

Abstract
Background
Watercress (Nasturtium officinale R. Br.) is an aquatic herb species that is a rich source of secondary metabolites such as glucosinolates. Among these glucosinolates, watercress contains high amounts of gluconasturtiin (2-phenethyl glucosinolate) and its hydrolysis product, 2-phennethyl isothiocyanate, which plays a role in suppressing tumor growth. However, the use of N. officinale as a source of herbal medicines is currently limited due to insufficient genomic and physiological information.
Results
To acquire precise information on glucosinolate biosynthesis in N. officinale, we performed a comprehensive analysis of the transcriptome and metabolome of different organs of N. officinale. Transcriptome analysis of N. officinale seedlings yielded 69,570,892 raw reads. These reads were assembled into 69,635 transcripts, 64,876 of which were annotated to transcripts in public databases. On the basis of the functional annotation of N. officinale, we identified 33 candidate genes encoding enzymes related to glucosinolate biosynthetic pathways and analyzed the expression of these genes in the leaves, stems, roots, flowers, and seeds of N. officinale. The expression of NoMYB28 and NoMYB29, the main regulators of aliphatic glucosinolate biosynthesis, was highest in the stems, whereas the key regulators of indolic glucosinolate biosynthesis, such as NoDof1.1, NoMYB34, NoMYB51, and NoMYB122, were strongly expressed in the roots. Most glucosinolate biosynthetic genes were highly expressed in the flowers. HPLC analysis enabled us to detect eight glucosinolates in the different organs of N. officinale. Among these glucosinolates, the level of gluconasturtiin was considerably higher than any other glucosinolate in individual organs, and the amount of total glucosinolates was highest in the flower.
Conclusions
This study has enhanced our understanding of functional genomics of N. officinale, including the glucosinolate biosynthetic pathways of this plant. Ultimately, our data will be helpful for further research on watercress bio-engineering and better strategies for exploiting its anti-carcinogenic properties.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3792-5) contains supplementary material, which is available to authorized users.
Keywords: Nasturtium officinale, Watercress, Transcriptome, Glucosinolates
Background
Nasturtium officinale R. Br. is an aquatic perennial herb that generally grows in around clear, cold water. It is primarily found in Europe, North and South America, and Asia, where it is commonly known as “watercress.” In some regions, N. officinale is considered an aquatic weed and is consumed as a fresh salad plant or soup garnish, or used in other recipes [1, 2]. It is well documented that N. officinale is recognized as a valuable traditional medicinal plant, because of its numerous health-benefiting constituents, such as vitamins B, C, and E, pro-vitamin A, folic acid, carotenoids, glucosinolates, and many minerals, including Ca, Fe, I, and S [3, 4]. In particular, watercress contains high amounts of gluconasturtiin (2-phenethyl glucosinolate), which is hydrolyzed by myrosinase to produce 2-phennethyl isothiocyanate [5, 6]. This latter metabolite has been demonstrated to suppress carcinogen activation through the inhibition of phase I enzymes and induction of phase II enzymes [7]. Recent study has shown that watercress accessions from the University of South-ampton germplasm collection contain various gluconasturtiin contents and antioxidant (AO) capacity [8]. In addition, N. officinale is now known to play a role in the prevention of several other diseases including diabetes, inflammatory diseases [9], and lymphocyte DNA damage [10].
Glucosinolates are sulfur-rich anionic secondary metabolites derived from glucose and amino acids. Approximately 200 different glucosinolates are known to occur naturally in plants [11, 12] and are found almost exclusively within the order Brassicales. These compounds play roles in defense against pests and have various biological activities related to human health [13–17]. Glucosinolates can be classified into three main groups, depending on the content of modified amino acids: aliphatic glucosinolates, derived from methionine, isoleucine, leucine, or valine; aromatic glucosinolates, derived from tyrosine or phenylalanine; and indole glucosinolates, derived from tryptophan [18]. Biosynthesis occurs in three independent phases: (i) side chain elongation of precursor amino acids with an additional methylene group, (ii) partial amino acid conversion to form the core structure, and (iii) secondary modification of the amino acid side chain [19]. Several glucosinolate biosynthetic genes are generally involved in these three independent phases of glucosinolate biosynthesis. Elongation of methionine is initiated by METHYLTHIOALKYLMALATE SYNTHASE (MAM), BILE ACID TRANSPORTER5 (BAT5), and BRANCHED-CHAIN AMINOTRANSFERASE (BCAT) [20–23]. Core structure formation of glucosinolates is accomplished in five steps via oxidation by cytochrome P450 of CYP79 and CYP83, followed by C-S lyase, S-glucosyltransferase, and sulfotransferase [24–26]. Finally, secondary modification is mediated by several genes, including GS-OX, GS-AOP, GS-OH, BZO1, and CYP81F2 [19]. Furthermore, various transcription factors are implicated in the regulation of glucosinolate biosynthesis. HIGH ALIPHATIC GLUCOSINOLATE1 (HAG1)/MYB28, HAG2/MYB76, and HAG3/MYB29 are the main regulators of aliphatic glucosinolate biosynthesis [27, 28], whereas HIGH INDOLIC GLUCOSINOLATE1 (HIG1)/MYB51, HIG2/MYB122, and ALTERED TRYPTOPHAN REGULATION1 (ATR1)/MYB34 regulate indolic glucosinolate biosynthesis. Among these regulators, AtMYB34 and AtMYB51 play major roles in indolic glucosinolate biosynthesis and AtMYB122 is presumed to play an accessory role in indolic glucosinolate biosynthesis [29]. IQD1, a nuclear-localized calmodulin-binding protein, controls the biosynthesis of aliphatic and indolic glucosinolates [30]. AtDof1.1 induces the transcription of CYP83B1 and increases the levels of aliphatic and indolic glucosinolates [31].
Whole transcriptome sequencing technologies have been widely utilized as powerful tools for high-throughput genotyping because they are inexpensive, rapid, accurate, and reproducible [32, 33]. Among next-generation sequencing (NGS) technologies, the Illumina sequencing platform [34] has been successfully used for de novo transcriptome sequencing of numerous species, such as rice (Oryza sativa) [35], maize (Zea mays) [36], soybean (Glycine max) [37], sweet potato (Ipomoea batatas) [38], barley (Hordeum vulgare) [39], chickpea (Cicer arietinum) [40], tea plant (Camellia sinensis) [41], and Chinese bayberry (Myrica rubra) [42].
In this study, we used an Illumina NextSeq500 sequencer to analyze the transcriptome of N. officinale seedlings and generated 69,570,892 raw reads that were assembled into 69,635 transcripts. The N. officinale transcriptome showed highest species similarity and annotation ratio to Arabidopsis thaliana. From the transcriptome data, we identified several candidate genes that encode enzymes related to glucosinolate biosynthetic pathways. To validate the spatial distribution of glucosinolate-related genes, we analyzed the expression of glucosinolate biosynthesis genes and transcription factors in different organs of N. officinale using quantitative real-time RT-PCR. Metabolite profiling using HPLC-UV analysis identified eight different glucosinolates in the different organs of N. officinale, and the total glucosinolate contents were found to be highest in flowers. Among the eight identified glucosinolates, the level of gluconasturtiin was considerably higher than that of any other glucosinolate, irrespective of the organ. Taken together, the data obtained from this comprehensive transcriptomic and metabolomic profiling will provide an invaluable resource for a better understanding of glucosinolate biosynthetic pathways, as well as strategies for exploiting the anti-carcinogenic properties in N. officinal.
Methods
Plant material and growth conditions
Nasturtium officinale seeds were purchased from Asia Seeds Co., Ltd (Seoul, Korea) and grown under field conditions at the experimental greenhouse of Chungnam National University (Daejeon, Korea). Different organs were harvested from mature plants at approximately 2 months after sowing. The samples were immediately frozen in liquid nitrogen and then stored at -80 °C for RNA isolation or freeze-dried for subsequent analysis by high performance liquid chromatography (HPLC).
