAT | ||
| Lyz1/Lyz2 | NM_013590.4 (lysozyme 1) | AGCCGATACTGGTGTAATGATG |
| NM_017372.3 (lysozyme 2) | GCACATTGTATGGCTGCAGTG | |
| Muc2 | NM_023566.2 (mucin 2) | GGTGACTGTGACTGTTTCTGC |
| CTTCAGGTCCTCATCATAGATG | ||
| Mmp7 | NM_010810.4 (matrix metallopeptidase 7) | CACTCTAGGTCATGCCTTCGC |
| GGTGGCAGCAAACAGGAAGTTC | ||
| Ang4 | NM_177544.4 (angiogenin, RNase A family, member 4) | AACTCTGGCTCAGAATGAAAG |
| GGCGAGGTTAGCTTTCTTTC | ||
| Fabp6 | NM_008375.2 (fatty acid binding protein 6, ileal [gastrotropin]) | GAATTACGATGAGTTCATGAAGC |
| TTGCCAATGGTGAACTTGTTGC | ||
| Slc51a | NM_145932.3 (organic solute transporter alpha [Osta]) | TCTCCATCTTGGCTAACAGTG |
| GATAGTACATTCGTGTCAGCAC | ||
| IL1b | NM_008361.4 (interleukin 1β) | ACGGACCCCAAAAGATGAAG |
| TTCTCCACAGCCACAATGAG | ||
| Tnf | NM_013693.3 (tumor necrosis factor) | CCACCACGCTCTTCTGTCTAC |
| AGGGTCTGGGCCATAGAACT | ||
| Ptgs2 | NM_011198.3 (prostaglandin-endoperoxide synthase 2 [Cox2]) | AAGACAGATCATAAGCGAGGAC |
| TACCTGAGTGTCTTTGACTGTG | ||
| Ccnd1 | NM_007631.2 (cyclin D1) | AGCATGCACAGACCTTTGTGG |
| GATGACTCTGGAAAGAAAGTGC | ||
| Rplp0 | NM_007475.5 (ribosomal protein, large, P0 [36B4]) | TCTGGAGGGTGTCCGCAAC |
| CTTGACCTTTTCAGTAAGTGG | ||
| Bacteria | ||
| Bacteroides | 16S rRNA | GAGAGGAAGGTCCCCCAC |
| CGCTACTTGGCTGGTTCAG | ||
| Bacteroidetes | 16S rRNA | CGATGGATAGGGGTTCTGAGAGGA |
| GCTGGCACGGAGTTAGCCGA | ||
| Firmicutes | 16S rRNA | GGAGYATGTGGTTTAATTCGAAGCA |
| AGCTGACGACAACCATGCAC | ||
| Enterobacteriaceae | 16S rRNA | GTGCCAGCMGCCGCGGTAA |
| GCCTCAAGGGCACAACCTCCAAG | ||
| Lactobacillus | 16S rRNA | AGCAGTAGGGAATCTTCCA |
| CACCGCTACACATGGAG | ||
| SFB | 16S rRNA | CGGAGCATGTGGTTTAATTC |
| GCTGTCTTCGCTAAAGTGCTC | ||
| Universal bacteria | 16S rRNA | CGGTGAATACGTTCCCGG |
| TACGACTACCTTGTTACGACTT |
The top sequence of each set corresponds to the forward primer and the bottom sequence to the reverse primer.
Mouse infections.
Salmonella enterica serovar Typhimurium strain SL1344 (Smr) and a streptomycin-resistant derivative of Citrobacter rodentium DS100 (a kind gift from B. Vallance, CFRI, Vancouver, Canada) were used in this study. Bacteria were grown overnight at 37°C in LB supplemented with 100 μg/ml streptomycin. Inocula were prepared in sterile 100 mM HEPES–0.9% NaCl (pH 8.0). Mice were infected orally with 5 × 107 Salmonella or 5 ×108 Citrobacter cells/per animal. For Salmonella counts, tissues were collected 3 days after infection and homogenized using a Mixer Mill MM400 (Retsch GmbH) followed by plating of serial dilutions in LB plates containing 100 μg/ml streptomycin. For Citrobacter quantification, feces were collected at the times indicated and treated as described for Salmonella.
