

Abstract
Human amniotic fluid stem cells (hAFS) have shown a distinct secretory profile and significant regenerative potential in several preclinical models of disease. Nevertheless, little is known about the detailed characterization of their secretome. Herein we show for the first time that hAFS actively release extracellular vesicles (EV) endowed with significant paracrine potential and regenerative effect. c‐KIT+ hAFS were isolated from leftover samples of amniotic fluid from prenatal screening and stimulated to enhance EV release (24 hours 20% O2 versus 1% O2 preconditioning). The capacity of the c‐KIT+ hAFS‐derived EV (hAFS‐EV) to induce proliferation, survival, immunomodulation, and angiogenesis were investigated in vitro and in vivo. The hAFS‐EV regenerative potential was also assessed in a model of skeletal muscle atrophy (HSA‐Cre, Smn F7/F7 mice), in which mouse AFS transplantation was previously shown to enhance muscle strength and survival. hAFS secreted EV ranged from 50 up to 1,000 nm in size. In vitro analysis defined their role as biological mediators of regenerative, paracrine effects while their modulatory role in decreasing skeletal muscle inflammation in vivo was shown for the first time. Hypoxic preconditioning significantly induced the enrichment of exosomes endowed with regenerative microRNAs within the hAFS‐EV. In conclusion, this is the first study showing that c‐KIT+ hAFS dynamically release EV endowed with remarkable paracrine potential, thus representing an appealing tool for future regenerative therapy. Stem Cells Translational Medicine 2017;6:1340–1355
Keywords: Fetal stem cells, Paracrine communication, Extracellular vesicles, Exosomes, Proliferation, Apoptosis, miRNA, Tissue regeneration
Significance Statement.
This is the first study providing a first characterization of the human amniotic fluid stem cell (hAFS)‐derived extracellular vesicles (EV). hAFS are fetal mesenchymal progenitors with a distinct cardioprotective and angiogeneic secretory profile that can be easily isolated, expanded, and cryopreserved, hence representing a suitable candidate for future clinical applications. Stem cell‐secreted EV are mediators of intercellular communication and have been described as active components of paracrine regenerative effects. Here we showed that EV from hAFS mediate significant prosurvival, proliferative, and anti‐inflammatory effects and may represent an ideal approach for next generation therapy. Therefore, AFS might be exploited as “drug‐stores” to produce EV endowed with paracrine regenerative potential that could ultimately be harnessed as advanced medicinal product.
Introduction
Stem cell‐derived paracrine effects have emerged as promising strategy for the reactivation of endogenous mechanisms of repair and regeneration in several disease models 1, 2, 3. Mounting evidence has shown that transplanted stem cells can release trophic signals that influence the microenvironment. This has led to a significant shift in paradigms, from exploring the stem cell genome to analyzing the stem cell “secretome,” the latter of which includes the entirely of growth factors and chemoattractant molecules produced by stem cells. In this scenario, growing interest has focused on the characterization of stem cell‐secreted extracellular vesicles (EV). EV are membrane‐bound cellular components enriched with soluble, bioactive factors (proteins, lipids, etc.) and RNA (mainly regulatory microRNA—miRNA). They elicit a wide range of effects while mediating horizontal intercellular transfer of genetic information on the responder cell, consequently modulating its function. EV are secreted as microsized (microvesicles: 100–1,000 nm) and nanosized (exosomes: 30–150 nm) particles, thus acting as key biological effectors of paracrine signaling 4. Microvesicles are released as shedding vesicles by direct budding of the plasma membrane, while exosomes are produced in endosomal multivesicular compartments (multivesicular body, MVB) and secreted as the MVB fuses with the plasma membrane 5. Recent studies have shown that the beneficial effects observed following stem cell transplantation in several preclinical models of experimental ischemic disease and injury could be mediated by stem cell‐EV. These include the activation of antiapoptotic and prosurvival pathways eliciting angiogenic, anti‐inflammatory, and antifibrotic responses, and the stimulation of resident endogenous progenitors, overall enhancing organ function 6. In particular, many studies have reported the potential efficacy of EV from adult mesenchymal stem cells (MSC) in providing cardioprotection against acute myocardial infarction (MI) 7, 8, 9, in enhancing wound healing 10, counteracting graft‐versus‐host‐disease 11, reducing renal injury 12, mediating liver regeneration 13 and stimulating neural plasticity following stroke 14. Since cell‐free delivery of bioactive cargos by EV recapitulates the same beneficial responses of stem cell transplantation, they offer remarkable benefits over conventional cell therapy as immunologically unresponsive agents 15.
Human amniotic fluid‐derived stem cells (hAFS) are broadly characterized as multipotent mesenchymal progenitors expressing pluripotency markers and high self‐renewal potential similar to embryonic stem cells, without being tumorigenic or causing any ethical concern 16. Because of their fetal, but non‐embryonic origin, hAFS overcome many ethical concerns and can be easily obtained upon the expression of the stem marker c‐KIT from leftover or discarded amniotic fluid samples collected during either amniocentesis 16, 17 or eligible cesarean delivery 18. We previously demonstrated that c‐KIT+ hAFS exert remarkable cardioprotective paracrine effects reducing the infarct size in a rat acute model of MI by approximately 14% 19, possibly via the secretion of the cardioactive peptide thymosin beta 4, which, in turn, can reactivate epicardial progenitor cells to give rise de novo to fully mature cardiomyocytes 20. Moreover, when injected in an established model of necrotizing enterocolitis, rodent c‐KIT+ AFS have been shown to improve survival and clinical status, while maintaining the gut structure and function via paracrine modulation of resident stromal cells expressing cyclooxygenase 2 (COX2) 21. Similar remarkable results have been also obtained in a model of skeletal muscle atrophy carrying a homozygous deletion of the exon 7 within the survival motor neuron (Smn) gene, specifically directed to new forming skeletal muscle fibers (HSA‐Cre, SmnF7/F7 mice), where mouse c‐KIT+ AFS confirmed their regenerative potential by providing significant improvement in muscle strength and in the survival rate of the affected animals, together with the replenishment of the depleted skeletal muscle niche 22.
Despite c‐KIT+ AFS paracrine potential and distinct proteomic profile being confirmed by different independent studies 19, 23, 24, at present the functional properties and the role of their secreted EV (hAFS‐EV) have not been elucidated. Consistent with the observation that hAFS are progenitors with embryonic, stem cell‐like properties, they are likely to possess a powerful paracrine potential given their early developmental stage. Hence, our hypothesis is based on the idea that the correlation between the therapeutic efficacy of the hAFS secretome and their fetal origin might be underpinned by their EV production and regenerative potential.
Here we aim to provide a first characterization of the regenerative potential of the hAFS‐EV, as a new promising tool for a future cell‐free therapy at the frontiers of regenerative medicine.
Materials and Methods
Cell Isolation and Culture
hAFS were from leftover samples of amniotic fluid obtained via amniocentesis upon written informed consent, as previously described 16, 17. All procedures were performed in compliance with the Helsinki Declaration and the local ethical committee (IRCCS AOU San Martino‐IST, P.R. 428REG2015). Normal adult human dermal fibroblasts (HDF), the mouse myoblast C2C12 cell line, and primary human peripheral blood mononuclear cells (hPBMCs) were used for in vitro experiments. For more details, please refer to Supporting Information.
hAFS Preconditioning
hAFS were cultured for 24 hours in serum‐free (SF) medium (Minimum Essential Medium Eagle alpha, with 1% l‐glutamine and 1% penicillin/streptomycin) under normoxic (20% O2 and 5% CO2 at 37°C) or hypoxic (1% O2 and 5% CO2 at 37°C in a hypoxic incubator, Eppendorf, Hamburg, Germany; https://www.eppendorf.com/) conditions. The hAFS‐conditioned medium (hAFS‐CM) was collected and processed for hAFS‐EV isolation.
Characterization of hAFS After Hypoxic Preconditioning
The expression of specific stem cell markers was assessed by immunostaining using an anti‐Stage‐Specific Embryonic Antigen‐4 (SSEA4) antibody (Abcam, Cambridge, United Kingdom; www.abcam.com) and an anti‐NANOG antibody (Epitomics, Cambridge, United Kingdom). NANOG mRNA levels were also evaluated by both qualitative reverse transcription‐polymerase chain reaction (RT‐PCR) and real time qRT‐PCR. hAFS protein content was analyzed by Western Blot (WB) for human hypoxia inducible factor‐1 alpha (HIF‐1α BD Bioscience, East Rutherford, New Jersey, http://www.bdbiosciences.com/eu/solrSearch?text=hypoxia+1+alpha&x=0&y=0) and βACTIN (Santa Cruz Biotechnology, Dallas, Texas, https://www.scbt.com/scbt/home?&_requestid=235153). hAFS immunophenotype and viability was assessed by fuorescence‐activated cell sorting (FACS). For more details, refer to Supporting Information.
Isolation and Characterization of hAFS‐EV
A scheme of hAFS‐EV isolation is shown in Supporting Information Figure S1. hAFS‐EV were isolated by ultracentrifugation 5 from hAFS‐CM obtained from cells cultured for 24 hours under SF normoxic (hAFS‐CMNormo) or hypoxic (hAFS‐CMHypo) conditions. hAFS and hAFS‐EV were analyzed by transmission electron microscopy (TEM). hAFS‐EV were characterized by nanoparticle tracking technology using a NanoSight LM10 (Malvern Instruments, Malvern, United Kingdom, http://www.malvern.com/en/?gclid=CJ746ZrK9NECFcMy0wodhJwFxA) to analyze particles released by 106 cells. The concentration of membrane‐bound protein on the surface of freshly isolated, intact hAFS‐EV was measured using BiCinchoninic acid (BCA) assay (Thermo Fisher Scientific, Waltham, Massachusetts, http://www.thermofisher.com/it/en/home.html). WB on hAFS and hAFS‐EV was performed for the expression of TSG101 (Abcam, Cambridge, United Kingdom, http://www.abcam.com), ALIX (Santa Cruz Biotechnology, Dallas, Texas, https://www.scbt.com/scbt/home?&_requestid=235153), GRP94 (Abcam, Cambridge, United Kingdom; www.abcam.com), and human βACTIN (Santa Cruz Biotechnlology, Dallas, Texas, https://www.scbt.com/scbt/home?&_requestid=235153). hAFS‐EV were also evaluated by FACS for the presence on their surface of the MSC antigen CD105 (eBioscience, Waltham, Massachusetts, https://www.ebioscience.com/), the conventional exosomal markers CD81, CD9, CD63, Annexin V (AnnV), and the costimulatory molecules CD80 and CD86 (all BD Bioscience, East Rutherford, New Jersey, https://www.ebioscience.com/). For more details, refer to Supporting Information.
Uptake Analysis of hAFS‐EV by Target Cells and Functional In Vitro Studies
hAFS‐EV were labeled with the PKH67 Green Fluorescent Cell Linker (Sigma, Milano, Italy, https://www.sigmaaldrich.com/), following manufacturer's instructions. 1 μg/104 cells (for HDF and C2C12 cells) and 1 μg/105 cells (for hPBMC) PKH67+ hAFS‐EVNormo and PKH67+ hAFS‐EVHypo were incubated for 3 hours with the target cells in SF medium. To assess the biological effects driven by hAFS‐EV on HDF and C2C12 cells, three different concentrations (1 μg/104, 2 μg/104, and 4 μg/104 cells) of either hAFS‐EVNormo or hAFS‐EVHypo were used. To analyze cell proliferation, HDF were primed with hAFS‐EV and then analyzed by BromodeoxyUridine (BrdU) colorimetric assay (Roche, Basel, Switzerland, http://www.roche.com/) according to the manufacturer's instructions. To define the antiapoptotic effect of hAFS‐EV, C2C12 were incubated with hAFS‐EV and exposed to oxidative damage. Cell viability was measured by 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐Diphenyltetrazolium Bromide (MTT) assay using a 150 μg/ml MTT solution (Sigma‐Aldrich, Milano, Italy, https://www.sigmaaldrich.com/).