Illumina sequencing of the transcriptome
Total RNA was isolated from frozen seedlings of N. officinale using the RNeasy Mini Kit (Qiagen, USA) and cleaned by ethanol precipitation. We removed rRNAs in total RNA using the ribo-zero rRNA removal kit (Epicentre, RZPL11016) and constructed a cDNA library for RNA sequencing using the TruSeq stranded total RNA sample prep kit-LT set A and B (Illumina, RS-122-2301 and 2302) according to the manufacturer’s protocols (Illumina, San Diego, CA, USA). The cDNA library was sequenced in 76 bp length paired-end (PE) reads in an Illumina NextSeq500 sequencer (Illumina Inc., San Diego, CA, USA) to produce 69,570,892 raw sequencing reads.
De novo assembly and annotation of the watercress transcriptome
The quality-trimmed reads of watercress RNAs were assembled as contigs of the watercress transcriptome using the Trinity software package (http://trinityrnaseq.github.io/) [43]. The Trinity program combines the overlapping reads of a given length and quality into longer contig sequences without gaps. The characteristic properties, including N50 length, average length, maximum length, and minimum length of the assembled contigs were calculated using Transrate software (http://hibberdlab.com/transrate) [44]. We clustered the watercress transcriptome contigs based on sequence similarity using CD-HIT-EST software (http://weizhongli-lab.org/cd-hit) [45]. To infer the biological functions of watercress transcripts, we performed a homology search of the transcripts in the various public protein and nucleotide databases. A BLASTX search was performed using the National Center for Biotechnology Information (NCBI) (http://blast.ncbi.nlm.nih.gov) nr and Clusters of Orthologous Group (COG) (http://www.ncbi.nlm.nih.gov/COG) protein databases, BRAD (http://brassicadb.org/brad) Brassica rapa protein database, TAIR (TAIR10, http://www.arabidopsis.org) Arabidopsis thaliana protein database, and the EBI Swiss-Prot (UniProt) database. A BLASTN search was performed using the NCBI nucleotide database. The best scored hit from the BLASTX and BLASTN results passed the cutoff of e-value < 10−5 and was selected for annotation of query transcripts for each database search. Transcript lists and sequences are presented in Additional files 1 and 2. The functional category distributions of watercress transcripts in terms of Gene Ontology (GO) and COG were evaluated using the results of the homology search. COG functional category information attached to the hit COG proteins was used for determining COG functional category distribution, and GO information attached to the hit UniProt proteins was collected and re-analyzed using the WEGO tool (http://wego.genomics.org.cn) [46] in terms of the level for the three GO categories.
Differentially expressed gene analysis
To quantify watercress transcript expressions, we aligned preprocessed quality-trimmed reads on the watercress transcript sequences and calculated the expression values with the aligned read counts for each transcript. Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2) software [47] was used to align the quality-trimmed reads on the transcript sequences, and eXpress (http://bio.math.berkeley.edu/eXpress) software [48] was used to evaluate gene expression, in terms of fragments per kilobase of exon per million mapped fragments (FPKM), from the aligned results. The FPKM method provides a comparison between genes within a sample or between samples by normalizing the amount of sequencing for samples and gene length bias during gene expression evaluation.
Identification of candidate genes related to glucosinolate biosynthetic pathways
We searched for candidate genes involved in glucosinolate biosynthetic pathways using functional annotation data based on the orthologous gene names. In addition, the glucosinolate biosynthetic genes of Arabidopsis obtained from TAIR were used as queries to search for homologous sequences in the watercress transcriptome database. Following this, each sequence was confirmed by the BLAST program in the NCBI GenBank database.
Quantitative real-time RT-PCR
For quantitative real-time RT-PCR, gene-specific primer sets were designed for each gene using the Primer3 website (http://frodo.wi.mit.edu/primer3/). Real-time RT-PCR was performed in a CFX96 real time system (BIO-RAD Laboratories, USA) using 2x Real-Time PCR Smart mix (BioFACT, Korea) under the following conditions: 95 °C for 15 min, followed by 40 cycles of 95 °C for 15 s, annealing for 15 s at 56 °C, and elongation for 20 s at 72 °C. PCR products were analyzed using Bio-Rad CFX Manager 2.0 software. Gene expression was normalized to that of the UBC9 gene, used as a housekeeping gene. The results of the real-time RT-PCR assay were calculated as the mean of three different biological experiments using seeds and different plant organs (all leaves, stems, roots, and flowers) of individual plants. Real-time RT-PCR product sizes and primer sequences are shown in Additional file 3: Table S1.
Extraction and HPLC analysis of glucosinolates
HPLC-UV analysis of glucosinolates was performed according to previously described methods with some modification [49, 50]. Glucosinolates were extracted with 70% (v/v) methanol from 100 mg lyophilized powder in a water bath at 70 °C for 5 min. After centrifugation at 12,000 × g for 10 min, the supernatant was loaded onto a mini-column packed with DEAE-Sephadex A-25 (Sigma-Aldrich Co., Ltd., St. Louis, MO, USA). After an overnight reaction at ambient temperature, the desulfo-glucosinolates were eluted with 1.5 mL of high-purity water and filtered through a 0.45 μm hydrophilic PTFE syringe filter (Ø, 13 mm; Advantec, Tokyo, Japan) in a vial. Desulfo-glucosinolates were quantified using a 1260 series HPLC system (Agilent Technologies, CA, USA) equipped with an Inertsil ODS-3 (C18) column 150 × 3.0 mm i.d., particle size 3 μm (GL Science, Tokyo, Japan). HPLC analysis was performed with a flow rate of 0.4 mL min−1 at a column temperature of 40 °C and a wavelength of 227 nm. The solvent systems employed were (A) water and (B) 100% acetonitrile. The gradient program used was as follows: 0 min, 0% solvent B; 0–2 min, 0% solvent B; 2–7 min, 10% solvent B; 7–16 min, 31% solvent B; 16–19 min, 31% solvent B; 19–21 min, 0% solvent B; 21–27 min, 0% solvent B (total 27 min). The individual glucosinolates were determined by their HPLC peak area ratios and response factors (ISO 9167-1, 1992) with reference to a desulfo-sinigrin external standard.
Results
Sequencing and de novo assembly of the N. officinale transcriptome
As shown in Fig. 1, watercress can grow to a height of 50 to 120 cm and has slender hollow stems and small round leaves (Fig. 1a). Tiny white flowers are formed in clusters and become small pods containing two rows of seeds (Fig. 1b). To provide an overview of the N. officinale transcriptome, we performed RNA-sequencing analysis of N. officinale seedlings using the Illumina NextSeq500 platform (Fig. 1c). After removal of adaptor sequences, 69,570,892 reads comprising a total of 5,287,387,792 nucleotides were obtained for assembly (Table 1). These reads were assembled using the Trinity program, resulting in 123,433 contigs with an average length of 724 nt and an N50 of 994 nt. After clustering with CD-Hit-EST, the contigs were assembled into 69,635 transcripts with a mean size of 681 nt and N50 of 930 nt. The size distribution of the transcripts exhibited the following pattern: 25.51% (17,770) of the transcripts were less than 300 nt, 55.38% (38,564) of the transcripts ranged from 300 to 1000 nt in length, 18.09% (12,603) of the transcripts ranged from 1000 to 3000 nt, and 1.0% (698) were more than 3000 nt in length (Additional file 4: Figure S1).
Fig. 1.

Photographs of mature plant (a), inflorescence (b), and seedling (c) of N. officinale
Table 1.
Summary of the transcriptome of N. officinale
| Raw reads | Contigs | Transcripts | |
|---|---|---|---|
| Total length (bp) | 5,287,387,792 | 89,449,846 | 47,428,745 |
| Number of sequences | 69,570,892 | 123,433 | 69,635 |
| Average length (bp) | 76 | 724 | 681 |
| Median length (bp) | 76 | 501 | 453 |
| Max length (bp) | 76 | 16,627 | 16,627 |
| Min length (bp) | 76 | 224 | 224 |
| N50 (bp) | 76 | 994 | 930 |
Functional annotation and classification of N. officinale transcripts
For functional annotation, the transcripts were identified based on the BLASTX algorithm (available at the NCBI website) against the non-redundant (NR) protein database and nucleotide (NT) database with an E-value cutoff of 1 x 10−5 (Table 2). Of the total 69,635 transcripts, 57,550 transcripts (82.65%) had BLAST hits to known proteins in the NR database and 61,020 transcripts (87.63%) had BLAST hits to nucleotides in the NT database. In addition, some transcripts were aligned to public databases, including 46,249 (66.42%) transcripts in the SWISS-PROT protein database, 60,335 (86.64%) transcripts in the Brassica database (BRAD), 61,369 (88.13%) transcripts in the Arabidopsis information resource (TAIR) database, 16,530 (23.74%) transcripts in the Clusters of Orthologous Group (COG) database, and 45,402 (65.20%) transcripts in Gene Ontology (GO) database. In total, 64,876 transcripts were identified, representing approximately 93.17% of all assembled transcripts.
