

Abstract
Successful expansion of bone marrow (BM) hematopoietic stem and progenitor cells (HSPCs) would benefit many HSPC transplantation and gene therapy/editing applications. However, current expansion technologies have been limited by a loss of multipotency and self‐renewal properties ex vivo. We hypothesized that an ex vivo vascular niche would provide prohematopoietic signals to expand HSPCs while maintaining multipotency and self‐renewal. To test this hypothesis, BM autologous CD34+ cells were expanded in endothelial cell (EC) coculture and transplanted in nonhuman primates. CD34+C38− HSPCs cocultured with ECs expanded up to 17‐fold, with a significant increase in hematopoietic colony‐forming activity compared with cells cultured with cytokines alone (colony‐forming unit‐granulocyte‐erythroid‐macrophage‐monocyte; p < .005). BM CD34+ cells that were transduced with green fluorescent protein lentivirus vector and expanded on ECs engrafted long term with multilineage polyclonal reconstitution. Gene marking was observed in granulocytes, lymphocytes, platelets, and erythrocytes. Whole transcriptome analysis indicated that EC coculture altered the expression profile of 75 genes in the BM CD34+ cells without impeding the long‐term engraftment potential. These findings show that an ex vivo vascular niche is an effective platform for expansion of adult BM HSPCs. Stem Cells Translational Medicine 2017;6:864–876
Keywords: Hematopoietic stem progenitor cell, Bone marrow transplantation, Endothelial cells, Gene therapy, CD34+ cells
Significance Statement.
Transplantation of gene‐corrected bone marrow (BM) blood‐producing stem cells could be used to treat hematologic disease. Expansion of genetically corrected stem cells before reinfusion into the patient would improve engraftment, thus providing an effective therapy for genetic diseases. The results of the present study show that BM stem cells grown on endothelial cells engraft at high levels in the monkey. These results show that endothelial cells support stem cell expansion and are safe for use in transplantation studies and could thus be translated for use in the clinic for expansion of modified stem cells in gene therapy applications in patients.
Introduction
Genetic diseases affecting the bone marrow (BM) and hematopoiesis can be treated by transplantation of gene‐corrected autologous or matched allogeneic hematopoietic stem and progenitor cells (HSPCs) or allogeneic HSPCs from umbilical cord blood (CB) donors. For standard of care, HSPCs are mobilized from BM and CD34+ cells collected from apheresis product [1]. Efficacy of gene‐modified HSPC transplantation (HSPCT) requires a threshold engraftment level, which is proportional to the CD34+ cell dose [1]. Many patients have predictive factors associated with poor mobilization [2, 3] and would thus be ineligible for HSPC gene therapy. Allogeneic HSPCT is an alternative to autologous HSPC gene therapy; however, identification of a human leukocyte antigen‐matched donor is often challenging. The side effects from allogeneic HSPCT can also be substantial and life‐threatening [4]. Therefore, development of a safe and effective BM HSPC expansion platform would increase the number of patients eligible for autologous HSPC gene therapy.
Several groups have developed human CB expansion platforms, including immobilized Notch ligand [5, 6], prostaglandin E2 (PGE2) [7, 8, 9], angiopoietins [10, 11], cytokines [12], and small molecules [13, 14]. In clinical trials, these were used for allogeneic CB, in which an expanded CB unit facilitated neutrophil recovery but might not engraft long term. To date, no expansion methods have been reported for clinical use with adult BM HSPCs.
We developed a vascular niche from human endothelial cells (ECs) that expands CB HSPCs with retained repopulation potential [15, 16]. The focus of the present study was to evaluate the vascular niche for BM HSPC expansion. We found that EC‐expanded gene‐modified BM HSPCs reconstituted long‐term hematopoiesis in the nonhuman primate. Our results indicate unprecedented marking in T lymphocytes, erythrocytes, and platelets, without selection. These findings reveal that EC‐mediated expansion of BM HSPCs is both safe and effective and, thus, has the potential to improve gene‐modified HSPCT for the treatment of lymphoid (HIV, severe combined immunodeficiency [SCID]) and erythroid (hemoglobinopathies) hematologic diseases.