Data were acquired on a VersaMax (GE Intelligent Platforms, Charlottesville, Virginia, https://www.moleculardevices.com/systems/microplate-readers/absorbance-readers/versamax-elisa-microplate-reader) plate reader. The immunomodulatory potential of hAFS‐EV was evaluated on hPBMC stimulated with 5 μg/ml phytohemagglutinin (PHA, Sigma Chemicals Co., Milano, Italy, https://www.sigmaaldrich.com/) and 5 μg/ml pokeweed mitogen (PWM, Sigma Chemicals Co., Milano, Italy, https://www.sigmaaldrich.com/) for 1 hour, to induce T‐ and B‐lymphocytes expansion 25, 26, followed by incubation with 1 μg/105 cells of hAFS‐EVNormo or hAFS‐EVHypo for 5 days. For more details, refer to Supporting Information.
In Vivo Angiogenic Matrigel Plug Assay
Adult C57Bl/6J male mice were subcutaneously injected with ice‐cold unpolymerized Matrigel (Corning, New York; https://www.corning.com/worldwide/en/products/life-sciences/products/surfaces/matrigel-matrix.html) solution containing 10 μg of either hAFS‐EVNormo or hAFS‐EVHypo. All the animal work was performed in compliance with ethical national and international (EU Directive 2010/63/EU) standards and according to institutional guidelines from the Animal Facility of IRCCS AOU San Martino‐IST, Genova. Mice were sacrificed 3 weeks after injection and Matrigel plugs were collected for histological and functional evaluation of the host endothelial infiltrating cells. Real time qRT‐PCR was also performed for the expression of VegfA, Pecam1, Cdh5 (VE‐Cadherin), and beta‐2‐microglobulin (B2M) genes. For more details, refer to Supporting Information.
Mouse Model of Muscular Atrophy
Twelve‐week‐old HSA‐Cre, SmnF7/F7 mice 22 (n = 8 per time point analysis) were randomized to receive either control phosphate‐buffered saline (PBS) or hAFS‐EV, as represented in Figure 5A. One microgram of hAFS‐EVNormo and hAFS‐EVHypo was injected in the right and left Tibialis Anterior (TA) muscles, respectively. Mice were sacrificed 1 and 7 days after treatment and muscle tissue was processed for immunostaining and real time qRT‐PCR. All the animal work was performed in compliance with ethical national and international (EU Directive 2010/63/EU) standards according to institutional guidelines from the University of Padova Animal Care and Use Committee (CEASA, protocol 67/2011). For more details, refer to Supporting Information.
Figure 5.

hAFS‐EV decrease muscle inflammation in a mouse model of muscle atrophy. (A): In vivo procedure. (B): IgG (red), CD68 (red), and LAMININ (green) expression in the Tibialis Anterior muscle of untreated (CTRL) and 1 μg hAFS‐EVNormo‐ and hAFS‐EVHypo‐treated mice at 1 and 7 days post injection (p.i.); scale bar: 100 μm and 50 μm, respectively. (C): IgG‐infiltrated fibers at 1 and 7 days p.i. (CTRL 1 Day: 14.9% ± 3.9%; hAFS‐EVNormo 1 Day: 2.9% ± 1.4%; hAFS‐EVHypo 1 Day: 1.6% ± 0.7%; *, p < .05 p = .031 and p = .016; CTRL 7 Days: 16.1% ± 4.3%; hAFS‐EVNormo 7 Days: 3.8% ± 1.0%; hAFS‐EVHypo 7 Days: 2.0% ± 0.7%; *, p < .05, p = .035) and corresponding mean fluorescent area (CTRL 1 Day: 8.33% ± 3.0%; hAFS‐EVNormo 1 Day: 0.71% ± 0.11%; hAFS‐EVHypo 1 Day: 0.42% ± 0.1%; **, p < .01, p = .002; CTRL 7 Days: 8.90% ± 3.2%; hAFS‐EVNormo 7 Days: 1.64% ± 0.44%; hAFS‐EVHypo 7 Days: 0.80% ± 0.15%; **, p < .01, p = .006 and p = .003). (D): Central nucleated fibers at 1 and 7 days p.i.: CTRL 1 Day: 9.4% ± 1.6%; hAFS‐EVNormo 1 Day: 4.0% ± 1.3%; hAFS‐EVHypo 1 Day: 2.6% ± 1.3%, *, p < .05, p = .015; CTRL 7 Days: 9.8% ± 1.7%; hAFS‐EVNormo 7 Days: 13.1% ± 3.5%; hAFS‐EVHypo 7 Days: 3.7% ± 0.9%; #, p < .05, p = .033, as hAFS‐EVHypo versus hAFS‐EVNormo. (E): Ratio of Nos2 over Arg2 by real time qRT‐PCR; *, p < .05, p = .045 and p = .036 for hAFS‐EVNormo and hAFS‐EVHypo;. A.U. (F): Real time qRT‐PCR for Il‐1α (***, p < .001, p = .0002 and p = .001; *, p < .05, p = .046 and p = .0163, for hAFS‐EVNormo and hAFS‐EVHypo) and Il‐10 (****, p < .0001, **, p < .01, p = .004). Abbreviations: A.U. arbitrary units; CD, cluster of differentiation; DAPI, 4′,6‐diamidino‐2‐phenylindole; EV, extracellular vesicles; hAFS, human amniotic fluid stem cells; Hypo, hypoxic; Normo, normoxic; SF, serum‐free.
Small Non‐Coding RNA Profiling of hAFS‐EV
Total hAFS‐EV RNA was extracted using Qiazol Lysis Reagent (QIAGEN, Hilden, Germany, http://www.qiagen.com). Analysis of the small non‐coding RNA content was performed on a 2100 Bioanalyzer using the Small RNA Kit and the Agilent 2100 expert software (all from Agilent Technologies, Santa Clara, California, http://www.agilent.com/home).
Real Time qRT‐PCR Analysis of the hAFS‐EV microRNA Content
Total RNA from hAFS and hAFS‐EV was extracted using miRNAeasy Mini Kit following the manufacturer's instructions. miScript II RT Kit was used for cDNA synthesis and selected miScript Primer Assay and miScript SYBR® Green PCR Kits were used for quantitative RT‐PCR analysis on a 7500 Fast Real‐Time PCR System (Thermo Fisher Scientific, Waltham, Massachusetts, http://www.thermofisher.com/it/en/home.html). The following human miRNAs (all from QIAGEN, Hilden, Germany; www.qiagen.com) were evaluated: miR‐223, miR‐146a, miR‐let7c, miR‐21, miR‐126, miR‐146b, miR‐199a‐3p, and miR‐210. Gene expression levels were normalized to the corresponding miRNA expression in hAFS using the 2‐ddCt method.
Analysis of Direct Transfer of Exosomal RNA from the hAFS‐EV into Target Cells
RNA content in hAFS‐EV was labeled using the SYTO® RNASelectTM Green Fluorescent Nucleic Acid Stain (Thermo Fisher Scientific, Waltham, Massachusetts, http://www.thermofisher.com/it/en/home.html) following the manufacturer's instructions. One microgram of SYTO®+ hAFS‐EVNormo or SYTO®RNASelect+ hAFS‐EVHypo was incubated for 3 hours with 104 HDF or C2C12 cells in SF medium and analyzed after 1 hour by FACS. For more details, refer to Supporting Information.
Real time qRT‐PCR Evaluation of Specific miRNA Enrichment and Their Target Genes in the Target Cells
HDF and C2C12 cells were incubated with 4 μg/104 cells of either hAFS‐EVNormo or hAFS‐EVHypo in SF medium, following the same protocol used for cell proliferation and viability assays. After 1 hour incubation in complete medium, total RNA from HDF and C2C12 cells was isolated using Qiazol Lysis Reagent (QIAGEN, Hilden, Germany, www.qiagen.com). Analysis of selected miRNAs was performed as described above; the miRNA target genes MNT (HDF) and Casp8ap2 (C2C12) were analyzed by real time qRT‐PCR. Gene expression levels were normalized to GAPDH (HDF) and B2M (C2C12) as endogenous controls using the 2‐ddCt method. For more details, refer to Supporting Information.
Statistical Analysis
Results are presented as mean ± SEM of at least three (n = 3) independent experiments. Analyses were performed using GraphPad Prism Version 6.0a (GraphPad Software); p values < .05 were considered statistically significant. For more details, refer to Supporting Information.
Results
Hypoxic Preconditioning Does Not Significantly Alter hAFS Stemness
To enrich the hAFS secretome with EV, cells were subject to a 24‐hour preconditioning in SF medium under normoxic (20% O2) or hypoxic conditions (1% O2), the latter of which has been shown to enhance secretion of EV from stem cells 9, 27, 28, 29. hAFS were analyzed under the following conditions: (a) control normoxic (20% O2 in complete medium, CTRL); (b) SF normoxic (20% O2 in SF medium, SF); (c) control hypoxic (1% O2 in complete medium, CTRL), and (d) SF hypoxic (1% O2 in SF medium, SF). hAFS did not show any substantial morphological alteration under hypoxic versus normoxic conditions, both in CTRL and SF culture (Fig. 1A). Also, the expression of the stem markers SSEA4 and NANOG (Fig. 1A) was not significantly altered. SSEA4 was consistently expressed by more than 83% of hAFS, regardless of the culture settings (Fig. 1C). While NANOG mRNA was not significantly affected by preconditioning (Fig. 1D), protein expression was heterogeneously distributed in hAFS, yet without any evident difference compared to the control condition (Fig. 1A). HIF‐1α expression confirmed that 24‐hours treatment was sufficient for the hAFS to become responsive to the hypoxic microenvironment (Fig. 1B). hAFS immunophenotype remained unaltered, lacking expression for hematopoietic and mature lineage antigens—such as CD45, CD34, human leucocyte antigen (HLA) Class II, and CD31—and showing reasonably high to medium‐high expression of the canonical mesenchymal markers CD73, CD90, CD44, and CD105. A small, but non‐significant decrease in CD73 and CD105 expression was detected in hypoxic and SF conditions. hAFS cells showed variable CD146 expression, as previously reported 30, 31, while they maintained robust expression of HLA Class I (Fig. 1E; Supporting Information Table S1). hAFS viability was variable: although the SF condition affected hAFS more than hypoxic stimulation, this was not statistically significant and likely dependent on hAFS donor variation (Fig. 1F; Supporting Information Table S1).
Figure 1.