Microscopy.
For histological analysis, tissue sections were fixed in 10% buffered formalin, embedded in paraffin, and stained with H&E. Unstained sections were used for immunofluorescence microscopy as previously described (50) and for immunohistochemistry (IHC) using a Dako EnVision+system-HRP (DAB) kit (Dako, K4007). The antibodies were against Reg3g (Aviva System Biology; OAAB03017 [1:100]), Reg3b (R&D System; AF5110 [1:50]), β-catenin (BD Transduction Laboratories; 610154 [1:800]), and lysozyme (DakoCytomation; A0099 [1:600]).
Analysis of microbial populations.
Total DNA was extracted from 5- to 7-mm ileum sections using a Qiagen stool extraction kit following the manufacturer's instructions, including a 95°C lysis step. The DNA was quantified using an ND1000 (Nanodrop, Wilmington, DE) and normalized to 40 ng/μl. qPCR mixtures included 40 ng DNA and 5 μM each forward and reverse primers (Table 1) with 2× EvaGreen master mix (Bio-Rad, Mississauga, Ontario, Canada). The efficiency of primer sets was determined to be between 85 and 115%. qPCRs were performed on a CFX96 (Bio-Rad) under the following conditions: initial denaturation of 98°C for 5 min, followed by 40 cycles of 98°C for 5 s and 55/60°C for 5 s, followed by a 65 to 95°C melt curve analysis. The data were analyzed with CFX Manager 3.0 software (Bio-Rad) by the relative quantity method, with bacterial populations normalized to total bacteria detected using the universal bacteria primer set.
Isolation and quantification of ileal mucosal immune cells.
Ileal sections were opened longitudinally, washed twice in PBS, and separated into 1- to 2-mm pieces, each in 50 to 100 μl of DMEM–10% fetal calf serum (FCS). The sections were transferred to a tube containing 15 ml DMEM–300 U/ml collagenase (Sigma; C2139) and gently dissociated by agitation for 2 h at 100 rpm at 37°C. The cell suspension was filtered through a 70-μm-pore strainer to separate individual cells, and the filtrate was centrifuged for 8 min at 1,000 × g. The supernatant was discarded, and the pellet was suspended in 10 ml of 30% Percoll (Sigma; P4937) and incubated for 1 min at room temperature. The Percoll cell suspension was centrifuged for 30 min at 800 × g at room temperature with the brake on low, and the upper Percoll layer containing fat and cellular debris was discarded. The rest of the suspension was treated with ACK lysis buffer (Invitrogen; A1049201) for 2 min to eliminate red blood cells. The reaction was stopped with PBS, and the cells were washed twice by centrifugation. Isolated cells were resuspended in PBS–2% FCS and stained with isotype control antibodies or fluorochrome-conjugated antibodies to cell surface antigens (phycoerythrin [PE]-conjugated rat anti-mouse CD4 [clone GK1.5; ebiosciences], allophycocyanin [APC]-conjugated rat anti-mouse Ly-6G [clone RB6-8C5; ebiosciences]), biotin-conjugated rat anti-mouse CD68 [clone FA-11; AbD Serotec]), streptavidin-APC-conjugated eFluor 780 (ebiosciences), and fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse Igκ [clone 187.1; BD Biosciences]). After incubation with the antibodies for 30 min at 4°C, the cells were washed with PBS–2% FCS and then examined using a FACSCanto flow cytometer (Becton Dickinson). Fluorescence-activated cell sorter (FACS) results were analyzed using FlowJo (Tree Star, Inc.).
Statistical analyses.
Data were analyzed with GraphPad Prism 6.0 (GraphPad Software, Inc., San Diego, CA). Statistical comparisons were done using the Mann-Whitney U test.