Table 2.
Summary of annotations of the N. officinale transcripts
| Number of BLASTed transcripts | Ratio (%) | |
|---|---|---|
| All transcripts | 69,635 | 100 |
| Transcripts BLASTed against NR | 57,550 | 82.65 |
| Transcripts BLASTed against NT | 61,020 | 87.63 |
| Transcripts BLASTed against SWISS-PROT | 46,249 | 66.42 |
| Transcripts BLASTed against BRAD | 60,335 | 86.64 |
| Transcripts BLASTed against TAIR | 61,369 | 88.13 |
| Transcripts BLASTed against COG | 16,530 | 23.74 |
| Transcripts BLASTed against GO | 45,402 | 65.20 |
| All annotated transcripts | 64,876 | 93.17 |
NR NCBI non-redundant protein database, NT NCBI nucleotide database, SWISS-PROT SwissProt protein database, BRAD Brassica rapa protein database, TAIR Arabidopsis protein database, COG Clusters of Orthologous Group, GO Gene Ontology
The E-value distribution of the transcripts in the NR databases showed that 54.6% of aligned transcripts had strong similarity with an E-value <1e-60, whereas the remaining 45.4% of the homologous sequences ranged from 1e-5 to 1e-60 (Fig. 2a). The similarity distribution in the NR database showed that 81% of the sequences had a similarity higher than 80.7 and 19.3% of the sequences had a similarity lower than 80% (Fig. 2b). In the species distribution, the N. officinale transcriptome showed 20.5% similarity with that of Arabidopsis thaliana, with lower similarities to other species, including Camelina sativa (19.4%), Arabidopsis lyrata (18.6%), Eutrema salsugineum (12.9%), Capsella rubella (12.8%), Brassica napus (5.7%), Arabis alpina (4.1%), Brassica rapa (2.4%), and others (3.6%) (Fig. 2c). Most BLAST hits (approximately 96.4%) were to sequences from the Brassicaceae family. The N. officinale transcriptome showed highest species similarity and annotation ratio to A. thaliana, which is an important plant model species. Arabidopsis is a member of Brassicaceae family such as N. officinale and contains 25,498 genes encoding proteins from 11,000 families [51]. Arabidopsis and N. officinale have similar morphology and significant sequence homology, indicating the correlation between mouse-ear cress and watercress.
Fig. 2.
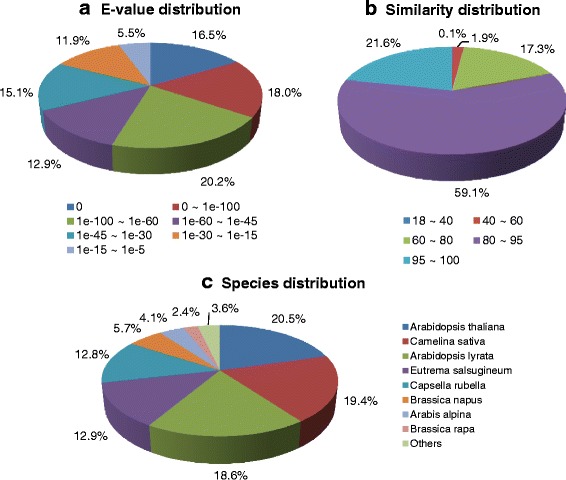
Classification of NR annotation results of the N. officinale transcripts. a E-value distribution, b Similarity distribution, and c Species distribution
COG analysis showed that 16,530 of the total transcripts were classified into 26 molecular families (Fig. 3). Among these categories, the largest category was “translation, ribosomal structure and biogenesis” containing 1850 transcripts (11.19%), followed by “carbohydrate transport and metabolism” (1586, 9.59%), “signal transduction mechanisms” (1573, 9.51%), “post-translational modification, protein turnover, chaperones” (1494, 9.03%), and “general functional prediction only” (1437, 8.69%). “Extracellular structures” (7, 0.04%) was the smallest category, and 489 transcripts were found in the clusters of the “secondary metabolite biosynthesis, transport and catabolism” category. GO analysis revealed that 45,402 of the total assembled transcripts were distributed in 56 sub-categories under three main GO functional categories: cellular components (143,456, 34.35%), molecular function (63,191, 15.13%), and biological process (210,884, 50.50%) (Fig. 4). In the three main categories, the dominant groups of sub-categories were “cellular process” (37,328, 82.2%) and “metabolic process” (35,372, 77.9%) in the biological processes, “cell” (39,487, 87%) and “cell part” (39,436, 86.9%) in cellular components, and “binding” (29,335, 64.6%) and “catalytic” (22,882, 50.4%) in molecular functions.
Fig. 3.
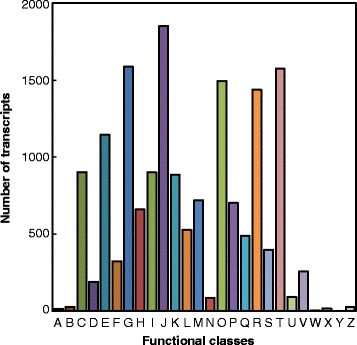
COG functional classification of the N. officinale transcripts. A total of 16,530 transcripts (23.74% of total) were annotated and divided into 26 subcategories. a RNA processing and modification; b Chromatin Structure and dynamics; c Energy production and conversion; d Cell cycle control, cell division, chromosome partitioning; e Amino acid transport and metabolism; f Nucleotide transport and metabolism; g Carbohydrate transport and metabolism; h Coenzyme transport and metabolism; i Lipid transport and metabolism; j Translation, ribosomal structure and biogenesis; k Transcription; l Replication, recombination and repair; m Cell wall/membrane/envelope biogenesis; n Cell motility; o Post-translational modification, protein turnover, chaperones; p Inorganic ion transport and metabolism; q Secondary metabolite biosynthesis, transport and catabolism; r General functional prediction only; s Function unknown; t Signal transduction mechanisms; u Intracellular trafficking, secretion, and vesicular transport; v Defense mechanisms; w Extracellular structures; x Phage-derived proteins, transposases and other mobilized components; y Nuclear structure; z Cytoskeleton
Fig. 4.
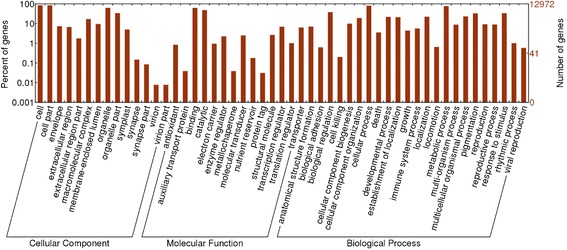
GO annotation of the N. officinale transcripts. A total of 45,402 transcripts (65.20% of total) were annotated and classified into three major categories (biological process, cellular component, and molecular function) and 60 subcategories
Expression analysis of glucosinolate-related genes in different organs of N. officinale
Brassica rapa has 102 putative glucosinolate genes, which are orthologs of 52 glucosinolate genes in A. thaliana. The homologous glucosinolate genes in B. rapa and A. thaliana share 59%-91% nucleotide sequence identity [52]. To identify the expression of genes that encode enzymes related to the glucosinolate biosynthetic pathways, we analyzed the N. officinale transcriptome dataset. On the basis of the functional annotation of the N. officinale transcriptome, we found that seven glucosinolate transcription factors and 26 glucosinolate biosynthetic genes were highly similar to those of species belonging to the Brassicaceae such as A. thaliana, B. oleracea, and B. rapa (Table 3). NoMYB28 was shown to have 83% similarity with A. thaliana MYB28 (NP_200950.1), 77% similarity with B. rapa MYB28 (ADQ92843.1), and 74% similarity with B. oleracea MYB28 (CBI71385.1). Similarly, other N. officinale glucosinolate biosynthetic genes showed more than 67% similarity with other orthologous genes. By comparing other orthologous genes, we identified several full-length cDNA clones encoding MYB28, MYB29, BCAT4, MAM1, CYP79F1, CYP83A1, GGP1, SUR1, UGT74B1, ST5b, ST5a, ST5c, FMO GS-OX5, CYP79B2, GSTF9, and IGMT, and partial-length cDNA clones encoding MYB34, MYB51, MYB122, IQD1-1, Dof1.1, MAM3, GSTF11, GSTF20, UGT74C1, FMO GS-OX2, CYP79B3, CYP83B1, GSTF10, CYP81F2, CYP81F3, PEN2, and TGG2.