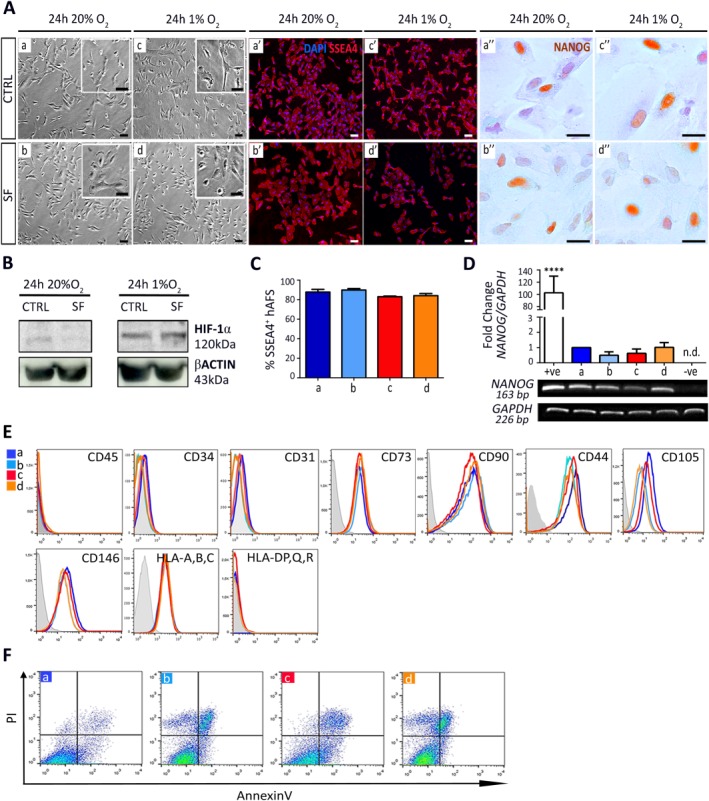
Hypoxic preconditioning does not significantly affect hAFS stemness and phenotype. (A): Bright field, SSEA4 and NANOG immunostaining images of hAFS under: (a), (a′), and (a′′) 24 hours 20% O2 CTRL conditions; (b), (b′), and (b′′) 24 hours 20% O2 SF conditions; (c), (c′), and (c′′) 24 hours 1% O2 preconditioning in CTRL medium; (d), (d′), and (d′′) 24 hours 1% O2 preconditioning in SF condition; scale bar 50 µm. (B): Western Blot for HIF‐1α expression by hAFS after 24 hours of 20% O2 or 1% O2 preconditioning in complete (CTRL) or SF medium. (C): SSEA4+ hAFS under: (a) 24 hours 20% O2 CTRL condition: 87.8% ± 2.8%; (b) 24 hours 20%O2 SF condition: 89.9% ± 1.5%; (c) 24 hours 1% O2 preconditioning in CTRL medium: 83.0% ± 0.7%; and (d) 24 hours 1% O2 preconditioning in SF condition: 84.1% ± 2.2%; values expressed as mean ± SEM. (D): Real time qRT‐PCR and qualitative RT‐PCR analysis for NANOG and GAPDH expression of hAFS under 24 hours: (a) 20% O2 CTRL and (b) 20% O2 SF conditions, (c) 1% O2 CTRL and (d) 1% O2 SF conditions; +ve: positive control, human embryonic stem cells (****, p < .0001); −ve: negative control: HDF; n.d. (E): hAFS immunophenotype under 24 hours 20% O2 in CTRL (a), or SF conditions (b), or under 24 hours 1% O2 preconditioning, in either CTRL (c), or SF conditions (d). (F): Apoptosis analysis by Annexin V and PI of hAFS under 24 hours: (a) 20% O2 CTRL and (b) 20% O2 SF conditions, (c) 1% O2 CTRL and (d) 1% O2 SF conditions. Abbreviations: CD, cluster of differentiation; hAFS, human amniotic fluid stem cells; HIF‐1α, hypoxia inducible factor‐1 alpha; HLA, human leucocyte antigen; n.d., not determined; PI, propidium iodide; SF, serum‐free.
hAFS Secrete EV in Their Conditioned Medium
TEM analysis revealed that hAFS can actively secrete EV ranging in size from 50 to 1,000 nm (Fig. 2A). We further investigated hAFS‐EV secretion for 24 hours under SF conditions to avoid any contamination by Fetal Bovine Serum (FBS)‐EV (1% O2 hypoxic versus 20% O2 normoxic) 5. The hAFS‐CM ultracentrifuged pellet contained hAFS‐EV between 50 and 200 nm (Fig. 2B), thus including both exosomes and microvesicles 5. The vesicle electron density and the absence of nuclear or cytoplasmic residues in both the EV released in situ by the cells (Fig. 2A), and the hAFS‐EV isolated by ultracentrifugation (Fig. 2B), demonstrated that apoptotic bodies were not significantly present. Nanoparticle tracking (Fig. 2C) showed a reasonably high number of particle in both hAFS‐EVNormo and hAFS‐EVHypo (283.33 ± 64.75 × 106 particle/ml and 283.83 ± 79.85 × 106 particle/ml, respectively, p = .65). Both hAFS‐EVNormo and hAFS‐EVHypo particle dimension showed average size of 180 nm, likely due to clustering effects previously described for other progenitor‐derived EV 32. BCA assay was performed to evaluate the protein content on the EV surface. Hypoxic preconditioning led to a significant enrichment of proteins on the hAFS‐EV surface (4.19 ± 0.43 μg/106 cells) with almost twofold increase compared to normoxic cells (2.35 ± 0.26 μg/106 cells, Fig. 2D, ***p < .001). WB confirmed the expression of the microvesicle and exosome canonical markers TSG101 and ALIX 5 on the hAFS‐EV (Fig. 2E). Notably, hAFS‐EV did not express GRP94, indicating no contamination from the endoplasmic reticulum associated with apoptotic blebs 33 (Fig. 2E). FACS analysis revealed a significant exosomal subpopulation expressing the markers CD81, CD9, AnnV 5, 34, and CD63 5 within the hAFS‐EV (Fig. 2F and Supporting Information Fig. S2B). In particular, hypoxic preconditioning led to a significant enrichment of CD81+, CD9+, and CD63+ exosomes in hAFS‐EVHypo compared to the hAFS‐EVNormo (1.5‐, 1.3‐, and 2.4‐fold, *p < .05 and **p < .01, respectively), despite similar total secreted particle content. We also labeled hAFS‐EV using the CellTrace Far Red DDAO‐SE kit to distinguish intact vesicles from cell debris, as recently described by others 35. CellTrace‐positive hAFS‐EV were further analyzed for the expression of the MSC antigen CD105, showing that approximately 60% of them maintained this marker 17 (61.3% ± 2.3% and 59.4% ± 12.3% in hAFS‐EVNormo and hAFS‐EVHypo, respectively, p = .8985, Supporting Information Fig. S2A). hAFS‐EV did not express costimulatory molecules CD80 and CD86 36, suggesting immune tolerance (Supporting Information Fig. S2B).
Figure 2.
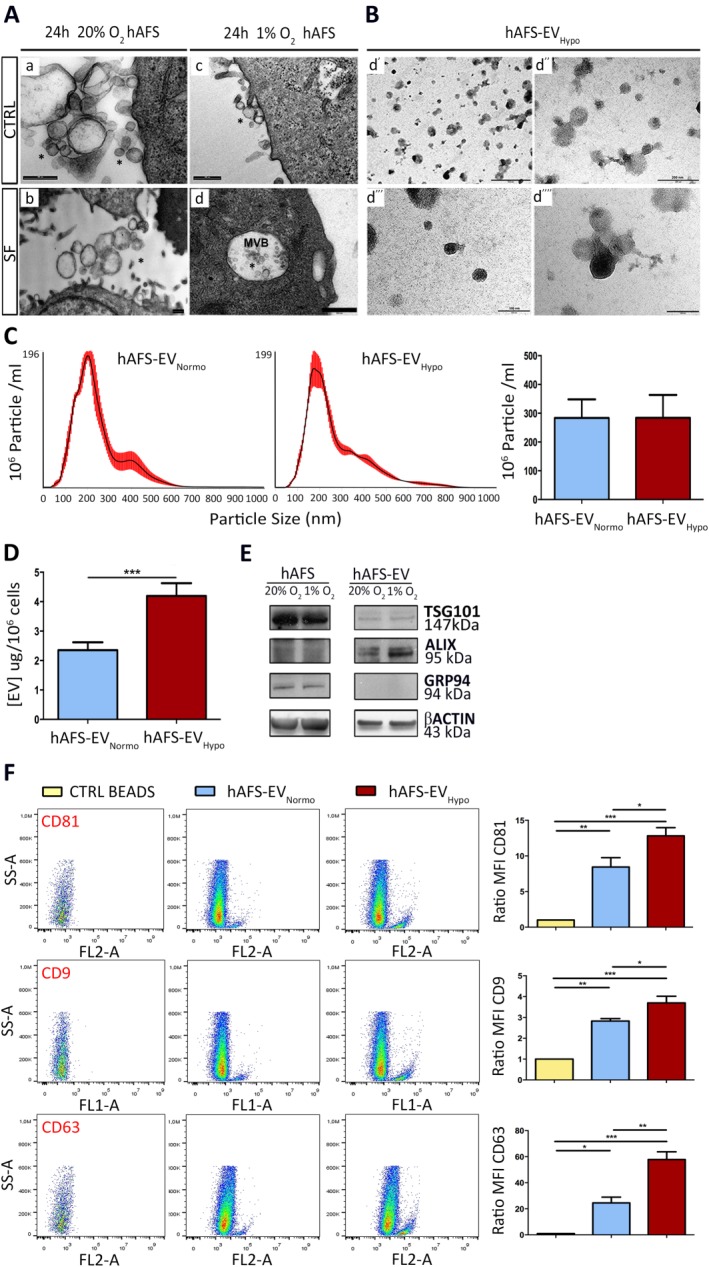
hAFS secrete EV containing microvesicles and exosomes. (A): Transmission electron microscopy (TEM) analysis of hAFS under 24 hours 20% O2 CTRL (a) or 20% O2 SF conditions (b) and 1% O2 CTRL (c) or 1% O2 SF (d) conditions; scale bars 500 nm. (B): TEM Analysis of 1% O2 hAFS‐EV (hAFS‐EVHypo); scale bar 500 nm in (d′); 200 nm in (d′′); and 100 nm (d′′′ and d′′′′). (C): Nanosight analysis measuring the amount of particles within hAFS‐EVNormo (283.3 ± 64.8 × 106 particle/ml) and hAFS‐EVHypo (283.8 ± 79.9 × 106 particle/ml) released by 106 cells (right panel); values are expressed as mean ± SEM. Representative images of the graphical output are reported in the left panel. (D): BCA assay of the protein concentration of hAFS‐EVHypo (4.2 ± 0.4 μg/106 cells, ***, p < .001, p = .0005) over hAFS‐EVNormo (2.4 ± 0.3 μg/106 cells) released by 106 cells. (E): Western Blot analysis of TSG101, ALIX, GRP94, and βACTIN by hAFS and hAFS‐EV, both under 20% O2 and 1% O2 SF conditions. (F): FACS analysis of hAFS‐EVNormo and hAFS‐EVHypo bound to the ExoCapTM Capture Beads compared to control empty beads (CTRL BEADS) for the exosomal markers CD81 (*, p < .05, p = .049; **, p < .01, p = .005; ***, p < .001, p = .0004), CD9 (*p < .05, p = .047; **, p < .01, p = .0015; ***, p < .001, p = .0002), and CD63 (*, p < .05, p = .02; **, p < .01, p = .004; ***, p < .001, p = .0002). Representative dot plots are illustrated in the left panel, the evaluation of the MFI ratio on the right panels. Abbreviations: CD, cluster of differentiation; EV, extracellular vesicles; hAFS, human amniotic fluid stem cells; Hypo, hypoxic; MFI, mean fluorescent intensity; Normo, normoxic; SF, serum‐free.