Materials and Methods
CD34+ Cell Isolation and Transduction
Cell isolation, transduction, and transplantation for nonhuman primate controls were conducted as described previously [17, 18]. Marrow aspiration was performed, and CD34+ cells were isolated [19]. For ex vivo studies, CD34+ cells were cryopreserved and thawed directly onto ECs, as described. For in vivo studies with EC‐expanded gene‐modified cells and for unexpanded controls, CD34+ cells were collected from three naïve animals and transduced as described.
The CD34+ cell fraction was isolated from steady‐state bone marrow or primed bone marrow [17, 18] and plated into StemSpan‐Serum‐Free Expansion Medium (SFEM) (StemCell Technologies, Vancouver, BC, http://www.stemcell.com) with 100 ng/ml each of human stem cell factor (SCF), thrombopoietin (TPO), and FLT3 ligand (FL; all from PeproTech, Rocky Hill, NJ, http://www.peprotech.com). After overnight culture, the cells were transduced in media with 4 µg/ml protamine sulfate and 1 µg/ml cyclosporine (Cyclosporin A [Novartis, www.novartis.com]) in nontissue culture‐treated T‐75 flasks coated with 2 µg/cm2 RetroNectin reagent (CH‐296; Clontech, Mountain View, CA, http://www.clontech.com) at 10 × 106 cells per milliliter in 10 ml.
For transduction, CD34+ cells were cultured overnight and transduced. The lentivirus vector SMPGW used in the present study has been previously described [18]. In brief, the concentrated lentivirus vector (4.5 × 108 transduction units per milliliter) expresses human P140K‐MGMT, regulated by spleen focus‐forming virus promoter, and enhanced green fluorescent protein (GFP), regulated by human phosphoglycerate kinase promoter. Lentivirus vector (multiplicity of infection [MOI], 10) was added and cultured overnight. The second dose of lentivirus (MOI, 10) was added the next morning and transduced for an additional 8 hours (total transduction: MOI, 10 × 2). The animals underwent myeloablative irradiation (linear accelerator source, hyperfractionated over 2 days). Transduced cells were collected, washed, and infused into the irradiated animals. For ex vivo EC expansion with primed CD34+ cells, the cells were collected from 3 naïve donors as described previously [17, 18], cryopreserved, thawed, and plated onto ECs as described.
CD34+ Cell EC Coculture, Preparation for Infusion, and Transplantation
E4ORF1+ human ECs were generated by Jason M. Butler [15, 20] or Angiocrine Bioscience (VeraVec ECs; Angiocrine Bioscience, New York, NY, http://www.angiocrinebioscienc.com). Two weeks before transplantation, ECs were thawed (3 million cells per flask) and passaged 1:2 (3 × 106 cells per T‐75 flask). Gene‐modified BM CD34+ cells were thawed into StemSpan‐SFEM (1% P/S, 100 ng/ml SCF, TPO, FL; StemCell Technologies). Cocultures were fed every 3 days and passed 1:2 into fresh ECs if the cell concentration was >2 × 106 cells per milliliter.
On the day of transplantation, the cells were resuspended in StemSpan SFEM containing 10 µM PGE2 (Cayman Chemical, Ann Arbor, MI, http://www.caymanchem.com), placed on ice, and vortexed every 30 minutes for 2 hours. The infusion product (autologous gene‐modified HSPC and human EC mixture) was filtered and suspended in 2% autoserum in saline. The animals underwent total body irradiation, 1,020 cGy, administered from a linear accelerator at 7 cGy/minute, as four equally divided doses 12 hours apart. Within 24 hours of radiation, cocultured HSPCs/ECs were intravenously infused into the donor animal. Hematopoietic recovery based on the complete blood count (platelet count >20,000 per microliter and absolute neutrophil count [ANC] >500) was defined as described previously [21].
Real‐Time Quantitative Polymerase Chain Reaction Analysis
In vivo gene marking was analyzed by TaqMan 5′ nuclease real‐time quantitative polymerase chain reaction (PCR) assay as described previously [17]. Genomic DNA samples from primate white blood cells (WBCs) were analyzed in triplicate with a lentivirus‐specific primer/probe combination (forward, 5′‐TGAAAGCGAAAGGGAAACCA; reverse, 5′‐CCGTGCGCGCTTCAG; probe, 5′‐AGCTCTCTCGACGCAGGACTCGGC [Integrated DNA Technologies (IDT), www.idtdna.com]), with a GFP‐specific primer/probe combination (forward, 5′‐CTGCACCACCGGCAA‐3′; reverse, 5′‐GTAGCGGCTGAAGCACTG‐3′; probe, CCACCCTGACCTACGGCGTG) in a separate reaction with a β‐globin‐specific primer/probe combination (forward, 5′‐CCTATCAGAAAGTGGTGGCTGG; reverse, 5′‐TTGGACAGCAAGAAAGTGAGCTT; probe, 5′‐TGGCTAATGCCCTGGCCCACAAGTA [IDT]) to adjust for equal loading volume of genomic DNA per reaction.