Supplementary Material
ACKNOWLEDGMENTS
We thank F. Boudreau for the IEC-6/L1 cells and for critical reading of the manuscript. We thank B. B. Finlay and B. Vallance for the Salmonella SL1344 and C. rodentium DS100 strains, respectively.
S.T. holds a PhD scholarship from the Fonds de Recherche du Québec-Nature et Technologies (FRQNT). A.M. and S.R. are members of the Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke (CRCHUS). This work was supported by a Discovery grant to A.M. (401949-2011) from the Natural Sciences and Engineering Research Council of Canada (NSERC).
All authors had access to the study data and reviewed and approved the final manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00942-16.
REFERENCES
- 1.Hofmann AF. 1999. Bile acids: The Good, the Bad, and the Ugly. News Physiol Sci 14:24–29. [DOI] [PubMed] [Google Scholar]
- 2.Keulemans YC, Mok KS, de Wit LT, Gouma DJ, Groen AK. 1998. Hepatic bile versus gallbladder bile: a comparison of protein and lipid concentration and composition in cholesterol gallstone patients. Hepatology 28:11–16. doi: 10.1002/hep.510280103. [DOI] [PubMed] [Google Scholar]
- 3.Alnouti Y, Csanaky IL, Klaassen CD. 2008. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 873:209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiang JY. 2009. Bile acids: regulation of synthesis. J Lipid Res 50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofmann AF, Hagey LR. 2008. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci 65:2461–2483. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridlon JM, Kang DJ, Hylemon PB. 2006. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Hofmann AF, Small DM. 1967. Detergent properties of bile salts: correlation with physiological function. Annu Rev Med 18:333–376. doi: 10.1146/annurev.me.18.020167.002001. [DOI] [PubMed] [Google Scholar]
- 8.Garidel P, Hildebrand A, Knauf K, Blume A. 2007. Membranolytic activity of bile salts: influence of biological membrane properties and composition. Molecules 12:2292–2326. doi: 10.3390/12102292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binder HJ, Filburn B, Floch M. 1975. Bile acid inhibition of intestinal anaerobic organisms. Am J Clin Nutr 28:119–125. [DOI] [PubMed] [Google Scholar]
- 10.Begley M, Gahan CG, Hill C. 2005. The interaction between bacteria and bile. FEMS Microbiol Rev 29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 11.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. 2013. Pleiotropic roles of bile acids in metabolism. Cell Metab 17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA. 2006. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woof JM, Mestecky J. 2005. Mucosal immunoglobulins. Immunol Rev 206:64–82. doi: 10.1111/j.0105-2896.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 14.Hooper LV, Macpherson AJ. 2010. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol 10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 15.Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM. 2014. Intestinal permeability—a new target for disease prevention and therapy. BMC Gastroenterol 14:189. doi: 10.1186/s12876-014-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arrieta MC, Bistritz L, Meddings JB. 2006. Alterations in intestinal permeability. Gut 55:1512–1520. doi: 10.1136/gut.2005.085373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee S, Hooper LV. 2015. Antimicrobial defense of the intestine. Immunity 42:28–39. doi: 10.1016/j.immuni.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 18.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. 2010. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. 2008. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A 105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. 2003. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422:522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 21.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. 1999. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 22.