Table 3.
Comparison of glucosinolate-related genes of N.officinale with the most orthologous genes
| Genes | Length (amino acid) | Sequence form | Orthologous genes (Accession no.) | Identity (%) |
|---|---|---|---|---|
| NoMYB28 | 370 | Full-length | Arabidopsis thaliana MYB28 (NP_200950.1) | 83 |
| Brassica rapa MYB28 (ADQ92843.1) | 77 | |||
| Brassica oleracea MYB28 (CBI71385.1) | 74 | |||
| NoMYB29 | 350 | Full-length | Arabidopsis thaliana MYB29 (NP_196386.13) | 78 |
| Brassica juncea MYB29-2 (AFY09821.1) | 75 | |||
| Brassica oleracea MYB29 (AKD49017.1) | 71 | |||
| NoMYB34 | 53 | Partial-length | Brassica oleracea MYB34 (BAM78216.1) | 98 |
| Brassica rapa MYB34-3 (ADV17461.1) | 98 | |||
| Arabidopsis thaliana MYB34 (NP_200897.1) | 96 | |||
| NoMYB51 | 122 | Partial-length | Eruca vesicaria MYB51 (AGS49160.1) | 81 |
| Arabidopsis thaliana MYB51 (NP_173292.1) | 90 | |||
| Brassica rapa MYB51-1 (ACR48187.1) | 80 | |||
| NoMYB122 | 35 | Partial-length | Arabidopsis thaliana MYB122 (NP_177548.1) | 94 |
| Arabidopsis lyrata MYB122 (XP_002887524.1) | 94 | |||
| Brassica rapa MYB122 (XP_009106064.1) | 91 | |||
| NoIQD1-1 | 81 | Partial-length | Arabidopsis thaliana IQD1 (NP_187582.1) | 86 |
| Camelina sativa IQD1 (XP_010464624.1) | 81 | |||
| Brassica rapa IQD1 (XP_009123236.1) | 76 | |||
| NoDof1.1 | 256 | Partial-length | Arabidopsis thaliana Dof 1.1(NP_850938.1) | 78 |
| Camelina sativa Dof1.1 (XP_010487779.1) | 79 | |||
| Brassica rapa Dof1.1 (XP_009110928.1) | 68 | |||
| NoBCAT4 | 352 | Full-length | Brassica oleracea BCAT4 (AJF21970.1) | 83 |
| Brassica rapa BCAT4 (ACR10245.1) | 82 | |||
| Arabidopsis lyrata BCAT4 (XP_002885325.1) | 79 | |||
| NoMAM1 | 500 | Full-length | Arabidopsis thaliana MAM1 (NP_197692.1) | 81 |
| Camelina sativa MAM1 (XP_010454580.1) | 80 | |||
| Brassica rapa MAM1 (XP_009130133.1) | 75 | |||
| NoMAM3 | 302 | Partial-length | Camelina sativa MAM3 (XP_010428949.1) | 82 |
| Boechera divaricarpa MAM3 (CAJ55514.1) | 82 | |||
| Arabidopsis thaliana MAM3 (NP_197693.1) | 81 | |||
| NoCYP79F1 | 539 | Full-length | Brassica oleracea CYP79F1 (ACB59213.1) | 84 |
| Brassica napus CYP79F1 (AGO59948.1) | 83 | |||
| Brassica rapa CYP79F1 (ACR10252.1) | 83 | |||
| NoCYP83A1 | 502 | Full-length | Arabidopsis thaliana CYP83A1 (NP_193113.1) | 88 |
| Raphanus sativus CYP83A1 (AHB11194.1) | 88 | |||
| Brassica oleracea CYP83A1 (AIK28472.1) | 87 | |||
| NoGSTF11 | 214 | Partial-length | Arabidopsis lyrata GSTF11 (XP_002882279.1) | 91 |
| Camelina sativa GSTF11 (XP_010463766.1) | 91 | |||
| Arabidopsis thaliana GSTF11 (NP_186969.1) | 88 | |||
| NoGSTF20 | 94 | Partial-length | Arabidopsis thaliana GSTF20 (NP_177958.1) | 91 |
| Camelina sativa GSTF20 (XP_010472052.1) | 91 | |||
| Brassica oleracea GSTF20 (XP_013592747.1) | 88 | |||
| NoGGP1 | 250 | Full-length | Brassica rapa GGP1 (XP_009108982.1) | 87 |
| Brassica oleracea GGP1 (XP_013598031.1) | 88 | |||
| Camelina sativa GGP1 (XP_010438166.1) | 87 | |||
| NoSUR1 | 376 | Full-length | Arabidopsis thaliana SUR1 (NP_179650.1) | 90 |
| Eruca vesicaria SUR1 (AGS49169.1) | 89 | |||
| Brassica rapa SUR1 (ACH41755.1) | 89 | |||
| NoUGT74B1 | 459 | Full-length | Arabidopsis thaliana UGT74B1 (XP_010477734) | 88 |
| Camelina sativa UGT74B1 (XP_010477734) | 85 | |||
| Brassica rapa UGT74B1 (XP_009115475.1) | 84 | |||
| NoUGT74C1 | 237 | Partial-length | Arabidopsis thaliana UGT74C1 (NP_180738.1) | 89 |
| Camelina sativa UGT74C1 (XP_010469678.1) | 89 | |||
| Brassica oleracea UGT74C1 (XP_013637126.1) | 87 | |||
| NoST5b | 341 | Full-length | Arabidopsis thaliana ST5b (NP_177549.1) | 84 |
| Camelina sativa ST5b (XP_010416243.1) | 80 | |||
| Brassica rapa ST5b (XP_009106065.1) | 80 | |||
| NoST5a | 337 | Full-length | Arabidopsis thaliana ST5a (NP_177550.1) | 96 |
| Brassica rapa ST5a (ACR10265.1) | 91 | |||
| Camelina sativa ST5a (XP_010419185.1) | 96 | |||
| NoST5c | 350 | Full-length | Arabidopsis thaliana ST5c (NP_173294.1) | 89 |
| Camelina sativa ST5c (XP_010459484.1) | 87 | |||
| Brassica rapa ST5c (ACR10273.1) | 85 | |||
|
NoFMO
GS-OX2 |
122 | Partial-length | Camelina sativa FMO GS-OX2 (XP_010473477.1) | 85 |
| Brassica oleracea FMO GS-OX2 (XP_013612296.1) | 80 | |||
| Brassica rapa FMO GS-OX2 (XP_009113068.1) | 79 | |||
|
NoFMO
GS-OX5 |
456 | Full-length | Arabidopsis thaliana FMO GS-OX5 (NP_172678.3) | 86 |
| Brassica oleracea FMO GS-OX5 (FMO GS-OX5) | 86 | |||
| Brassica rapa FMO GS-OX5 (XP_009110664.1) | 85 | |||
| NoCYP79B2 | 541 | Full-length | Arabidopsis lyrata CYP79B2 (XP_002866896.1) | 94 |
| Brassica oleracea CYP79B2 (ADW54459.1) | 94 | |||
| Eruca vesicaria CYP79B2 (AGM16417.1) | 93 | |||
| NoCYP79B3 | 144 | Partial-length | Brassica rapa CYP79B3 (ACR10255.1) | 91 |
| Arabidopsis lyrata CYP79B3 (XP_002878610) | 90 | |||
| Brassica napus CYP79B3 (AAN76810.1) | 73 | |||
| NoCYP83B1 | 448 | Partial-length | Brassica oleracea CYP83B1 (ADW54460.1) | 96 |
| Arabidopsis thaliana CYP83B1 (NP_194878.1) | 95 | |||
| Raphanus sativus CYP83B1 (AHB11193.1) | 96 | |||
| NoGSTF9 | 213 | Full-length | Brassica rapa GSTF9 (XP_009132756.1) | 97 |
| Brassica oleracea GSTF9 (XP_013636508.1) | 97 | |||
| Arabidopsis thaliana GSTF9 (NP_180643.1) | 96 | |||
| NoGSTF10 | 48 | Partial-length | Brassica rapa GSTF10 (XP_009132757.1) | 90 |