Hematopoietic Colony‐Forming Assays
Colony‐forming assays were performed [22]. Cells were plated into MethoCult 4230 (StemCell Technologies), supplemented with 100 ng/ml SCF, TPO, granulocyte colony‐stimulating factor (G‐CSF; Amgen, Thousand Oaks, CA, http://www.amgen.com), granulocyte macrophage colony‐stimulating factor, interleukin‐3 (IL‐3), IL‐6, and 4 U/ml erythropoietin (all from Peprotech), and scored 10 days later.
Cytospin Analysis
A total of 100,000 cells were placed in cytocentrifuge, distributed onto slides, Wright‐stained, mounted, and analyzed.
Flow Cytometry Analysis
The antibodies used were from BD Biosciences (San Jose, CA, http://www.bdbiosciences.com), unless indicated otherwise. WBCs were stained with antibodies: mouse anti‐human CD34‐PE (clone 563; catalog no. 550761), CD34‐APC (clone 563; catalog no. 561209), CD45‐APC (clone D058‐1283; catalog no. 561290), CD45‐V450 (clone D058‐1283; catalog no. 561291), and nonhuman primate‐specific CD38‐APC (Nonhuman Primate Reagent Resource, NIH, Bethesda, MD, http://www.nhpreagents.org). ECs and HSPC‐EC cocultures were also stained with KDR‐PE (catalog no. FAB357P; R&D Systems, Minneapolis, MN, http://www.rndsystems.com), CD31‐V450 (clone WM59; catalog no. 561653), CD31‐PE (clone WM59; catalog no. 555446), CD31‐APC (clone WM59; catalog no. 17‐0319; eBioscience Inc., San Diego, CA, http://www.ebioscience.com), CD144‐APC (VE‐cadherin; clone 16B1; catalog no. 17‐1449; eBioscience), and CD144‐Alexa Fluor 700 (clone 16B1; catalog no. 56‐1449; eBioscience), and Tra‐1‐85‐APC (catalog no. FAB3195A; R&D Systems). ECs were distinguished from HSPCs by forward and side scatter, expression of the pan‐human antigen CD147 (encoded by the human basigin gene), and identified by immunostaining with the Tra‐1‐85 antibody. For in vivo studies, WBCs were costained with CD13‐PE (clone L138; catalog no. 347837), CD14‐PeCy7 (clone M5E2; catalog no. 560919), CD3‐APC (clone SP34‐2; catalog no. 557597), CD20 BrilliantViolet605 (clone 2H7; catalog no. 302333; Biolegend, San Diego, CA, http://www.biolegend.com), CD4‐Alexa Fluor 700 (clone L200; catalog no. 560836), and CD8‐Pacific‐Blue (clone RPA‐T8; catalog no. 558207). For HSPC ontogeny analysis, CD34+ cells and EC cocultures were stained with the following antibodies: CD34‐PE, CD38‐APC, CD90‐PeCy7 (Thy1.1, clone 5E10; catalog no. 561558), CD45RA‐APC‐H7 (clone 5H9; catalog no. BDB561212), and CD49f‐PacificBlue (clone GoH3). Flow cytometry analysis was conducted on a BD LSR II flow cytometer machine (BD Bioscience). Fluorescence‐activated cell sorting (FACS) was conducted on a FACSAria II (BD Bioscience).