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. 2000. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 23.Menendez A, Willing BP, Montero M, Wlodarska M, So CC, Bhinder G, Vallance BA, Finlay BB. 2013. Bacterial stimulation of the TLR-MyD88 pathway modulates the homeostatic expression of ileal Paneth cell alpha-defensins. J Innate Immun 5:39–49. doi: 10.1159/000341630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farin HF, Karthaus WR, Kujala P, Rakhshandehroo M, Schwank G, Vries RG, Kalkhoven E, Nieuwenhuis EE, Clevers H. 2014. Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell-derived IFN-gamma. J Exp Med 211:1393–1405. doi: 10.1084/jem.20130753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer-Hoffert U, Hornef MW, Henriques-Normark B, Axelsson LG, Midtvedt T, Putsep K, Andersson M. 2008. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 57:764–771. doi: 10.1136/gut.2007.141481. [DOI] [PubMed] [Google Scholar]
- 26.McGuckin MA, Linden SK, Sutton P, Florin TH. 2011. Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9:265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 27.Bowcutt R, Forman R, Glymenaki M, Carding SR, Else KJ, Cruickshank SM. 2014. Heterogeneity across the murine small and large intestine. World J Gastroenterol 20:15216–15232. doi: 10.3748/wjg.v20.i41.15216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dupont A, Heinbockel L, Brandenburg K, Hornef MW. 2014. Antimicrobial peptides and the enteric mucus layer act in concert to protect the intestinal mucosa. Gut Microbes 5:761–765. doi: 10.4161/19490976.2014.972238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clevers HC, Bevins CL. 2013. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol 75:289–311. doi: 10.1146/annurev-physiol-030212-183744. [DOI] [PubMed] [Google Scholar]
- 30.Burger-van Paassen N, Loonen LM, Witte-Bouma J, Korteland-van Male AM, de Bruijn AC, van der Sluis M, Lu P, Van Goudoever JB, Wells JM, Dekker J, Van Seuningen I, Renes IB. 2012. Mucin Muc2 deficiency and weaning influences the expression of the innate defense genes Reg3beta, Reg3gamma and angiogenin-4. PLoS One 7:e38798. doi: 10.1371/journal.pone.0038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balakrishnan A, Polli JE. 2006. Apical sodium dependent bile acid transporter (ASBT, SLC10A2): a potential prodrug target. Mol Pharm 3:223–230. doi: 10.1021/mp060022d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. 2011. The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis 29:37–44. doi: 10.1159/000324126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lou G, Ma X, Fu X, Meng Z, Zhang W, Wang YD, Van Ness C, Yu D, Xu R, Huang W. 2014. GPBAR1/TGR5 mediates bile acid-induced cytokine expression in murine Kupffer cells. PLoS One 9:e93567. doi: 10.1371/journal.pone.0093567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dvorak K, Chavarria M, Payne CM, Ramsey L, Crowley-Weber C, Dvorakova B, Dvorak B, Bernstein H, Holubec H, Sampliner RE, Bernstein C, Prasad A, Green SB, Garewal H. 2007. Activation of the interleukin-6/STAT3 antiapoptotic pathway in esophageal cells by bile acids and low pH: relevance to Barrett's esophagus. Clin Cancer Res 13:5305–5313. doi: 10.1158/1078-0432.CCR-07-0483. [DOI] [PubMed] [Google Scholar]
- 35.Graf D, Kohlmann C, Haselow K, Gehrmann T, Bode JG, Haussinger D. 2006. Bile acids inhibit interleukin-6 signaling via gp130 receptor-dependent and -independent pathways in rat liver. Hepatology 44:1206–1217. doi: 10.1002/hep.21368. [DOI] [PubMed] [Google Scholar]
- 36.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Becker C. 2009. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi SM, McAleer JP, Zheng M, Pociask DA, Kaplan MH, Qin S, Reinhart TA, Kolls JK. 2013. Innate Stat3-mediated induction of the antimicrobial protein Reg3gamma is required for host defense against MRSA pneumonia. J Exp Med 210:551–561. doi: 10.1084/jem.20120260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa-Kawakita M, Laurent-Puig P, Kahn A, Robine S, Perret C, Romagnolo B. 2005. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development 132:1443–1451. doi: 10.1242/dev.01700. [DOI] [PubMed] [Google Scholar]