Analysis of hAFS‐EV Biological Effects on Target Cells
The role of both hAFS‐EVHypo and hAFS‐EVNormo as biological mediators of paracrine effects was assessed on different target cells.
Analyses on HDF, C2C12, and hPBMC incubated for 3 hours with either PKH67+ hAFS‐EVNormo or hAFS‐EVHypo showed similar internalization efficiency (65.4% ± 4.4% and 63.5% ± 5.6% PKH67+HDF, p = .94; 75.9% ± 3.4% and 83.7% ± 1.1% PKH67+ C2C12, p = .11, and 9.3% ± 2.6% and 12.5% ± 2.5% PKH67+ hPBMC, p = .54, respectively, Fig. 3A). Following 4 hours incubation with 1 μg/104, 2 μg/104, and 4 μg/104 cells hAFS‐EVNormo, proliferation of BrDU‐labeled HDF showed a dose‐dependent, 2‐ to almost 3.5‐fold increase, compared to untreated cells (****p < .0001). hAFS‐EVHypo treatment further stimulated HDF proliferation by 2.5‐ to 4.3‐fold change compared to untreated cells (****p < .0001), and with higher dose‐effect response compared to hAFS‐EVNomro (## p < .01; # p < .5, and ### p < .001, Fig. 3B; Supporting Information Table S2). C2C12 mouse myoblast cell line underwent oxidative stress by treatment with 1 mM H2O2 for 2 hours in the presence of hAFS‐EV. MTT analysis showed that cells exposed to H2O2 had 65% lower cell viability (****p < .0001). Following 3 hours of pretreatment with hAFS‐EV, C2C12 cells significantly recovered from injury, with improved cell viability and increased proliferation (Fig. 3C). hAFS‐EVNormo mediated remarkable dose‐dependent antiapoptotic effects enhancing cell survival up to two‐ and sixfold compared to control cells and to cells not receiving any treatment before undergoing oxidative stress, respectively (****p < .0001). hAFS‐EVHypo exerted an even more effective cytoprotective role by increasing cell viability in a dose‐dependent manner up to 3‐ and eightfold (Fig. 3C; Supporting Information Table S2, **** p < .0001). Moreover, the prosurvival influence exerted by hAFS‐EVHypo was significantly more effective than that observed with hAFS‐EVNormo (#### p < .0001).
Figure 3.
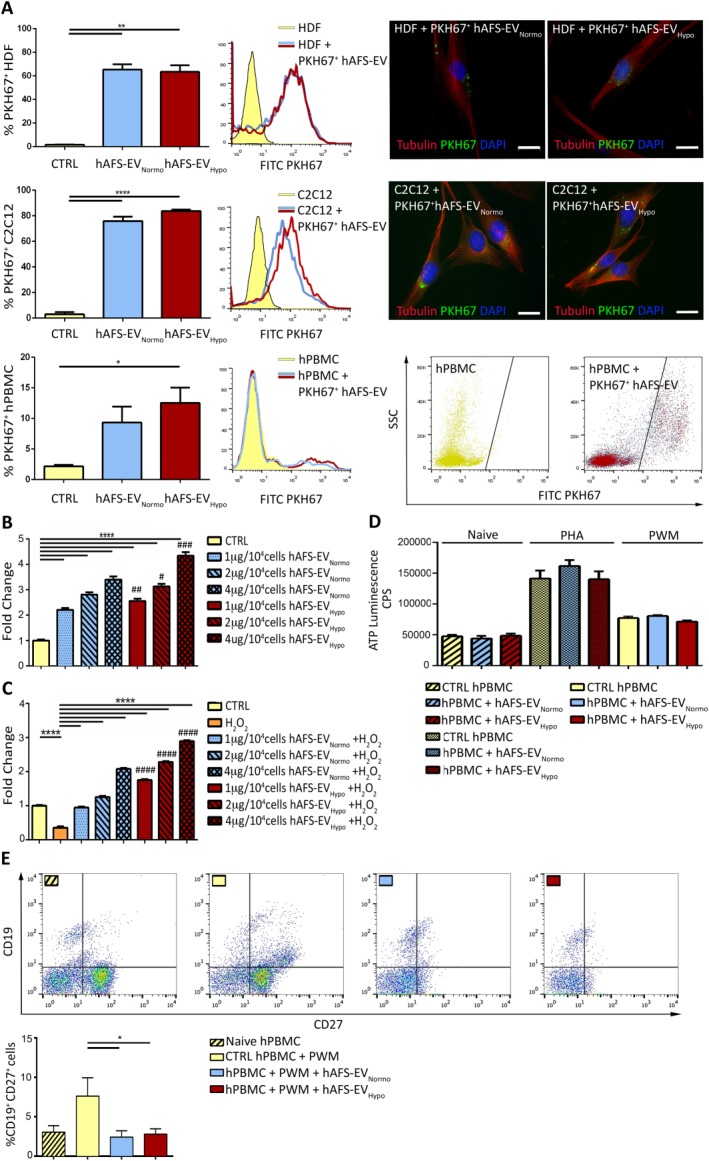
hAFS‐EV mediate paracrine effects on target cells. (A): PKH67+ hAFS‐EV uptake by HDF, C2C12, and hPBMC by FACS (values normalized to mode on the y‐axis) and immunostaining (scale bar: 20 µm). PKH67+ cells after treatment with hAFS‐EVNormo and hAFS‐EVHypo were: 65.4% ± 4.4% and 63.5% ± 5.6% HDF (**, p < .01, p = .0033 and p = .0036, respectively); 75.9% ± 3.4% and 83.7% ± 1.1% C2C12 (****, p < .0001); 9.3% ± 2.6% and 12.5% ± 2.5% hPBMC (*, p < .05, p = .017). (B): BrdU Enzyme‐Linked ImmunoSorbent Assay (ELISA) on HDF treated with 1 µg/104, 2 µg/104, and 4 µg/104 cells of hAFS‐EVNormo and hAFS‐EVHypo versus untreated cells (CTRL); ****, p < .0001; #, p referring to hAFS‐EVHypo versus hAFS‐EVNormo: #, p < .05 (p = .033); ##, p < .01 (p = .0077); ###, p < .001 (p = .0002). Values expressed as the fold change of sample absorbance read at 370 nm (reference wavelength approximately 492 nm). (C): MTT assay on C2C12 primed with 1 µg/104, 2 µg/104, and 4 µg/104 cells of hAFS‐EVNormo or hAFS‐EVHypo and exposed to oxidative stress versus untreated cells (CTRL) or damaged cells without hAFS‐EV stimulation (H2O2). ****, p < .0001; #, p referring to hAFS‐EVHypo versus hAFS‐EVNormo: ####, p < .0001. Values expressed as the fold change of the sample absorbance read at 560 and 670 nm. (D): hPBMC proliferation under unstimulated (Naive) and activating (PHA or PWM) conditions with 1 μg/105 cells of hAFS‐EVNormo and hAFS‐EVHypo versus untreated cells (CTRL hPBMC); CPS. (E): FACS analysis of mature CD19+CD27+ B cells treated with 1 μg/105 cells of hAFS‐EVNormo or hAFS‐EVHypo. Naive hPBMC: 3.48% ± 0.86%; CTRL hPBMC + PWM: 8.48% ± 2.63%; hPBMC + PWM + hAFS‐EVNormo: 2.32% ± 0.96%; hPBMC + PWM + hAFS‐EVHypo: 2.36% ± 0.62%. *, p < .05 (p = .045 and p = .047). Abbreviations: CD, cluster of differentiation; CPS, counts per second; DAPI, 4′,6‐diamidino‐2‐phenylindole; EV, extracellular vesicles; hAFS, human amniotic fluid stem cells; HDF, human dermal fibroblast; hPBMC, human peripheral blood mononuclear cell; Hypo, hypoxic; MVB, multivesicular body; Normo, normoxic; PHA, phytohemagglutinin; PWM, pokeweed mitogen; SF, serum‐free.
To understand the immunomodulatory potential of hAFS‐EV, hPBMC were stimulated by PHA 25 and PWM 26 in order to activate T‐ and B‐lymphocytes, followed by incubation with either hAFS‐EVNormo or hAFS‐EVHypo. The target cell response was evaluated by analyzing cell proliferation and selective maturation into T reg and memory B cells by the expression of CD4/CD25 (high) 37 and CD19/CD27 38, respectively. hAFS‐EV were internalized by hPBMC with lower efficiency compared to other target cells used in this study (Fig. 3A). Both hAFS‐EVNormo and hAFS‐EVHypo did not significantly affect hPBMC proliferation in control conditions (naïve hPBMC, p = .6417), or after stimulation (PHA and PWM, p = .3632 and p = .5120, Fig. 3D; Supporting Information Table S2). hAFS‐EV did not influence the polarization of T reg (Supporting Information Fig. S3A, p = .4616). However, both hAFS‐EVNormo and hAFS‐EVHypo exerted significant modulatory effect by reducing the maturation of CD27+CD19+ memory B cell in response to PWM (*p < .05, Fig. 3E).
A mouse Matrigel plug assay was used to determine the proangiogenic effect of hAFS‐EVNormo and hAFS‐EVHypo in vivo. Histological analysis and CD31 immunostaining revealed that in the hAFS‐EV‐loaded Matrigel plugs infiltrating host cells were detectable (Fig. 4A, 4D). Real time qRT‐PCR analysis suggested a certain degree of angiogenic potential of hAFS‐EV compared to vehicle‐loaded (PBS) control plugs, as shown by upregulation of the vascular marker Cdh5, Pecam1, and VegfA on the cells colonizing the plug (Fig. 4C), although not in a significant manner. Nevertheless, the evaluation of hemoglobin content showed a 2.6‐fold increase in the hAFS‐EVHypo‐treated plugs (33.73 ± 7.97 μg/μl) compared to the control (13.06 ± 1.04 μg/μl, *p < .05) and the hAFS‐EVNormo ones (14.63 ± 1.79 μg/μl, Fig. 4B). Hence, hAFS‐EVHypo were able to, at least to some extent, attract and stimulate host endothelial cells.
Figure 4.

hAFS‐EV exert a certain degree of angiogenic potential in a mouse Matrigel plug assay. (A): Representative images of Matrigel plugs loaded with 10 μg of hAFS‐EVNormo or hAFS‐EVHypo and recovered after 3 weeks from C57Bl/6J mice; scale bar 5 mm. (B): Evaluation of blood hemoglobin content on the Matrigel plugs loaded with 10 μg of hAFS‐EVNormo or hAFS‐EVHypo versus the PBS control *, p < .05 (p = .04). (C): Real time qRT‐PCR analysis on the recovered plugs for Cdh5, VegfA, and Pecam1 expression as normalized to B2M expression. (D): Representative images of histological evaluation of vessel formation within the Matrigel plug by infiltrating host cells via hematoxylin and eosin and CD31 staining, scale bar: 50 µm. CTRL: vehicle loaded (PBS) Matrigel plug; hAFS‐EVNormo: hAFS‐EVNormo loaded Matrigel plug; hAFS‐EVHypo: hAFS‐EVHypo loaded Matrigel plug. Abbreviations: CD, cluster of differentiation; EV, extracellular vesicles; hAFS, human amniotic fluid stem cells; Hypo, hypoxic; Normo, normoxic.