Retroviral Integration Site Analysis
Mapping of integration sites (IS) was conducted on WBCs and sorted subsets, as described previously [22, 23]. Genomic DNA was extracted from leukocytes using the Qiagen blood DNA mini kit (Qiagen, Hilden, Germany, http://www.qiagen.com) in accordance with the manufacturer’s instructions. Lentivirus long terminal repeat (LTR)‐genome junctions were amplified by modified genomic sequencing‐PCR. Libraries were subjected to ion torrent semiconductor sequencing, and sequence reads were analyzed using custom Perl scripts (Dr. Grant Trobridge, Washington State University, Pullman, WA). Genomic sequences were mapped to the rhesus macaque genome (rheMac3) using a standalone version of Blat available from the UCSC Genome Browser (UCSC Genome Informatics Group, Center for Biomolecular Science & Engineering, Santa Cruz, CA, http://www.genome.ucsc.edu). Sequences corresponding to the same integration locus were grouped together to determine the total number of unique integration sites (clones) identified. The relative contributions of each clone were determined by the number of IS‐associated sequence reads corresponding to that clone. A quality control check was performed to reveal clones overrepresented by PCR bias by comparing the number of IS‐associated sequence reads with the number of different fragment lengths observed for each genomic locus.
RNA‐Sequence Library Construction, Sequencing, and Analysis
RNA isolation, library construction, sequencing, and analysis were performed as described previously [24]. CD34+ cells from donors in the transplantation studies were subdivided into two subgroups. In group 1, unexpanded CD34+ cells went into TRIzol (Thermo Fisher Scientific Life Sciences, Waltham, MA, http://www.thermofisher.com) for RNA extraction. In group 2, CD34+ cells plated into EC coculture for 1 week, a fraction of which were FACS sorted to deplete the human ECs (by exclusion of human CD147+ cells by staining with Tra‐1‐85 antibody that does not cross‐react with monkey cells) and sorting on the CD34+ fraction (the cells were ∼98% CD34+ after 1 week of coculture). The second fractions were injected into nonhuman primates. All original RNA‐sequence data were uploaded to the NCBI Gene Expression Omnibus database (National Center for Biotechnology Information, NIH, Bethesda, MD, http://www.ncbi.nlm.nih.gov/geo/) with a public release date of September 1, 2015.
Study Approval
Juvenile macaques (Macaca nemestrina) were housed at the University of Washington Regional Primate Research Center under conditions approved by the American Association for the Accreditation of Laboratory Animal Care. Healthy macaques were randomly assigned to the present study. The experiments were conducted under protocols approved by the institutional review board and the animal care and use committees of the University of Washington. Transplantation, priming (mobilization), and cell collection procedures and protocols have been previously described [17, 18].
Results
ECs Expand Macaque Primed and Steady‐State BM CD34+ Cells
We previously showed that human ECs support the expansion of mouse HSPCs [20] and human CB CD34+ cells [16] capable of hematopoietic reconstitution. We also showed that ECs support specification of hematopoietic progenitor cells from induced pluripotent stem cells through a Notch pathway mechanism [22]. In the present study, we hypothesized that a vascular niche would support expansion of BM HSPCs for reconstitution of nonhuman primates.
To evaluate whether the vascular niche would support expansion of adult HSPCs, we compared the expansion of steady state BM CD34+ cells obtained from multiple naïve donors and primed/mobilized CD34+ cells collected from animals after treatment with G‐CSF/SCF as described previously [17, 18]. BM CD34+CD38− cell expansion peaked on day 7, and expansion of primed CD34+CD38− cells peaked on day 10 (Fig. 1A). Endothelial coculture supported 15‐fold expansion of steady‐state BM CD34+CD38− cells, which was significantly higher compared with cells cultured in cytokines (10‐fold expansion) or primed CD34+ cells cultured with ECs (7‐fold expansion). Endothelial‐expanded BM CD34+ cells maintained higher colony‐forming potential compared with cytokine‐expanded BM CD34+ cells or EC‐expanded primed CD34+ cells (p < .005; unpaired two‐tailed t test; Fig. 1B).
Figure 1.