- 39.van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, Taketo MM, Clevers H. 2005. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol 7:381–386. doi: 10.1038/ncb1240. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Altorki NK, Wu YC, Soslow RA, Subbaramaiah K, Dannenberg AJ. 2001. Duodenal reflux induces cyclooxygenase-2 in the esophageal mucosa of rats: evidence for involvement of bile acids. Gastroenterology 121:1391–1399. doi: 10.1053/gast.2001.29781. [DOI] [PubMed] [Google Scholar]
- 41.de Ledinghen V, Liu H, Zhang F, Lo CR, Subbaramaiah K, Dannenberg AJ, Czaja MJ. 2002. Induction of cyclooxygenase-2 by tumor promoters in transformed and cytochrome P450 2E1-expressing hepatocytes. Carcinogenesis 23:73–79. doi: 10.1093/carcin/23.1.73. [DOI] [PubMed] [Google Scholar]
- 42.Pai R, Tarnawski AS, Tran T. 2004. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell 15:2156–2163. doi: 10.1091/mbc.E03-12-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng JP, Ceryak S, Aratsu Z, Jones L, Epstein L, Bouscarel B. 2006. Biphasic regulation by bile acids of dermal fibroblast proliferation through regulation of cAMP production and COX-2 expression level. Am J Physiol Cell Physiol 291:C546–C554. doi: 10.1152/ajpcell.00011.2006. [DOI] [PubMed] [Google Scholar]
- 44.Lim K, Han C, Xu L, Isse K, Demetris AJ, Wu T. 2008. Cyclooxygenase-2-derived prostaglandin E2 activates beta-catenin in human cholangiocarcinoma cells: evidence for inhibition of these signaling pathways by omega 3 polyunsaturated fatty acids. Cancer Res 68:553–560. doi: 10.1158/0008-5472.CAN-07-2295. [DOI] [PubMed] [Google Scholar]
- 45.Mahmoud NN, Dannenberg AJ, Bilinski RT, Mestre JR, Chadburn A, Churchill M, Martucci C, Bertagnolli MM. 1999. Administration of an unconjugated bile acid increases duodenal tumors in a murine model of familial adenomatous polyposis. Carcinogenesis 20:299–303. doi: 10.1093/carcin/20.2.299. [DOI] [PubMed] [Google Scholar]
- 46.Kabat AM, Srinivasan N, Maloy KJ. 2014. Modulation of immune development and function by intestinal microbiota. Trends Immunol 35:507–517. doi: 10.1016/j.it.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jakobsson HE, Rodriguez-Pineiro AM, Schutte A, Ermund A, Boysen P, Bemark M, Sommer F, Backhed F, Hansson GC, Johansson ME. 2015. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep 16:164–177. doi: 10.15252/embr.201439263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsolis RM, Xavier MN, Santos RL, Baumler AJ. 2011. How to become a top model: impact of animal experimentation on human Salmonella disease research. Infect Immun 79:1806–1814. doi: 10.1128/IAI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 50.Menendez A, Arena ET, Guttman JA, Thorson L, Vallance BA, Vogl W, Finlay BB. 2009. Salmonella infection of gallbladder epithelial cells drives local inflammation and injury in a model of acute typhoid fever. J Infect Dis 200:1703–1713. doi: 10.1086/646608. [DOI] [PubMed] [Google Scholar]
- 51.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. 2009. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183:6251–6261. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 52.Cash HL, Whitham CV, Behrendt CL, Hooper LV. 2006. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. 2011. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141:1773–1781. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 54.Tourret J, Willing BP, Dion S, MacPherson J, Denamur E, Finlay BB. 2017. Immunosuppressive treatment alters secretion of ileal antimicrobial peptides and gut microbiota, and favors subsequent colonization by uropathogenic Escherichia coli. Transplantation 101:74–82. doi: 10.1097/TP.0000000000001492. [DOI] [PubMed] [Google Scholar]