Regenerative Paracrine Potential of hAFS‐EV in a Mouse Model of Skeletal Muscle Atrophy
TA muscle fibers of HSA‐Cre, SmnF7/F7 mice treated with hAFS‐EVNormo and hAFS‐EVHypo showed remarkable decrease of immunoglobulin (IgG) and CD68+ cell infiltration compared to control animals, 1 and 7 days after injection (Fig. 5B). This effect correlated with a drastic reduction of damaged infiltrated fibers (approximately 80% and 89% in the first 24 hours after hAFS‐EVNormo and hAFS‐EVHypo administration, *p < .05, Fig. 5C). In mice treated with hAFS‐EVHypo, this effect was maintained up to 7 days after injection by a 79% decrease of the IgG‐damaged fibers (*p < .05, Fig. 5C). This trend was further confirmed by the evaluation of the percentage of the infiltrated fiber mean fluorescent area. Indeed, hAFS‐EVNormo and hAFS‐EVHypo treatments were equally effective in reducing the extension of the IgG‐infiltrated area within the muscle fibers by 91.5% and 95%, respectively (**p < .01) at day 1 post injection, and by 80.25% and 91% at day 7 post injection (**p < .01, Fig. 5C). Compared to controls, the number of central nucleated TA fibers was significantly reduced by 72% 1 day after receiving intramuscular injection of hAFS‐EVHypo (*p < .05, Fig. 5D). This effect was no longer observed after 7 days, although hAFS‐EVHypo maintained a significant lower amount of central nucleated fibers compared to hAFS‐EVNormo (71% less, # p < .05, Fig. 5D). Real time qRT‐PCR of the ratio between the inflammatory markers Nos2 and Arg1 39 showed a substantial decrease in the activation of proinflammatory M1 macrophages (ratio Nos2/Arg1>1), in favor of pro‐resolving M2 phenotype (ratio Nos2/Arg1<1). After 1 day, both hAFS‐EVNormo and hAFS‐EVHypo induced macrophage polarization by decreasing the Nos2/Arg1 ratio by approximately fourfold compared to untreated animals (*p < .05, Fig. 5E). This effect began to decrease 7 days after hAFS‐EV injection. Proinflammatory cytokines followed a similar pattern of expression. Following hAFS‐EV injection, the expression of the proinflammatory Il‐1α 40 decreased by almost 50% at 1 day (***p < .001) and 7 days (*p < .05) after treatment, with similar effects between hAFS‐EVNormo and hAFS‐EVHypo (Fig. 5F). Likewise, Il‐6 41 levels dropped significantly by 87% and 65% (***p < .001 and **p < .01, respectively) within the first 24 hours after hAFS‐EV administration. hAFS‐EVHypo was more effective (*p < .05) as maintaining this modulation for up to 7 days after treatment (Supporting Information Fig. S4A). Proinflammatory Il‐4 42 expression decreased by 37% 1 day after hAFS‐EVHypo treatment (*p < .05). At the same time, within the first 24 hours after treatment, there was a significant upregulation of the pro‐resolving Il‐10, typically released by M2 macrophages 43 (1.6‐fold, **p < .01 and 2.8‐fold, ****p < .0001, respectively) with hypoxic EV exerting a stronger effect compared to normoxic ones (1.8‐fold increase, ****p < .0001, Fig. 5F). This trend was maintained also after a few days, although without reaching statistical significance. hAFS‐EV were also able to modulate the expression of the Pstg2 gene, encoding for the COX2 enzyme, in host cells. hAFS‐EVNormo and the hAFS‐EVHypo induced a significant down regulation of Pstg2 in mice 1 day (67% and 56%, respectively, **p < .01 and *p < .05), and 7 days (86% and 87%, respectively, ****p < .0001, Supporting Information Fig. S4A) after treatment.
MicroRNA Profiling of the hAFS‐EV
hAFS‐EVNormo and hAFS‐EVHypo showed to contain small non‐coding RNAs and miRNAs (20–40 nucleotides, Fig. 6A). In particular, hAFS‐EVHypo showed significant enrichment over the hAFS‐EVNormo of the following miRNAs (Fig. 6B): miR‐223 (***p < .001, 1.3‐fold), miR‐146a (**p < .01, 1.6‐fold), miR‐let7c (***p < .001, twofold), miR‐21 (***p < .001, twofold), miR‐126 (****p < .0001, 2.5‐fold), miR‐146b (****p < .0001, almost threefold), miR‐199a‐3p (**p < .01, almost threefold), miR‐210 (****p < .0001, 3.6‐fold).
Figure 6.
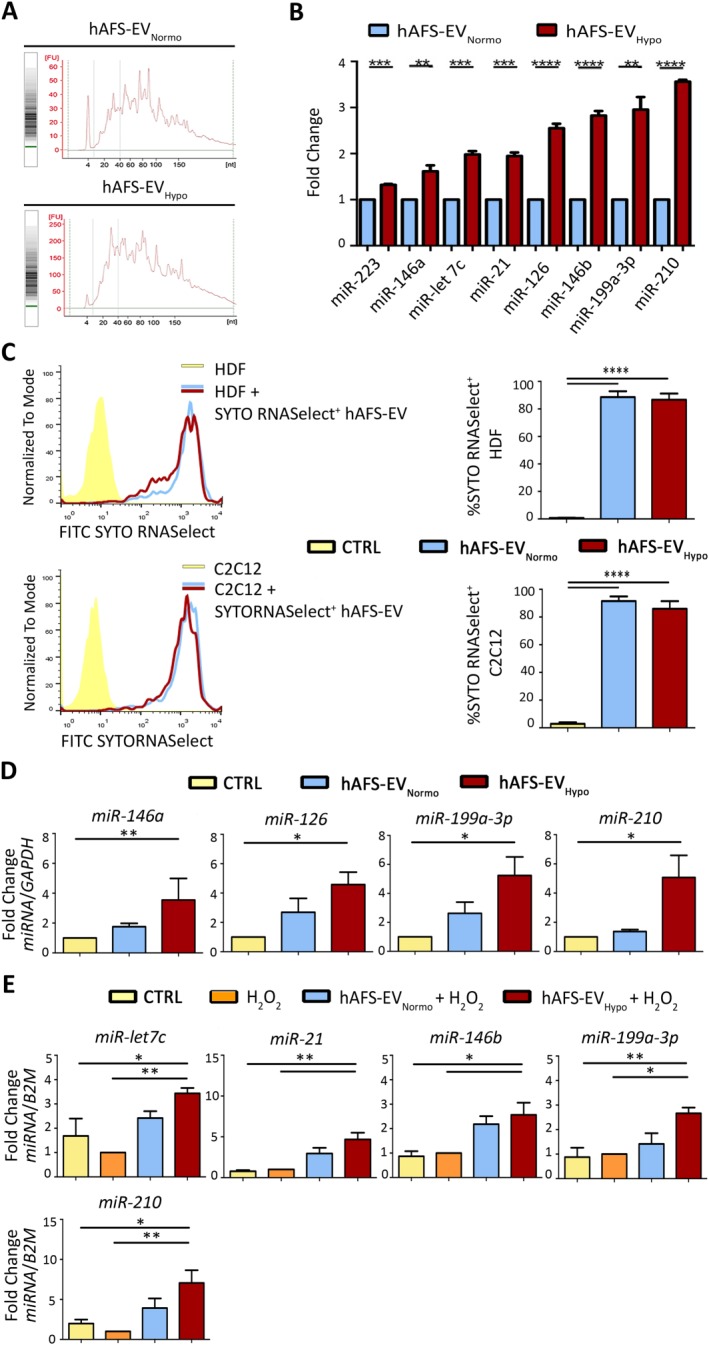
hAFS‐EV act as biological carrier of regenerative miRNAs which are significantly increased in the target cells following direct uptake. (A): Analysis of hAFS‐EV small non‐coding RNA content by Agilent technology. (B): Real time qRT‐PCR of hAFS‐EVHypo versus hAFS‐EVNormo showing significant enrichment of the following regenerative miRNAs: miR‐223 (***, p < .001, p = .0001), miR‐146a (**, p < .01, p = .009), miR‐let7c (***, p < .001, p = .0002), miR‐21 (***, p < .001, p = .0003), miR‐199a‐3p (**, p < .01, p = .002), miR‐126, miR‐146b, and miR‐210 (****, p < .0001). (C): Uptake analysis of hAFS‐EV fluorescently labeled RNA by HDF (upper panel) and C2C12 (lower panel) via FACS (on the left representative histogram graphs with corresponding values in the graphs on the right). 88.67% ± 4.21% and 86.73% ± 4.46% of HDF (****, p < .0001) and 91.5% ± 3.38% and 85.97% ± 5.51% of C2C12 cells (****, p < .0001) became fluorescent following incubation with 1 µg/104 cells of SYTO®RNASelect+ hAFS‐EVNormo and hAFS‐EVHypo, showing direct uptake of the marked RNA cargo. (D): Real time qRT‐PCR analysis for the enrichment of specific hAFS‐EV‐delivered miRNAs in the target proliferative HDF: miRNA‐146a, **, p < .01, p = .006; miRNA‐126 and miR‐199a‐3p (*, p < .05, p = .03), miR‐210 (*, p < .05, p = .04). (E): Real time qRT‐PCR analysis for the enrichment of specific hAFS‐EV‐delivered miRNAs in the target C2C12 with or without H2O2 treatment: miR‐let7c (**, p < .01, p = .005 and *, p < .05, p = .04); miR‐21 (**, p < .01, p = .002 and p = .001, respectively); miRNA‐146b (*, p < .05, p = .020 and p = .012); miRNA‐199a‐3p (*, p < .05, p = .013 and **, p < .01 p = .008); and miRNA‐210 (**, p < .01, p = .006 and *, p < .05 p = .02). Gene expression was normalized to human GAPDH and to mouse B2M as internal controls in the HDF and C2C12 cells, respectively. Abbreviations: EV, extracellular vesicles; FITC, fluorescein isothiocyanate; hAFS, human amniotic fluid stem cells; HDF, human dermal fibroblast; Hypo, hypoxic; Normo, normoxic.
Mechanism Insights of the hAFS‐EV Regenerative Potential via Direct Transfer of Exosomal miRNAs Into the Target Cells
To assess whether direct transfer of their nucleic acid cargo could drive the hAFS‐EV paracrine effect, we analyzed the uptake of fluorescently labeled exosomal RNA by target cells in vitro. Following incubation with SYTO®RNASelect+ hAFS‐EVNormo and SYTO®RNASelect+ hAFS‐EVHypo, we observed acquisition of fluorescence by 88.67% ± 4.21% and 86.73% ± 4.46% of HDF cells (****p < .0001 compared to untreated cells), and by 91.5% ± 3.38% and 85.97% ± 5.51% of C2C12 cells (****p < .0001 compared to untreated ones), respectively (Fig. 6C). This result indicates that hAFS‐EV can actively release their RNA content into the target cells in the very short time. Real time qRT‐PCR analysis on the responder HDF and C2C12 cells revealed significant enrichment of some of the eight regenerative miRNAs carried by hAFS‐EV (Fig. 6D, 6E). Following incubation with hAFS‐EVHypo, HDF cells showed a significant enrichment of four out of the eight miRNAs analyzed (Fig. 6D): miR‐146a (3.5‐fold, **p < .01), miR‐126 (4.6‐fold, *p < .05), miR‐199a‐3p (5.2‐fold, *p < .05) and miR‐210 (5.1‐fold, *p < .05); all remaining miRNAs (miR‐223, miR‐let7c, miR‐21, miR‐146b) were enhanced in the cells primed with the hAFS‐EV, although this increase was not significant (Supporting Information Fig. S5A). C2C12 cells primed with the hAFS‐EVHypo and exposed to H2O2, when compared to untreated cells, with or without oxidative damage, revealed higher amount of miR‐let7c (3.4‐ and 2.0‐fold, **p < .01 and *p < .05, respectively), miR‐21 (4.7‐and 6.0‐fold, **p < .01), miR‐146b (2.6‐ and 2.9‐fold, *p < .05), miR‐199a‐3p (2.7‐ and 3.0‐fold, *p < .05 and **p < .01), and miR‐210 (7.0‐ and 3.5‐fold, **p < .01 and *p < .05, respectively). Similar to HDF, all other miRNAs (miR‐223, miR‐146a, miR‐126) were enhanced in the cells primed with the hAFS‐EV, although this increase was not significant (Supporting Information Fig. S5B). Since miR‐210 was one of the mostly enriched miRNA following hAFS‐EVHypo uptake, we evaluated whether its target genes were modulated. Expression of MNT, an antagonist of the cell‐cycle promoter c‐MYC 44, and Casp8ap2, a proapoptotic gene 45, was analyzed in HDF and C2C12 cells, respectively, following treatment with hAFS‐EVHypo. While HDF cells reduced MNT expression by almost 60%, this decrease was not statistically significant. On the other hand, C2C12 cells were able to significantly decrease Casp8ap2 expression of approximately 83% (*p < .05, Supporting Information Fig. S5C), altogether supporting a substantial, functional role of the transferred miR‐210 in mediating antiapoptotic effect.