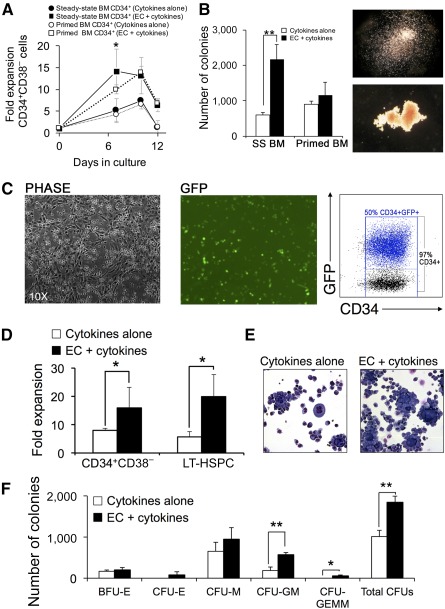
Human ECs support robust expansion of hematopoietic stem/progenitor cells. (A): Kinetics of expansion of SS and granulocyte colony‐stimulating factor/stem cell factor‐primed BM‐derived CD34+CD38− cells after coculture with cytokines with or without human ECs (n = 3 naïve donors per condition). (B): Analysis of hematopoietic colony‐forming cell (CFC) potential of BM‐derived CD34+ cells after 7‐day expansion with cytokines with or without EC. Right: Images of CFU‐M (top) and BFU‐E (bottom) colonies. (C): Phase contrast (left) and fluorescent (middle) images of P140K‐MGMT‐GFP transduced macaque SS BM CD34+ cells and EC after 7 days of coculture. Right: Flow cytometry analysis of GFP and CD34 coexpression in gene‐modified CD34+ cells after EC expansion. (D): Summary of P140K‐MGMT‐GFP lentivirus‐transduced SS BM CD34+ cell expansion by flow cytometry analysis for detection of CD34+CD38− and LT‐HSPC phenotype (CD34+CD49f+Thy1+CD38−CD45RA−; n = 3 naïve donors). (E): Microscopy showing morphology of Wright‐stained cytospin samples of gene‐modified CD34+ hematopoietic cells after coculture with cytokines with or without EC. (F): CFC analysis showing frequency and morphology of CFUs generated from gene‐modified CD34+ cells after expansion with cytokines with or without EC. Data are shown as the mean from the three experiments (donors) ± SD. CFC assays were conducted with three macaque donors and three biologic replicates per donor. Total CFUs are expressed per 105 cells plated in MethoCult (StemCell Technologies). The colony types included BFU‐E, CFU‐M, CFU‐GM, and CFU‐GEMM. Statistical analysis used the Student t test: ∗, p < .05; ∗∗, p < .005. Abbreviations: BM, bone marrow; BFU‐E, burst‐forming unit‐erythroid; CFU, colony‐forming unit; CFU‐E, colony‐forming unit‐erythroid burst; CFU‐GEMM, colony‐forming unit‐granulocyte‐erythroid‐monocyte‐macrophage; CFU‐GM, colony‐forming unit‐granulocyte‐macrophage; CFU‐M, colony‐forming unit‐macrophage; EC, endothelial cell; GFP = green fluorescent protein; LT‐HSPC, long‐term hematopoietic and progenitor stem cell; SS, steady‐state.
To assess the suitability of the vascular niche for gene therapy, we evaluated EC‐mediated expansion of gene‐modified BM CD34+ cells. Marrow CD34+ cells from naïve donors were transduced with lentivirus vector expressing GFP and the chemotherapy‐resistant variant of the methylguanine methyltransferase gene (P140K‐MGMT), which may be used to expand gene‐modified HSPCs in vivo by treatment with alkylating chemotherapy, in the event of low gene‐modified cell engraftment [17]. CD34+/EC coculture contained 97% CD34+ cells, and 50% of these cells were GFP+ (Fig. 1C). The 7‐day EC coculture supported an ∼10‐fold increase in gene‐modified CD34+CD38− cells and 17‐fold increase in long‐term (LT)‐HSPC‐like cells (CD34+CD90+CD49f+CD38−CD45RA−; p < .05, paired two‐tailed t test; Fig. 1D). Wright staining of cytospins from expanded HSPC samples revealed a greater number of blasts for EC‐expanded cells compared with cells expanded with cytokines alone (Fig. 1E). Colony‐forming cell (CFC) assays indicated that EC‐expanded cells gave rise to more mixed hematopoietic colonies (colony‐forming unit granulocyte macrophage [CFU‐GM], colony‐forming unit granulocyte‐erythroid‐macrophage‐monocyte [CFU‐GEMM]; paired two‐tailed t test, p < .005; Fig. 1F), which correlated with engraftment after HSPC transplantation [25, 26, 27].