- 55.Thompson JA, Oliveira RA, Djukovic A, Ubeda C, Xavier KB. 2015. Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep 10:1861–1871. doi: 10.1016/j.celrep.2015.02.049. [DOI] [PubMed] [Google Scholar]
- 56.Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, Schreiber JR, Varki NM, Kagnoff MF, Eckmann L. 2004. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun 72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wlodarska M, Willing B, Keeney KM, Menendez A, Bergstrom KS, Gill N, Russell SL, Vallance BA, Finlay BB. 2011. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect Immun 79:1536–1545. doi: 10.1128/IAI.01104-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Ampting MT, Loonen LM, Schonewille AJ, Konings I, Vink C, Iovanna J, Chamaillard M, Dekker J, van der Meer R, Wells JM, Bovee-Oudenhoven IM. 2012. Intestinally secreted C-type lectin Reg3b attenuates salmonellosis but not listeriosis in mice. Infect Immun 80:1115–1120. doi: 10.1128/IAI.06165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loonen LM, Stolte EH, Jaklofsky MT, Meijerink M, Dekker J, van Baarlen P, Wells JM. 2014. REG3gamma-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal Immunol 7:939–947. doi: 10.1038/mi.2013.109. [DOI] [PubMed] [Google Scholar]
- 60.Furci L, Baldan R, Bianchini V, Trovato A, Ossi C, Cichero P, Cirillo DM. 2015. New role for human alpha-defensin 5 in the fight against hypervirulent Clostridium difficile strains. Infect Immun 83:986–995. doi: 10.1128/IAI.02955-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamada N, Sakamoto K, Seo SU, Zeng MY, Kim YG, Cascalho M, Vallance BA, Puente JL, Nunez G. 2015. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe 17:617–627. doi: 10.1016/j.chom.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tam MA, Rydstrom A, Sundquist M, Wick MJ. 2008. Early cellular responses to Salmonella infection: dendritic cells, monocytes, and more. Immunol Rev 225:140–162. doi: 10.1111/j.1600-065X.2008.00679.x. [DOI] [PubMed] [Google Scholar]
- 63.Mastroeni P, Grant A, Restif O, Maskell D. 2009. A dynamic view of the spread and intracellular distribution of Salmonella enterica. Nat Rev Microbiol 7:73–80. doi: 10.1038/nrmicro2034. [DOI] [PubMed] [Google Scholar]
- 64.Tanabe H, Ayabe T, Bainbridge B, Guina T, Ernst RK, Darveau RP, Miller SI, Ouellette AJ. 2005. Mouse Paneth cell secretory responses to cell surface glycolipids of virulent and attenuated pathogenic bacteria. Infect Immun 73:2312–2320. doi: 10.1128/IAI.73.4.2312-2320.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 66.Mastroianni JR, Costales JK, Zaksheske J, Selsted ME, Salzman NH, Ouellette AJ. 2012. Alternative luminal activation mechanisms for Paneth cell alpha-defensins. J Biol Chem 287:11205–11212. doi: 10.1074/jbc.M111.333559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mastroianni JR, Ouellette AJ. 2009. Alpha-defensins in enteric innate immunity: functional Paneth cell alpha-defensins in mouse colonic lumen. J Biol Chem 284:27848–27856. doi: 10.1074/jbc.M109.050773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Breton J, Tennoune N, Lucas N, Francois M, Legrand R, Jacquemot J, Goichon A, Guerin C, Peltier J, Pestel-Caron M, Chan P, Vaudry D, do Rego JC, Lienard F, Penicaud L, Fioramonti X, Ebenezer IS, Hokfelt T, Dechelotte P, Fetissov SO. 2016. Gut commensal E. coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metab 23:324–334. doi: 10.1016/j.cmet.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 69.Suh E, Traber PG. 1996. An intestine-specific homeobox gene regulates proliferation and differentiation. Mol Cell Biol 16:619–625. doi: 10.1128/MCB.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lussier CR, Babeu JP, Auclair BA, Perreault N, Boudreau F. 2008. Hepatocyte nuclear factor-4alpha promotes differentiation of intestinal epithelial cells in a coculture system. Am J Physiol Gastrointest Liver Physiol 294:G418–G428. doi: 10.1152/ajpgi.00418.2007. [DOI] [PubMed] [Google Scholar]
- 71.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.