Proteomic Profiling of the hAFS‐EV
The proteomic characterization of both hAFS‐EVNormo and hAFS‐EVHypo allowed the identification of 498 proteins, some of which (74 out of 100) among those most expressed by exosomes, according to ExoCarta (http://www.exocarta.org, a dedicated database of exosomal proteins, RNA and lipids 46). Eighty‐four proteins out of the total 498 were enriched within the hAFS‐EVHypo compared to the hAFS‐EVNormo and significantly involved in biological pathways (****p < .0001, Supporting Information Fig. S6A) including metabolism of protein and RNA, including mRNA, gene expression and translation, metabolism and peptide chain elongation.
Discussion
Recently, different studies have provided significant insights into stem cell paracrine‐mediated repair as a new strategy for tissue regeneration 2. In addition to soluble molecules, cell‐released EV are emerging as potential candidates for future therapeutic approaches for a number of pathological conditions. Indeed, they have been demonstrated to play a pivotal role in the intercellular crosstalk underlying stem cell paracrine potential through the promotion of proliferation, survival, angiogenesis, and immunosuppression, just to name a few 47, 48, 49.
Here, for the first time, we provide robust evidence that hAFS dynamically secrete EV as functional mediators of proliferative, antiapoptotic, immunomodulatory, proangiogenic, and anti‐inflammatory effects.
Structural analyses revealed that hAFS actively release immunologically inert EV, as similarly indicated for other MSC‐EV 50, which are heterogeneous in size. These include nanosized exosomal particles identified by the expression of TSG101, ALIX, CD81, CD9, AnnV, and CD63 [5, 34], along with cell specific markers such as CD105. Similar to other stem cells 51, hAFS are likely to originate from a hypoxic niche within the amniotic fluid, and they demonstrated remarkable antiapoptotic effects when transplanted in an ischemia/reperfusion injury rat model 19. Therefore, we applied an in vitro 24 hours hypoxic preconditioning to enrich the cell secretome with regenerative EV. Consistent with previous observations on MSC‐EV 9, 27, 52, we observed that hypoxia stimulation did not alter hAFS phenotype or stemness, as confirmed by NANOG and SSEA4 expression, and it significantly enriched the exosomal fraction within the hAFS‐EV, whereby the total amount of secreted particles found in hAFS‐EVNormo and hAFS‐EVHypo were comparable. hAFS promptly reacted to the hypoxic environment by upregulating HIF‐1α, which has been previously associated to exosome release in both cardiomyocytes and cancer cells 52, 53.
Uptake analysis confirmed the role of hAFS‐EV as biological mediators of intercellular communication via rapid incorporation by the target cells. hAFS‐EVHypo in particular showed superior paracrine influence in mediating both proliferative and prosurvival effects against oxidative stress on human fibroblasts and murine myoblast cells, compared to hAFS‐EVNormo. Our results are coherent with previous findings, which reported that following stimulation with hypoxic CM from human amniotic fluid‐derived MSC, improvement in wound healing was described to occur via the enhancement of fibroblast activation 54. Data presented herein on the antiapoptotic effect during oxidative stress integrate and support our earlier results on the prosurvival potential of the hAFS‐CM 19 and endorse the idea that the hAFS secretome is a valuable source of cytoprotective factors. Nevertheless, those previous findings relied on indirect mechanisms based on soluble chemokines, cytokines and/or growth factors as the main players of therapeutic effects: here we report that EV may be another strategic component of their paracrine potential.
Since mild hypoxic priming was shown to sustain hAFS endothelial regenerative potential in rescuing blood flow in a rodent model of chronic hind‐limb ischemia and carotid injury 18, here we evaluated if hAFS‐EV could induce an angiogenic response in a Matrigel plug assay in vivo. Despite the fact that differences among control and hAFS‐EV treated groups were not significant in terms of expression of the angiogenic genes Cdh5, Pecam1, and VegfA from the host cells colonizing the plug, a positive trend was noticeable. Nonetheless, hAFS‐EVHypo demonstrated a substantial potential in triggering the maturation of new resident vessels as revealed by the evaluation of the blood hemoglobin content within the treated plugs. While the angiogenic potential of stem cell‐EV has been broadly investigated in vitro by several groups using capillary‐forming assay or proliferation of HUVEC 9, 32, 55 or other endothelial cells 27, at present very little has been reported using an in vivo Matrigel plug assay, with a rare exception 56. In our settings, hAFS‐EVHypo showed some degree of angiogenic potential, which might have been limited by dosage. Indeed, the only study reporting robust vascular potential of MSC‐EV in a Matrigel plug assay, used a much higher dose of EV (100 μg per plug, 10 times more than that used herein) 56. Moreover, we cannot exclude that the angiogenic potential of the hAFS secretome may reside within the soluble factor content (i.e., the whole of CM) rather than in the secreted vesicles.
Considering that MSC are well known for their immunomodulatory properties, we also investigated whether EV alone can recapitulate the paracrine potential exerted by hAFS in inhibiting the proliferation of allogeneic human PBMC and lymphocyte activation 25, 57, 58. hAFS‐EV did not affect proliferation of human PBMC under resting or challenged conditions and showed to be up taken with lower efficiency compared to other target cells. Moreover, we could not detect any alteration in the maturation of CD4+CD25high regulatory T reg by the hAFS‐EV. However, both normoxic and hypoxic hAFS‐EV equally mediated a significant reduction in B‐cell maturation. The immunomodulation competence of MSC‐CM in regulating T cell proliferation is well established as extensively reviewed in 59, 60. However, the potential of EV to recapitulate this feature is still debatable, since a general consensus has not been reached. Several reports have shown that MSC‐EV can induce tolerogenic signaling by inhibiting syngeneic and allogeneic T lymphocyte proliferation, while supporting T reg expansion 61, 62 and attenuating infiltration of neutrophils in a cardiac ischemia/reperfusion mouse model 7, or by enhancing the survival of allogeneic skin graft in mice 63. Yet, other studies revealed that MSC‐EV possess a lower immune‐modulatory effect on T cells in vitro compared to their cellular counterpart 64, while, on the other hand, they mediate significant immunosuppressive effects on memory B cell maturation 65. Of note, an independent study has recently shown that the immunoregulatory properties of hAFS vary according to gestational age, with first trimester hAFS exerting stronger inhibitory effects on activated T‐cell proliferation than less effective second and third trimester cells, which need to be primed with specific inflammatory stimuli Tumor Necrosis Factor‐alpha; Interferon‐gamma (TNF‐α, INF‐γ) to block B cell proliferation 57. Hence, further investigations on hAFS‐EV based on gestational age could help better understanding these mechanisms. As well, we cannot exclude whether increasing the hAFS‐EV dosage could also provide a substantial effect on both hPBMC proliferation and T reg activation, besides effectively targeting B cells.
Our in vitro findings were further validated in vivo in a preclinical model of skeletal muscle atrophy. HSA‐Cre, SmnF7/F7 mice have the exon 7 in the Smn deleted only in skeletal myofibers with Cre recombinase under the promoter of the human skeletal actin gene (HSA‐Cre). These mice have a high content of central nucleated fibers, leading to necrosis and infiltration of inflammatory cells, muscle degeneration, and impaired satellite cell potential 66. Since systemic administration of murine AFS in HSA‐Cre, SmnF7/F7 mice showed to enhance muscle strength, and support long‐term regeneration following injury 22, here we evaluated whether hAFS‐EV have a paracrine role in this disease model.
Adult bone marrow MSC‐derived exosomes recently showed to promote tissue regeneration by sustaining local angiogenesis and myogenesis in a skeletal muscle cardiotoxin injury model 67; here, for the first time, we report that fetal hAFS‐EV were effective in quenching muscle inflammation in a chronic inflammatory environment with genetic background. In some cases, hAFS‐EVHypo were more efficient than normoxic EV. Indeed hAFS‐EV reduced the extension of immunoglobulin infiltration within the muscle fibers, hence counteracting the development of necrotic fibers and modulated the expression of inflammatory (Il‐1α, Il‐6, Il‐4) and pro‐resolving (Il‐10) cytokines. Similarly, the exhaustion of the resident stem resources caused by the prolonged pathological activation of the satellite niche was considerably counteracted within the first 24 hours from treatment with a reduced amount of central nucleated fibers. Notably, hAFS‐EV were also able to modulate the expression of the Ptgs2 gene encoding for the proinflammatory enzyme COX2, which has been linked to muscle wasting and represents a potential therapeutic target for skeletal muscle atrophy 68. This is in line with the results of a previous study showing that rat AFS injected in a model of necrotizing enterocolitis exerted specific paracrine potential by acting on the host COX2‐expressing stromal cells 21. However, some of the beneficial effects revealed here, such balanced levels between proinflammatory M1 and pro‐resolving M2 phenotype and the modulation of some interleukins were reduced after 7 days. These findings are in accordance with a general consensus that stem cell paracrine effect is usually a prompt and short‐term response. Moreover, in our mouse model of genetically induced muscle atrophy both hAFS‐EVHypo and hAFS‐EVNormo were equally effective in delivering most of the regenerative paracrine effects described. Yet, we report a significantly greater ability of hAFS‐EVHypo to modulate the inflammatory cytokines, Il‐10, Il‐4, and Il‐6, when compared to hAFS‐EVNormo. In light of these considerations, further evaluation is needed to determine whether either more frequent or higher‐dose administration of hAFS‐EVHypo can boost their paracrine potential and maintain it over time.