| Brassica oleracea GSTF10 (XP_013622385.1) | 90 | |||
| Arabidopsis thaliana GSTF10 (NP_180644.1) | 88 | |||
| NoCYP81F2 | 170 | Partial-length | Arabidopsis thaliana CYP81F2 (NP_200532.1) | 89 |
| Arabidopsis lyrata CYP81F2 (XP_002864506.1) | 89 | |||
| Arabis alpina CYP81F2 (AEM44335.1) | 81 | |||
| NoCYP81F3 | 231 | Partial-length | Arabidopsis thaliana CYP81F3 (NP_568025.1) | 93 |
| Arabidopsis lyrata CYP81F3 (XP_002868991.1) | 93 | |||
| Brassica napus CYP81F3 (CDY44041.1) | 92 | |||
| NoIGMT | 373 | Full-length | Arabidopsis thaliana IGMT (NP_173534.1) | 91 |
| Camelina sativa IGMT (XP_010498564.1) | 90 | |||
| Brassica rapa IGMT (XP_009149591.1) | 91 | |||
| NoPEN2 | 405 | Partial-length | Arabidopsis thaliana PEN2 (NP_181977.1) | 90 |
| Arabis alpina PEN2 (AEM44334.1) | 90 | |||
| Brassica rapa PEN2 (XP_009143040.1) | 87 | |||
| NoTGG2 | 532 | Partial-length | Armoracia rusticana TGG2 (AAV71147.1) | 90 |
| Arabidopsis thaliana TGG2 (BAE98479.1) | 70 | |||
| Tarenaya hassleriana TGG2 (XP_010519862.1) | 67 |
The expression of glucosinolate-related transcription factors was analyzed in the leaves, stems, roots, flowers, and seeds of N. officinale by quantitative RT-PCR (Fig. 5). The expression of NoMYB28 and NoMYB29 was highest in the stems, which is consistent with the transcript levels of BrMYB28, BrMYB29-2, and BrMYB29-3 in the stems of B. rapa [53]. NoMYB34, NoMYB51, NoMYB122, and NoDof1.1 were more strongly expressed in the roots compared with other organs. Finally, the highest expression of NoIQD1-1, which is involved in both aliphatic and indolic glucosinolate biosynthesis, was observed in leaves. Most glucosinolate biosynthetic genes were more highly expressed in the flowers compared with the leaves, stems, roots, and seeds. However, NoMAM1, NoMAM3, NoCYP83A1, NoGSTU20, NoST5c, and NoFMO GS-OX2, which are involved in aliphatic glucosinolate biosynthesis, had the highest expression levels in stems, roots, leaves, seeds, roots, and leaves, respectively (Fig. 6). In addition, among the indolic glucosinolate biosynthetic genes, the highest expression levels of NoCYP79B3, NoGSTF10, NoCYP81F3, and NoPEN2 were observed in roots (Fig. 7).
Fig. 5.
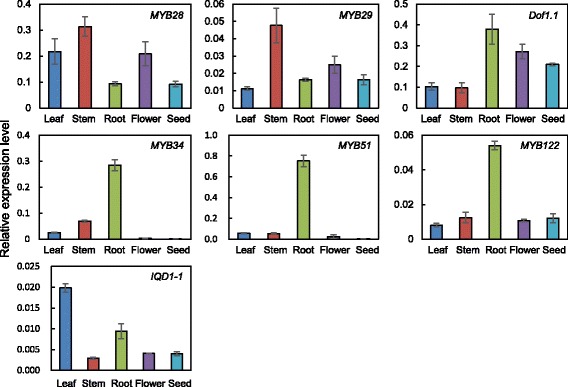
Expression of glucosinolate transcription factors in different organs of N. officinale. The expression level was measured in 2-month-old N. officinale. Relative expression level was plotted after normalization to UBC9. Mean values and SDs from triplicate biological experiments are plotted
Fig. 6.
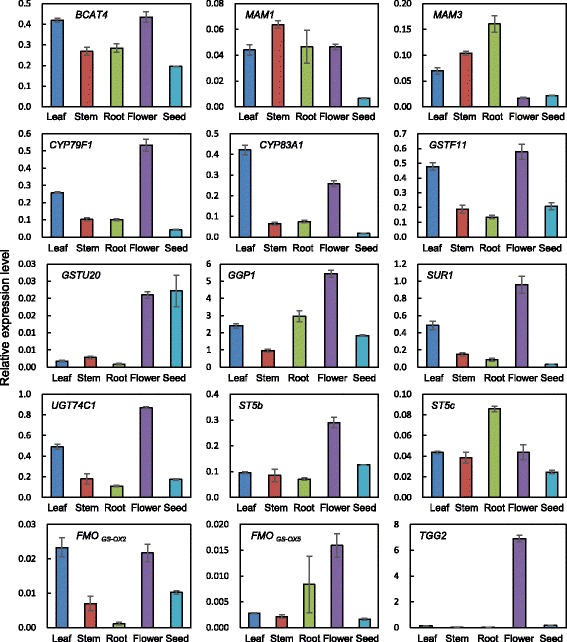
Expression of aliphatic glucosinolate biosynthetic genes in different organs of N. officinale. The expression level was measured in 2-month-old N. officinale. Relative expression level was plotted after normalization to UBC9. Mean values and SDs from triplicate biological experiments are plotted
Fig. 7.
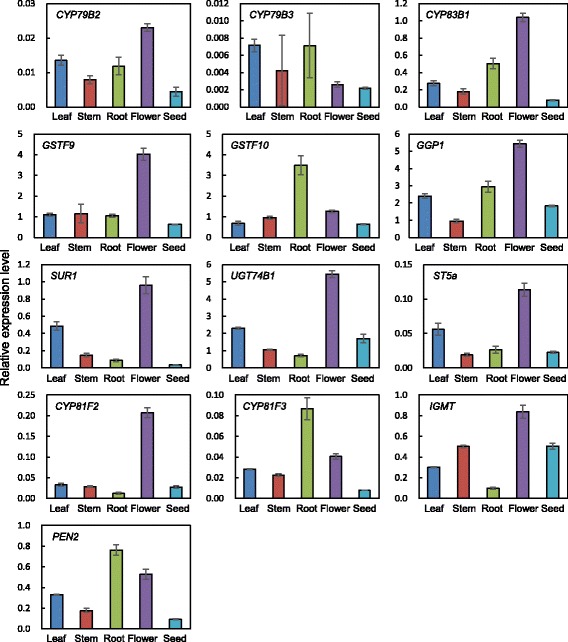
Expression of indolic glucosinolate biosynthetic genes in different organs of N. officinale. The expression level was measured in 2-month-old N. officinale. Relative expression level was plotted after normalization to UBC9. Mean values and SDs from triplicate biological experiments are plotted
Analysis of glucosinolate content in different organs of N. officinale
In HPLC analysis, we identified eight different glucosinolates in the different organs of N. officinale; glucoiberin, glucotropaeolin, 4-hydroxyglucobrassicin, glucosiberin, glucohirsutin, glucobrassicin, 4-methoxyglucobrassicin, and gluconasturtiin (Table 4). The levels of these glucosinolates distributed over the different organs of N. officinale (Table 5). The amount of total glucosinolates was highest in the flower, 6.1, 3.0, 2.3, and 1.2 times higher than that in the root, stem, leaf, and seed, respectively. Among the eight glucosinolates, the level of gluconasturtiin was considerably higher than any other glucosinolate, irrespective of the organ. In particular, the gluconasturtiin content in the flower and seed was considerably higher than that in other organs. The content of gluconasturtiin in the flower was 9.8, 2.9, 2.2, and 1.3 times higher than that in the root, stem, leaf, and seed, respectively. The content of glucotropaeolin was also highest in the flower, with concentrations 12.0, 4.5, and 2.3 times higher than that in the stem, leaf, and root, respectively. The second highest level of total glucosinolates was observed in the seed. The seed contains higher amounts of glucoiberin, glucosiberin, and glucohirsutin than the other organs of N. officinale. The level of glucoiberin was 7.9, 5.6, 3.3, and 1.8 times higher in the seed than in the root, leaf, stem, and flower, respectively. The content of glucosiberin was highest in the seed, with levels 8.6, 7.8, 6.8, and 1.4 times higher than those in the stem, root, leaf, and flower, respectively. The amount of glucohirsutin was highest in the seed, being 9.6, 7.6, 6.5, and 1.6 times higher than that in the stem, leaf, root, and flower, respectively. Although the total glucosinolate content was lowest in the root, the amount of 4-hydroxyglucobrassicin was highest in the root, with levels 36.8, 32.7, 26.7, and 2.9 times higher than that in the seed, stem, leaf, and flower, respectively. The root also contained the highest amount of glucobrassicin.
Table 4.
Glucosinolates identified by LC-ESI/MS in N. officinale
| Classification | Trivial name | Chemical formula | R side chain | Molecular weighta |
|---|---|---|---|---|
| Aliphatic | Glucoiberin | CH3SO(CH2)3 | 3-(methylsulfinyl)propyl | 343.18 |
| Glucosiberin | CH3SO(CH2)7 | 7-(methylsulfinyl)heptyl | 399.29 | |
| Glucohirsutin | CH3SO(CH2)8 | 8-(methylsulfinyl)octyl | 413.32 | |
| Indole | Glucobrassicin | C8H6NCH2 | 3-indolylmethyl | 368.17 |
| 4-Hydroxyglucobrassicin | 4-OHC8H6NCH2 | 4-hydroxy-3-indolylmethyl | 384.17 | |
| 4-Methoxyglucobrassicin | 4-(CH3O)C8H6NCH2 | 4-methoxy-3-indolylmethyl | 398.20 | |
| Aromatic | Gluconasturtiin | C6H5(CH2)2 | 2-phenylethyl | 343.16 |
| Glucotropaeolin | C6H5CH2 | Benzyl | 329.13 |
Molecular weighta: molecular weight of desulfo-glucosinolates
Table 5.
Glucosinolate contents in different organs of N. officinale
| Glucosinolates | Leaf | Stem | Root | Flower | Seed |
|---|---|---|---|---|---|
| Glucoiberin | 0.14 ± 0.01 | 0.24 ± 0.02 | 0.10 ± 0.02 | 0.43 ± 0.01 | 0.79 ± 0.75 |
| Glucotropaeolin | 0.08 ± 0.04 | 0.03 ± 0.03 | 0.16 ± 0.02 | 0.36 ± 0.05 | 0.33 ± 0.10 |
| 4-Hydroxyglucobrassicin | 0.11 ± 0.02 | 0.09 ± 0.00 | 2.94 ± 0.17 | 1.02 ± 1.56 | 0.08 ± 0.03 |
| Glucosiberin | 1.00 ± 0.03 | 0.79 ± 0.01 | 0.86 ± 0.10 | 4.91 ± 3.64 | 6.75 ± 1.58 |
| Glucohirsutin | 0.42 ± 0.00 | 0.33 ± 0.01 | 0.49 ± 0.05 | 2.01 ± 1.39 | 3.18 ± 0.79 |
| Glucobrassicin | 0.40 ± 0.02 | 0.53 ± 0.03 | 0.96 ± 0.06 | 0.75 ± 0.02 | 0.13 ± 0.03 |
| 4-Methoxyglucobrassicin | 0.19 ± 0.00 | 0.78 ± 0.04 | 0.69 ± 0.03 | 0.17 ± 0.01 | 0.13 ± 0.01 |
| Gluconasturtiin | 33.77 ± 0.73 | 25.20 ± 0.72 | 7.56 ± 0.26 | 73.90 ± 0.93 | 56.68 ± 7.45 |
| Total | 36.13 ± 0.84 | 27.98 ± 0.86 | 13.76 ± 0.71 | 83.55 ± 7.61 | 68.07 ± 10.73 |
Total glucosinolates were measured in 2-month-old N. officinale (μg g−1 dry weight). Each value represents the mean of three replicates and error bars are SDs
Discussion
Despite the health-benefiting importance and economical value of watercress, there is still limited genomic and physiological information available for N. officinale. In this study, we performed comparative analyses of the phytonutritional property of N. officinale, using both transcriptomic and metabolomics approaches. In our transcriptome analysis of N. officinale seedlings, we revealed total 69,635 transcripts and annotated 64,876 (93.17%) of total transcripts of N. officinale using public databases. On the basis of the annotations and sequence identities of N. officinale, we identified 33 candidate genes encoding enzymes related to glucosinolate biosynthetic pathways and analyzed the expression of these genes in the leaves, stems, roots, flowers, and seeds of N. officinale. Furthermore, we also profiled glucosinolate metabolic data via HPLC-UV analysis and identified eight glucosinolates in different organs of N. officinale. Among these eight glucosinolates, the level of gluconasturtiin was considerably higher than any other glucosinolate in individual organs. These transcriptomic and metabolomics results are highly consistent with those obtained in a recently published study by Voutsina et al. [8]. These authors performed RNA-sequencing analysis of 12 watercress accessions to investigate the genetic basis of two key watercress nutritional attributes: antioxidant (AO) capacity and glucosinolate (GLS) content. The transcriptome analysis of N. officinale yielded 80,800 transcripts (48,732 unigenes), of which 54,595 (67.6%) transcripts were annotated using a BLASTx search against Arabidopsis. Differentially expressed gene (DEG) analysis comparing watercress accessions with “high” and “low” AO and GLS revealed 145 and 94 differentially expressed loci for AO capacity and GLS, respectively. DEG analysis between the high and low GLS watercress identified links to GLS regulation and novel transcripts warranting further investigation. In the DEG analysis, they identified two differentially expressed shikimate pathway genes, c33663_g1_i2; similar to shikimate kinases and c37926_g1_i6; dehydroquinateshikimate dehydrogenase, acting upstream of the glucosinolate pathway. Our transcriptome data for N. officinale also revealed seven putative genes encoding glucosinolate transcription factors and 26 putative glucosinolate biosynthetic genes (Table 3). The seven putative genes encoding glucosinolate transcription factors, NoMYB28, NoMYB29, NOMYB34, NoMYB51, NoMYB122, NoDof1.1, and NoIQD1.1, are thought to act in glucosinolate biosynthesis regulation [27–31]. Intensive research on the relationship between shikimate pathway genes and glucosinolate biosynthetic genes in watercress will enhance our understanding of functional genomic approach, including glucosinolate biosynthetic pathways.
Many indole glucosinolate biosynthetic genes are specifically expressed at highest levels in the roots and flowers. In N. officinale, the accumulation patterns of indole glucosinolates, such as 4-hydroxyglucobrassicin and glucobrassicin, coincide with the expression patterns of the genes related to these indole glucosinolates. In contrast, the accumulation patterns of aliphatic glucosinolates did not coincide with the expression pattern of aliphatic glucosinolate-related genes in N. officinale. Most of the aliphatic glucosinolate biosynthetic genes were more highly expressed in the flowers compared with the leaves, stems, roots, and seeds, whereas the contents of aliphatic glucosinolates, such as glucoiberin, glucosiberin, and glucohirsutin, were relatively higher in the seed. Several genes involved in the regulation of glucosinolate biosynthetic pathways and external stimulations could be linked to accumulate the glucosinolate contents [54, 55]. Although there are many reasons for this discordance, the shift in developmental stage from flower to seed might possibly explain the discrepancy between gene expression pattern and metabolite content.