Recent reports have highlighted the specific role of stem cell‐EV as functional cargo for the horizontal transfer of genetic information, such as mRNAs and small non‐coding RNAs, including miRNAs 69, 70. We revealed that hAFS‐EV are endowed with regenerative miRNAs supporting several key biological effects, such as proliferation of fibroblasts and other differentiated cells (miR‐210 71, miR‐199a‐3p 72); angiogenesis (miR‐210 [73, 74] and miR‐126 [75, 76, 41], the so called “angio‐miRNAs” 77); inhibition of fibrosis (miR‐let7c 78); cardioprotection (miR‐210 [32, 79], miR‐146a 32, 80), and prosurvival effect via the activation of the ERK1/2‐Stat3 antiapoptotic signaling pathway (miR‐21 79); pro‐resolving activity by decreasing the levels of proinflammatory proteins like RANTES (miR‐146b 81) or PDCD4 (miR‐21 [82, 83]) and macrophage polarization toward a pro‐resolving, M2 phenotype (miR‐223 84). Coherent with our results on the paracrine effects elicited by hAFS‐EVHypo, hypoxic preconditioning led to a significant enrichment of miR‐210 (a consistent feature of hypoxia stimulation 85), miR‐199a‐3p, miR‐146b, miR‐126, miR‐21, and miR‐let7c.
Considering that miRNA cargo is a distinctive feature of exosomes, this evidence further strengthens the hypothesis that hypoxic preconditioning might represent an ideal strategy to significantly enrich hAFS‐EV with regenerative exosomes.
Since the results of our comparative proteomic did not identify any exclusive paracrine candidate molecule, we focused on the in vitro analysis of miRNA horizontal transfer into the target cells, to provide some mechanistic insights on the regenerative effects mediated by the hAFS‐EV. Indeed, we verified direct RNA trafficking from hAFS‐EV to responder cells via the labeling of C2C12 and HDF incorporating the fluorescently marked exosomal RNA cargo. Additionally, target cells showed significant, higher enrichment of some of the eight regenerative miRNAs mostly expressed by hypoxic hAFS‐EV. In particular, miR‐210 and miR‐199a‐3p, in association with others differentially enriched within C2C12 (miR‐let7c, miR‐21, and miR‐146b) or HDF (miR‐146a and miR‐126), were significantly and promptly enhanced following uptake of hAFS‐EVHypo over hAFS‐EVNormo. While we cannot ignore a distinct function of each miRNA in contributing to the beneficial paracrine effects mediated by the hAFS‐EV, we may have identified a putative regenerative signature with miR‐210 and miR‐199a‐3p pair. Indeed, the functional role of hypoxic miR‐210 in mediating hAFS‐EVHypo‐driven prosurvival effects was reinforced by a significant decrease of expression of its target gene Casp8ap2 in the responder C2C12 cells.
Therefore, we identified a candidate mechanism of action underlying hAFS‐EV paracrine effects by the specific transfer of hypoxic‐stimulated miRNAs. Further experiments, which are beyond the scope of this initial characterization, are needed to fully elucidate the specific role of single or combination of hAFS‐EVHypo miRNAs in addressing the different beneficial effects described here.
Conclusion
While functional analyses of microvesicles and exosomes secreted by both adult and perinatal stem cells (such as placental 48, amniotic 86, 87, and Wharton's jelly 88 MSC), have been reported, little is known about hAFS‐EV. To the best of our knowledge this is the first study that provides an initial description of the paracrine potential of second trimester c‐KIT+ hAFS‐EV by the horizontal transfer of specific miRNAs.
Although there are some aspects that need additional attention to make the EV dose‐effect response more efficient in vivo and to harness the full potential of the hAFS‐EVHypo, we believe that our results still offer meaningful insights on the characterization of the fetal stem cell secretome for future therapy.
In order to progress toward the clinical application and to exploit next generation stem cell‐EV therapy, the ideal stem cell source should be selected upon consideration of their paracrine potential and the feasibility of their isolation, together with their in vitro self‐renewal properties. hAFS unambiguously fulfill all criteria, since they can be easily isolated from left over samples of amniotic fluid, either during pregnancy via amniocentesis (fetal hAFS) or at term, during scheduled cesarean delivery as waste clinical material (perinatal hAFS). Their remarkable self‐renewal potential, along with the lack of ethical concern, ease of isolation, expansion, and cryopreservation, make them an optimal candidate over adult MSC, iPS, and embryonic stem cells as source for future paracrine therapy.
Author Contributions
C.B.: conception and design; collection and assembly of data; data analysis and interpretation; manuscript writing; final approval of manuscript; M. Piccoli: conception and design; collection and assembly of data; data analysis and interpretation; manuscript writing; L.B.: conception and design of some experiments; collection and assembly of data; A.P.: collection and assembly of data; data analysis and interpretation; A.A., E.P., D.R., and P.B.: collection and assembly of data; L.P.: collection and assembly of data; data analysis and interpretation; L.V.: provision of equipment; M.M., D.C., and R.C.: provision of study material; T.B.: provision of study material and financial support; M. Pozzobon: data analysis and interpretation; S.B: conception and design; financial support; data analysis and interpretation; manuscript writing; final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
This research was supported by Programma Giovani Ricercatori “Rita Levi Montalcini” 2012 from MIUR ‐ Italian Ministry of Education and Research (S.B.). M. Pozzobon is funded by University of Padova, Assegno Senior 2015, Medical Sciences area 07 and by Fondazione Istituto di Ricerca Pediatrica Città della Speranza, Padova, Italy; M. Piccoli is funded by Fondazione Istituto di Ricerca Pediatrica Città della Speranza, Padova, Italy. We also thank Dr. Renata Bocciardi, Prof. Roberto Ravazzolo (Medical Genetics Laboratory, IRCCS Giannina Gaslini, Genova, Italy), Dr. Angela Laporta, Prof. Elena Cattaneo (Department of Biosciences, University of Milan, Milan, Italy), and Dr. Alessandro Poggi (IRCCS AOU San Martino – IST) for kindly providing HDF cells, human ES cDNA and reagents for the hPBMC culture and analysis, respectively. Finally, the authors thank Dr. Antonietta Silini from Centro di Ricerca “E. Menni,” Fondazione Poliambulanza Istituto Ospedaliero in Brescia, Italy, for critical reading of the manuscript and help with editorial comments. Part of the Supporting Information Figure S1 and the Graphical Abstract have been produced using Servier Medical Art (www.servier.com).
References
- 1. Gnecchi M, Zhang Z, Ni A et al. Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res 2008;103:1204–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mirotsou M, Jayawardena TM, Schmeckpeper J et al. Paracrine mechanisms of stem cell reparative and regenerative actions in the heart. J Mol Cell Cardiol 2011;50:280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bollini S, Gentili C, Tasso R et al. The regenerative role of the fetal and adult stem cell secretome. J Clin Med J Clin Med 2013;2:302–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tetta C, Ghigo E, Silengo L et al. Extracellular vesicles as an emerging mechanism of cell‐to‐cell communication. Endocrine 2012;44:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lötvall J, Hill AF, Hochberg F et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014;3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang B, Yeo R, Tan K et al. Focus on extracellular vesicles: Therapeutic potential of stem cell‐derived extracellular vesicles. Int J Mol Sci 2016;17:pii: E174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arslan F, Lai RC, Smeets MB et al. Mesenchymal stem cell‐derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res 2013;10:301–312. [DOI] [PubMed] [Google Scholar]
- 8. Lai RC, Arslan F, Lee MM et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res 2010;4:214–222. [DOI] [PubMed] [Google Scholar]
- 9. Bian S, Zhang L, Duan L et al. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J Mol Med (Berl) 2014;92:387–397. [DOI] [PubMed] [Google Scholar]
- 10. Xu W, Qian H, Zhang B et al. HucMSC‐exosome mediated‐Wnt4 signaling is required for cutaneous wound healing. Stem Cells 2015;33:2158–2168. [DOI] [PubMed] [Google Scholar]
- 11. Kordelas L, Rebmann V, Ludwig A‐K et al. MSC‐derived exosomes: A novel tool to treat therapy‐refractory graft‐versus‐host disease. Leukemia 2014;28:970–973. [DOI] [PubMed] [Google Scholar]
- 12. Bruno S, Grange C, Collino F et al. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One 2012;7:e33115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan CY, Lai RC, Wong W et al. Mesenchymal stem cell‐derived exosomes promote hepatic regeneration in drug‐induced liver injury models. Stem Cell Res Ther 2014;5:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xin H, Li Y, Buller B et al. Exosome‐mediated transfer of miR‐133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012;30:1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bobis‐Wozowicz S, Kmiotek K, Sekula M et al. Human induced pluripotent stem cell‐derived microvesicles transmit RNAs and proteins to recipient mature heart cells modulating cell fate and behavior. Stem Cells 2015;33:2748–2761. [DOI] [PubMed] [Google Scholar]
- 16. De Coppi P, BartschG Jr., Siddiqui MM et al. Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol 2007;25:100–106. [DOI] [PubMed] [Google Scholar]
- 17. Pozzobon M, Piccoli M, Schiavo AA et al. Isolation of c‐Kit+ human amniotic fluid stem cells from second trimester. Methods Mol Biol 2013;1035:191–198. [DOI] [PubMed] [Google Scholar]
- 18. Schiavo AA, Franzin C, Albiero M et al. Endothelial properties of third‐trimester amniotic fluid stem cells cultured in hypoxia. Stem Cell Res Ther 2015;6:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bollini S, Cheung KK, Riegler J et al. Amniotic fluid stem cells are cardioprotective following acute myocardial infarction. Stem Cells Dev 2011;20:1985–1994. [DOI] [PubMed] [Google Scholar]
- 20. Smart N, Bollini S, Dubé KN et al. De novo cardiomyocytes from within the activated adult heart after injury 2011. Nature 2011;474:640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zani A, Cananzi M, Fascetti‐Leon F et al. Amniotic fluid stem cells improve survival and enhance repair of damaged intestine in necrotising enterocolitis via a COX‐2 dependent mechanism. Gut 2014;63:300–309. [DOI] [PubMed] [Google Scholar]
- 22. Piccoli M, Franzin C, Bertin E et al. Amniotic fluid stem cells restore the muscle cell niche in a HSA‐Cre, SmnF7/F7 mouse model. Stem Cells 2012;30:1675–1684. [DOI] [PubMed] [Google Scholar]
- 23. Mirabella T, Hartinger J, Lorandi C et al. Proangiogenic soluble factors from amniotic fluid stem cells mediate the recruitment of endothelial progenitors in a model of ischemic fasciocutaneous flap. Stem Cells Dev 2012;21:2179–2188. [DOI] [PubMed] [Google Scholar]
- 24. Mirabella T, Cilli M, Carlone S et al. Amniotic liquid derived stem cells as reservoir of secreted angiogenic factors capable of stimulating neo‐arteriogenesis in an ischemic model. Biomaterials 2011;32:3689–3699. [DOI] [PubMed] [Google Scholar]
- 25. Mirabella T, Poggi A, Scaranari M et al. Recruitment of host's progenitor cells to sites of human amniotic fluid stem cells implantation. Biomaterials 2011;32:4218–4227. [DOI] [PubMed] [Google Scholar]
- 26. Bekeredjian‐Ding I, Foermer S, Kirschning CJ et al. Poke weed mitogen requires Toll‐like receptor ligands for proliferative activity in human and murine B lymphocytes. PLoS One 2012;7:e29806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang H‐C, Liu X‐B, Huang S et al. Microvesicles derived from human umbilical cord mesenchymal stem cells stimulated by hypoxia promote angiogenesis both in vitro and in vivo. Stem Cells Dev 2012;21:3289–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ayers L, Nieuwland R, Kohler M et al. Dynamic microvesicle release and clearance within the cardiovascular system: Triggers and mechanisms. Clin Sci (Lond) 2015;129:915–931. [DOI] [PubMed] [Google Scholar]