Conclusions
In RNA sequencing analysis using an Illumina NextSeq500 sequencer, we identified a total 69,635 transcripts and annotated 64,876 transcripts, which provide basic information for further research on the secondary metabolites in N. officinale. Our transcriptome data reveal that several genes encoding enzymes related to glucosinolate biosynthetic pathways are well conserved in N. officinale and that these genes have high similarity to those in other cruciferous plants such as Arabidopsis thaliana, Brassica rapa, and Camelina sativa.
On the basis of our gene expression study and HPLC analysis, we identified that most glucosinolate biosynthetic genes are highly expressed in flowers and that the content of total glucosinolates was also higher in flowers than in other organs, indicating a positive correlation between the expression of glucosinolate-related genes and glucosinolate contents in different organs of N. officinale. The results of this research provide comprehensive information on the N. officinale genome and enhance our understanding of the glucosinolate biosynthesis pathways in this plant.
Additional files
EXCEL file including detailed analysis of transcripts in Nasturtium officinale. (XLSX 15290 kb)
EXCEL file including sequence of transcripts in Nasturtium officinale. (XLSX 36722 kb)
Primers used in this work. (DOCX 17.9 kb)
Length distribution of contigs and transcripts in N. officinale. (PPTX 53.1 kb)
Acknowledgements
None.
Funding
This study was supported by IPET (115054-02-2-SB010), funded by Ministry of Agriculture, Food, and Rural affairs (MAFRA), Republic of Korea.
Availability of data and materials
Our Illumina RNA sequencing data for N. officinale has been deposited in the NCBI Short Read Archive (SRA) database under accession number SRR3490957 and SRA experiment SRX1747064.
Authors’ contributions
Conception and design of the experiments: SUP Performed the experiments and analyzed the data: JJ, SJB, JSP, YP, MVA and NAA. Wrote the manuscript: JJ, SJB, and SUP. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The Nasturtium officinale seeds were obtained from Asia Seeds Co., Ltd (Seoul, Korea). Since the plant materials were collected at the experimental greenhouse of Chungnam National University (Daejeon, Korea) in compliance with Chungnam National University biosafety guidelines.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ATR1
Altered tryptophan regulation1
- BAT
Bile acid transporter
- BCAT4
Branched-chain aminotransferase4
- CYP83A1
Cytochrome P450 family 83 subfamily A polypeptide 1
- FMOGS-OX5
Flavin-monooxygenase glucosinolate S-oxygenase5
- GGP1
Class I glutamine amidotransferase-like superfamily protein
- GSTF9
Glutathione S-transferase PHI9
- HAG1
High aliphatic glucosinolate1
- HIG1
High indolic glucosinolate1
- I3C
Indole-3-carbinol
- MAM1
Methylthioalkylmalate synthase1
- ST5B
Sulfotransferase5b
- SUR1
Tyrosine transaminase family protein
- UGT74B1
UDP-glucosyl transferase74B1
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3792-5) contains supplementary material, which is available to authorized users.
Contributor Information
Jin Jeon, Email: jeonjin519@gmail.com.
Sun Ju Bong, Email: asop_258@naver.com.
Jong Seok Park, Email: jongseok@cnu.ac.kr.
Young-Kyu Park, Email: yk-park@lascience.co.kr.
Mariadhas Valan Arasu, Email: mvalanarasu@gmail.com.
Naif Abdullah Al-Dhabi, Email: naldhabi@ksu.edu.sa.
Sang Un Park, Phone: +82-42-821-5730, Email: supark@cnu.ac.kr.
References
- 1.Cruz RM, Vieira MC, Silva CL. Effect of heat and thermosonication treatments on watercress (Nasturtium officinale) vitamin C degradation kinetics. Innov Food Sci Emerg Technol. 2008;9:483–8. doi: 10.1016/j.ifset.2007.10.005. [DOI] [Google Scholar]
- 2.Cruz RM, Vieira MC, Silva CLM. Effect of heat and thermosonication treatments on peroxidase inactivation kinetics in watercress (Nasturtium officinale) J Food Eng. 2006;72:8–15. doi: 10.1016/j.jfoodeng.2004.11.007. [DOI] [Google Scholar]
- 3.Pourhassan-Moghaddam M, Zarghami N, Mohsenifar A, Rahmati-Yamchi M, Gholizadeh D, Akbarzadeh A, et al. Watercress-based gold nanoparticles: biosynthesis, mechanism of formation and study of their biocompatibility in vitro. Micro Nano Lett. 2014;9:345–50. doi: 10.1049/mnl.2014.0063. [DOI] [Google Scholar]
- 4.Gonçalves E, Cruz R, Abreu M, Brandão T, Silva CL. Biochemical and colour changes of watercress (Nasturtium officinale R. Br.) during freezing and frozen storage. J Food Eng. 2009;93:32–9. doi: 10.1016/j.jfoodeng.2008.12.027. [DOI] [Google Scholar]
- 5.Heaney RK, Fenwick GR. The analysis of glucosinolates in Brassica species using gas chromatography. Direct determination of the thiocyanate ion precursors, glucobrassicin and neoglucobrassicin. J Sci Food Agric. 1980;31:593–9. doi: 10.1002/jsfa.2740310612. [DOI] [Google Scholar]
- 6.Hecht SS. Chemoprevention by isothiocyanates. J Cell Biochem Suppl. 1995;22:195–209. doi: 10.1002/jcb.240590825. [DOI] [PubMed] [Google Scholar]
- 7.Pullar JM, Thomson SJ, King MJ, Turnbull CI, Midwinter RG, Hampton MB. The chemopreventive agent phenethyl isothiocyanate sensitizes cells to Fas-mediated apoptosis. Carcinogenesis. 2004;25:765–72. doi: 10.1093/carcin/bgh063. [DOI] [PubMed] [Google Scholar]
- 8.Voutsina N, Payne AC, Hancock RD, Clarkson GJ, Rothwell SD, Chapman MA, et al. Characterization of the watercress (Nasturtium officinale R. Br.; Brassicaceae) transcriptome using RNASeq and identification of candidate genes for important phytonutrient traits linked to human health. BMC Genomics. 2016;17:378. doi: 10.1186/s12864-016-2704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadeghi H, Mostafazadeh M, Sadeghi H, Naderian M, Barmak MJ, Talebianpoor MS, et al. In vivo anti-inflammatory properties of aerial parts of Nasturtium officinale. Pharm Biol. 2014;52:169–74. doi: 10.3109/13880209.2013.821138. [DOI] [PubMed] [Google Scholar]
- 10.Gill CI, Haldar S, Boyd LA, Bennett R, Whiteford J, Butler M, et al. Watercress supplementation in diet reduces lymphocyte DNA damage and alters blood antioxidant status in healthy adults. Am J Clin Nutr. 2007;85:504–10. doi: 10.1093/ajcn/85.2.504. [DOI] [PubMed] [Google Scholar]
- 11.Baenas N, Moreno DA, Garcia-Viguera C. Selecting sprouts of brassicaceae for optimum phytochemical composition. J Agric Food Chem. 2012;60:11409–20. doi: 10.1021/jf302863c. [DOI] [PubMed] [Google Scholar]
- 12.Baenas N, Garcia-Viguera C, Moreno DA. Biotic elicitors effectively increase the glucosinolates content in Brassicaceae sprouts. J Agric Food Chem. 2014;62:1881–9. doi: 10.1021/jf404876z. [DOI] [PubMed] [Google Scholar]
- 13.Podsędek A. Natural antioxidants and antioxidant capacity of Brassica vegetables: a review. LWT - Food Sci Technol. 2007;40:1–11. doi: 10.1016/j.lwt.2005.07.023. [DOI] [Google Scholar]
- 14.Talalay P, Fahey JW. Phytochemicals from cruciferous plants protect against cancer by modulating carcinogen metabolism. J Nutr. 2001;131:3027S–33. doi: 10.1093/jn/131.11.3027S. [DOI] [PubMed] [Google Scholar]
- 15.Dias JS. Nutritional quality and health benefits of vegetables: a review. Food Nutr Sci. 2012;3:1354–74. doi: 10.4236/fns.2012.310179. [DOI] [Google Scholar]
- 16.Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: metabolism and excretion in humans. Cancer Epidemiol Biomarkers Prev. 2001;10:501–8. [PubMed] [Google Scholar]
- 17.Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci U S A. 1997;94:10367–72. doi: 10.1073/pnas.94.19.10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halkier BA, Gershenzon J. Biology and biochemistry of glucosinolates. Annu Rev Plant Biol. 2006;57:303–33. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- 19.Sønderby IE, Geu-Flores F, Halkier BA. Biosynthesis of glucosinolates-gene discovery and beyond. Trends Plant Sci. 2010;15:283–90. doi: 10.1016/j.tplants.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Gigolashvili T, Yatusevich R, Rollwitz I, Humphry M, Gershenzon J, Flugge UI. The plastidic bile acid transporter 5 is required for the biosynthesis of methionine-derived glucosinolates in Arabidopsis thaliana. Plant Cell. 2009;21:1813–29. doi: 10.1105/tpc.109.066399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kroymann J, Textor S, Tokuhisa JG, Falk KL, Bartram S, Gershenzon J, et al. A gene controlling variation in Arabidopsis glucosinolate composition is part of the methionine chain elongation pathway. Plant Physiol. 2001;127:1077–88. doi: 10.1104/pp.010416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawada Y, Toyooka K, Kuwahara A, Sakata A, Nagano M, Saito K, et al. Arabidopsis bile acid:sodium symporter family protein 5 is involved in methionine-derived glucosinolate biosynthesis. Plant Cell Physiol. 2009;50:1579–86. doi: 10.1093/pcp/pcp110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Textor S, de Kraker JW, Hause B, Gershenzon J, Tokuhisa JG. MAM3 catalyzes the formation of all aliphatic glucosinolate chain lengths in Arabidopsis. Plant Physiol. 2007;144:60–71. doi: 10.1104/pp.106.091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brader G, Mikkelsen MD, Halkier BA, Tapio PE. Altering glucosinolate profiles modulates disease resistance in plants. Plant J. 2006;46:758–67. doi: 10.1111/j.1365-313X.2006.02743.x. [DOI] [PubMed] [Google Scholar]
- 25.Grubb CD, Abel S. Glucosinolate metabolism and its control. Trends Plant Sci. 2006;11:89–100. doi: 10.1016/j.tplants.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Wittstock U, Halkier BA. Glucosinolate research in the Arabidopsis era. Trends Plant Sci. 2002;7:263–70. doi: 10.1016/S1360-1385(02)02273-2. [DOI] [PubMed] [Google Scholar]
- 27.Gigolashvili T, Yatusevich R, Berger B, Müller C, Flügge UI. The R2R3‐MYB transcription factor HAG1/MYB28 is a regulator of methionine‐derived glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 2007;51:247–61. doi: 10.1111/j.1365-313X.2007.03133.x. [DOI] [PubMed] [Google Scholar]
- 28.Gigolashvili T, Engqvist M, Yatusevich R, Müller C, Flügge UI. HAG2/MYB76 and HAG3/MYB29 exert a specific and coordinated control on the regulation of aliphatic glucosinolate biosynthesis in Arabidopsis thaliana. New Phytol. 2008;177:627–42. doi: 10.1111/j.1469-8137.2007.02295.x. [DOI] [PubMed] [Google Scholar]
- 29.Frerigmann H, Gigolashvili T. MYB34, MYB51, and MYB122 distinctly regulate indolic glucosinolate biosynthesis in Arabidopsis thaliana. Mol Plant. 2014;7:814–28. doi: 10.1093/mp/ssu004. [DOI] [PubMed] [Google Scholar]
- 30.Levy M, Wang Q, Kaspi R, Parrella MP, Abel S. Arabidopsis IQD1, a novel calmodulin‐binding nuclear protein, stimulates glucosinolate accumulation and plant defense. Plant J. 2005;43:79–96. doi: 10.1111/j.1365-313X.2005.02435.x. [DOI] [PubMed] [Google Scholar]
- 31.Skirycz A, Reichelt M, Burow M, Birkemeyer C, Rolcik J, Kopka J, et al. DOF transcription factor AtDof1. 1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. Plant J. 2006;47:10–24. doi: 10.1111/j.1365-313X.2006.02767.x. [DOI] [PubMed] [Google Scholar]
- 32.Morozova O, Hirst M, Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet. 2009;10:135–51. doi: 10.1146/annurev-genom-082908-145957. [DOI] [PubMed] [Google Scholar]
- 33.Van Verk MC, Hickman R, Pieterse CM, Van Wees SC. RNA-Seq: revelation of the messengers. Trends Plant Sci. 2013;18:175–9. doi: 10.1016/j.tplants.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010;20:646–54. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekhon RS, Briskine R, Hirsch CN, Myers CL, Springer NM, Buell CR, et al. Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hao QN, Zhou XA, Sha AH, Wang C, Zhou R, Chen SL. Identification of genes associated with nitrogen-use efficiency by genome-wide transcriptional analysis of two soybean genotypes. BMC Genomics. 2011;12:525. doi: 10.1186/1471-2164-12-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao X, Gu YH, Wang HY, Zheng W, Li X, Zhao CW, et al. Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam] PLoS One. 2012;7:e36234. doi: 10.1371/journal.pone.0036234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Druka A, Muehlbauer G, Druka I, Caldo R, Baumann U, Rostoks N, et al. An atlas of gene expression from seed to seed through barley development. Funct Integr Genomics. 2006;6:202–11. doi: 10.1007/s10142-006-0025-4. [DOI] [PubMed] [Google Scholar]
- 40.Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics. 2011;12:131. doi: 10.1186/1471-2164-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng C, Chen M, Xu CJ, Bai L, Yin XR, Li X, et al. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genomics. 2012;13:19. doi: 10.1186/1471-2164-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc. 2013;doi:10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed]
- 44.Smith-Unna R, Boursnell C, Patro R, Hibberd JM, Kelly S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016;26:1134–44. doi: 10.1101/gr.196469.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–7. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71–3. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ISO norm. Rapeseed-Determination of glucosinolate content. Part 1: Method using high-performance liquid chromatography. ISO 9167-1, 1992:1–9.
- 50.Lee MK, Chun JH, Byeon DH, Chung SO, Park SU, Park S, et al. Variation of glucosinolates in 62 varieties of Chinese cabbage (Brassica rapa L. ssp. pekinensis) and their antioxidant activity. LWT-Food Sci Technol. 2014;58:93–101. doi: 10.1016/j.lwt.2014.03.001. [DOI] [Google Scholar]
- 51.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37:501–6. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 52.Wang H, Wu J, Sun S, Liu B, Cheng F, Sun R, et al. Glucosinolate biosynthetic genes in Brassica rapa. Gene. 2011;487:135–42. doi: 10.1016/j.gene.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 53.Kim YB, Li X, Kim SJ, Kim HH, Lee J, Kim H, et al. MYB transcription factors regulate glucosinolate biosynthesis in different organs of Chinese cabbage (Brassica rapa ssp. pekinensis) Molecules. 2013;18:8682–95. doi: 10.3390/molecules18078682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Booth EJ, Walker KC, Griffiths DW. A time-course study of the effect of sulphur on glucosinolates in oilseed rape (Brassica napus) from the vegetative stage to maturity. J Sci Food Agric. 1991;56:479–93. doi: 10.1002/jsfa.2740560408. [DOI] [Google Scholar]
- 55.Velasco P, Cartea ME, Gonzalez C, Vilar M, Ordas A. Factors affecting the glucosinolate content of kale (Brassica oleracea acephala group) J Agric Food Chem. 2007;55:955–62. doi: 10.1021/jf0624897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EXCEL file including detailed analysis of transcripts in Nasturtium officinale. (XLSX 15290 kb)
EXCEL file including sequence of transcripts in Nasturtium officinale. (XLSX 36722 kb)
Primers used in this work. (DOCX 17.9 kb)
Length distribution of contigs and transcripts in N. officinale. (PPTX 53.1 kb)
Data Availability Statement
Our Illumina RNA sequencing data for N. officinale has been deposited in the NCBI Short Read Archive (SRA) database under accession number SRR3490957 and SRA experiment SRX1747064.