- 29. Gray WD, French KM, Ghosh‐Choudhary S et al. Identification of therapeutic covariant microRNA clusters in hypoxia‐treated cardiac progenitor cell exosomes using systems biology. Circ Res 2015;116:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bottai D, Cigognini D, Nicora E et al. Third trimester amniotic fluid cells with the capacity to develop neural phenotypes and with heterogeneity among sub‐populations. Restor Neurol Neurosci 2012;30:55–68. [DOI] [PubMed] [Google Scholar]
- 31. Chiavegato A, Bollini S, Pozzobon M et al. Human amniotic fluid‐derived stem cells are rejected after transplantation in the myocardium of normal, ischemic, immuno‐suppressed or immuno‐deficient rat 2006 J Mol Cell Cardiol 2007;42:746–759. [DOI] [PubMed] [Google Scholar]
- 32. Barile L, Lionetti V, Cervio E et al. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res 2014;103:530–541. [DOI] [PubMed] [Google Scholar]
- 33. Caradec J, Kharmate G, Hosseini‐Beheshti E et al. Reproducibility and efficiency of serum‐derived exosome extraction methods. Clin Biochem 2014;47:1286–1292. [DOI] [PubMed] [Google Scholar]
- 34. Connolly KD, Guschina IA, Yeung V et al. Characterisation of adipocyte‐derived extracellular vesicles released pre‐ and post‐adipogenesis. J Extracell Vesicles 2015;24;4:29159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grisendi G, Finetti E, Manganaro D et al. Detection of microparticles from human red blood cells by multiparametric flow cytometry. Blood Transfus 2015;13:274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Judge TA, Wu Z, Zheng XG et al. The role of CD80, CD86, and CTLA4 in alloimmune responses and the induction of long‐term allograft survival. J Immunol 1999;162:1947–1951. [PubMed] [Google Scholar]
- 37. Mareschi K, Castiglia S, Sanavio F et al. Immunoregulatory effects on T lymphocytes by human mesenchymal stromal cells isolated from bone marrow, amniotic fluid, and placenta. Exp Hematol 2016;44:138–150. [DOI] [PubMed] [Google Scholar]
- 38. de Masson A, Le Buanec H, Bouaziz J‐D. Purification and immunophenotypic characterization of human B cells with regulatory functions. Methods Mol Biol 2014;1190:45–52. [DOI] [PubMed] [Google Scholar]
- 39. Heredia JE, Mukundan L, Chen FM et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013;153:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shamim EA, Rider LG, Miller FW. Update on the genetics of the idiopathic inflammatory myopathies. Curr Opin Rheumatol 2000;12:482–491. [DOI] [PubMed] [Google Scholar]
- 41. Fan J, Kou X, Yang Y et al. MircoRNA‐regulated proinflammatory cytokines in sarcopenia. Mediators Inflamm 2016;2016:1438686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deyhle MR, Gier AM, Evans KC et al. Skeletal muscle inflammation following repeated bouts of lengthening contractions in humans. Front Physiol 2016;6:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. AJP Regul Integr Comp Physiol 2010;298:R1173–R1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Z, Sun H, Dai H et al. MicroRNA miR‐210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009;8:2756–2768. [DOI] [PubMed] [Google Scholar]
- 45. Won Kim H, Haider HK, Jiang S et al. Ischemic preconditioning augments survival of stem cells via miR‐210 expression by targeting Caspase‐8‐associated protein 2. J Biol Chem 2009;284:33161–33168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keerthikumar S, Chisanga D, Ariyaratne D et al. ExoCarta: A web‐based compendium of exosomal cargo. J Mol Biol 2016;428:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rani S, Ryan AE, Griffin MD et al. Mesenchymal stem cell‐derived extracellular vesicles: Toward cell‐free therapeutic applications. Mol Ther 2015;23:812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tetta C, Consiglio AL, Bruno S et al. The role of microvesicles derived from mesenchymal stem cells in tissue regeneration; A dream for tendon repair? Muscles Ligaments Tendons J 2012;2:212–221. [PMC free article] [PubMed] [Google Scholar]
- 49. Camussi G, Deregibus MC, Cantaluppi V. Role of stem‐cell‐derived microvesicles in the paracrine action of stem cells. Biochem Soc Trans 2013;41:283–287. [DOI] [PubMed] [Google Scholar]
- 50. EL Andaloussi S, Mäger I, Breakefield XO et al. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat Rev Drug Discov 2013;12:347–357. [DOI] [PubMed] [Google Scholar]
- 51. Mohyeldin A, Garzón‐Muvdi T, Quiñones‐Hinojosa A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010;7:150–161. [DOI] [PubMed] [Google Scholar]
- 52. Salomon C, Ryan J, Sobrevia L et al. Exosomal signaling during hypoxia mediates microvascular endothelial cell migration and vasculogenesis. PLoS One 2013;8:e68451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu X, Deng L, Wang D et al. Mechanism of TNF‐α autocrine effects in hypoxic cardiomyocytes: Initiated by hypoxia inducible factor 1α, presented by exosomes. J Mol Cell Cardiol 2012;53:848–857. [DOI] [PubMed] [Google Scholar]
- 54. Jun EK, Zhang Q, Yoon BS et al. Hypoxic conditioned medium from human amniotic fluid‐derived mesenchymal stem cells accelerates skin wound healing through TGF‐β/SMAD2 and PI3K/Akt pathways. Int J Mol Sci 2014;15:605–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kang K, Ma R, Cai W et al. Exosomes secreted from CXCR4 overexpressing mesenchymal stem cells promote cardioprotection via Akt signaling pathway following myocardial infarction. Stem Cells Int 2015;2015:659890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang T, Jones TM, Naddell C et al. Adipose‐derived stem cells induce angiogenesis via microvesicle transport of miRNA‐31. Stem Cells Transl Med 2016;5:440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Di Trapani M, Bassi G, Fontana E et al. Immune regulatory properties of CD117(pos) amniotic fluid stem cells vary according to gestational age. Stem Cells Dev 2015;24:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moorefield EC, McKee EE, Solchaga L et al. Cloned, CD117 selected human amniotic fluid stem cells are capable of modulating the immune response. PLoS One 2011;6:e26535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gao F, Chiu SM, Motan DAL et al. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis 2016;7:e2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uccelli A, de Rosbo NK. The immunomodulatory function of mesenchymal stem cells: Mode of action and pathways. Ann NY Acad Sci 2015;1351:114–126. [DOI] [PubMed] [Google Scholar]
- 61. Mokarizadeh A, Delirezh N, Morshedi A et al. Microvesicles derived from mesenchymal stem cells: Potent organelles for induction of tolerogenic signaling. Immunol Lett 2012;147:47–54. [DOI] [PubMed] [Google Scholar]
- 62. Blazquez R, Sanchez‐Margallo FM, de la Rosa O et al. Immunomodulatory potential of human adipose mesenchymal stem cells derived exosomes on in vitro stimulated T cells. Front Immunol 2014;5:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang B, Yin Y, Lai RC et al. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev 2014;23:1233–1244. [DOI] [PubMed] [Google Scholar]
- 64. Conforti A, Scarsella M, Starc N et al. Microvescicles derived from mesenchymal stromal cells are not as effective as their cellular counterpart in the ability to modulate immune responses in vitro. Stem Cells Dev 2014;23:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Budoni M, Fierabracci A, Luciano R et al. The immunosuppressive effect of mesenchymal stromal cells on B lymphocytes is mediated by membrane vesicles. Cell Transplant 2013;22:369–379. [DOI] [PubMed] [Google Scholar]
- 66. Nicole S, Desforges B, Millet G et al. Intact satellite cells lead to remarkable protection against Smn gene defect in differentiated skeletal muscle. J Cell Biol 2003;161:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nakamura Y, Miyaki S, Ishitobi H et al. Mesenchymal‐stem‐cell‐derived exosomes accelerate skeletal muscle regeneration. FEBS Lett 2015;589:1257–1265. [DOI] [PubMed] [Google Scholar]
- 68. Dutt V, Gupta S, Dabur R et al. Skeletal muscle atrophy: Potential therapeutic agents and their mechanisms of action. Pharmacol Res 2015;99:86–100. [DOI] [PubMed] [Google Scholar]
- 69. Das S, Halushka MK. Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc Pathol 2015;24:199–206. [DOI] [PubMed] [Google Scholar]
- 70. Zhou Y, Zhou G, Tian C et al. Exosome‐mediated small RNA delivery for gene therapy. Wiley Interdiscip Rev RNA 2016;7:758–771. [DOI] [PubMed] [Google Scholar]
- 71. Bodempudi V, Hergert P, Smith K et al. miR‐210 promotes IPF fibroblast proliferation in response to hypoxia. Am J Physiol Lung Cell Mol Physiol 2014;307:L283–L294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Eulalio A, Mano M, Ferro MD et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012;492:376–381. [DOI] [PubMed] [Google Scholar]
- 73. Zeng L, He X, Wang Y et al. MicroRNA‐210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther 2014;21:37–43. [DOI] [PubMed] [Google Scholar]
- 74. Alaiti MA, Ishikawa M, Masuda H et al. Up‐regulation of miR‐210 by vascular endothelial growth factor in ex vivo expanded CD34+ cells enhances cell‐mediated angiogenesis. J Cell Mol Med 2012;16:2413–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cervio E, Barile L, Moccetti T et al. Exosomes for intramyocardial intercellular communication. Stem Cells Int 2015;2015:482171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang S, Aurora AB, Johnson BA et al. The endothelial‐specific microRNA miR‐126 governs vascular integrity and angiogenesis. Dev Cell 2008;15:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang S, Olson EN. AngiomiRs‐‐Key regulators of angiogenesis. Curr Opin Genet Dev 2009;19:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang B, Yao K, Huuskes BM et al. Mesenchymal stem cells deliver exogenous microRNA let7c via exosomes to attenuate renal fibrosis. Mol Ther 2016; 24:1290–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Haider KH, Idris NM, Kim HW et al. MicroRNA‐21 is a key determinant in IL‐11/Stat3 anti‐apoptotic signalling pathway in preconditioning of skeletal myoblasts. Cardiovasc Res 2010;88:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ibrahim AG, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep 2014;2:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Recchiuti A, Krishnamoorthy S, Fredman G et al. MicroRNAs in resolution of acute inflammation: Identification of novel resolvin D1‐miRNA circuits. FASEB J 2011;25:544–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sheedy FJ, Palsson‐McDermott E, Hennessy EJ et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR‐21. Nat Immunol 2010;11:141–147. [DOI] [PubMed] [Google Scholar]
- 83. Asangani IA, Rasheed SAK, Nikolova DA et al. MicroRNA‐21 (miR‐21) post‐transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008;27:2128–2136. [DOI] [PubMed] [Google Scholar]
- 84. Essandoh K, Li Y, Huo J et al. Mirna‐mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 2016. 46:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ivan M, Huang X. miR‐210: Fine‐tuning the hypoxic response. Adv Exp Med Biol 2014;772:205–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lange‐Consiglio A, Perrini C, Tasquier R et al. Equine amniotic microvesicles and their anti‐inflammatory potential in a tenocyte model in vitro. Stem Cells Dev 2016;25:610–621. [DOI] [PubMed] [Google Scholar]
- 87. Iglesias DM, El‐Kares R, Taranta A et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS One 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang G, Zou X, Miao S et al. The anti‐oxidative role of micro‐vesicles derived from human Wharton‐Jelly mesenchymal stromal cells through NOX2/gp91(phox) suppression in alleviating renal ischemia‐reperfusion injury in rats. PLoS One 2014;9:e92129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information