EC‐Expanded Gene‐Modified BM CD34+ Cells Engraft Without Toxicity
To determine the engraftment of EC‐expanded HSPCs, gene‐modified CD34+ cells/EC cocultures were transplanted into macaques (n = 3). EC coculture increased the CD34+CD38− cell dose by 12‐fold. The mean CD34+ cell dose per kilogram was 35 × 106 (range, 20–52 × 106 CD34+ cells per kilogram; Fig. 2A). Infusion of high doses of EC‐expanded CD34+ cells did not cause any adverse events or hypersensitivity reactions. Coinfusion of the ECs did not cause infusional toxicity (i.e., vomiting, hypotension). ECs were detected in the blood up to 4 days after infusion but not thereafter (Fig. 2B).
Figure 2.
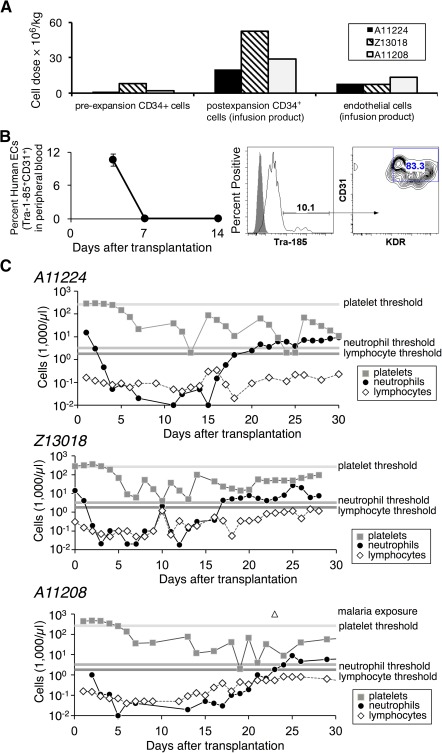
Hematopoietic reconstitution after transplantation with endothelial cell (EC) expanded CD34+ cells. (A): Cell doses used in autologous hematopoietic and progenitor stem cell transplantation in three nonhuman primates (animal identification nos.: A11224, Z13018, A11208). For each naïve animal, CD34+ cells were collected from bone marrow (BM), prestimulated with cytokines for 2 days, transduced with P140K‐green fluorescent protein lentivirus vector, cryopreserved, thawed, and then expanded in endothelial cell coculture for 7 days. Each animal received myeloablative conditioning (1,020 cGy) followed by intravenous infusion with a heterogeneous mixture of the transduced CD34+ cells and endothelial cell coculture. Cell doses are indicated per kilogram of body weight of BM‐derived transduced CD34+ cells before (pre‐expansion) and after (postexpansion infusion product) coculture with endothelial cells. The infusion products contained both the autologous CD34+ cells and the xenogeneic human EC dose. (B): Human ECs detected in peripheral blood up to 1 week after cell infusion. Data shown represent mean ± SD for n = 3 monkeys (A11224, Z13018, A11208). Representative flow cytometry analysis of peripheral blood sample 4 days after transplantation of CD34+/EC cocultures for detection of the human‐specific cell surface marker CD147 (Tra‐1‐85 antibody) and EC‐specific cell surface markers CD31 and KDR. (C): Hematopoietic recovery (over the first 30 days for each of the three indicated animals) as indicated by complete blood count analysis. Horizontal shaded lines represent minimal threshold of recover for (from top to bottom): platelets, neutrophils, lymphocytes. Abbreviation: EC, endothelial cell.
Analysis of hematologic recovery revealed that the ANC was >500 cell per microliter by day 17 for two animals [28]. The third animal achieved neutrophil engraftment on day 21; this delayed reconstitution, blood transfusion requirements, and failure to recover platelet counts were attributed to latent malarial infection. For animals A11224 and Z13018, platelet recovery (>20,000 cells per microliter) without transfusion occurred after neutrophil recovery, consistent with the kinetics of recovery observed after clinical mobilized peripheral blood HSPCT (range, 16–20 days) [29]. These data show that ECs do not cause infusional toxicity and do not persist and indicate that EC‐expanded CD34+ cells exhibit similar engraftment kinetics to patients undergoing HSPCT.
Long‐Term Multilineage Engraftment of EC‐Expanded Gene‐Modified CD34+ Cells
To confirm that EC‐expanded gene‐modified CD34+ cells contribute to hematopoiesis, transgene marking was analyzed in blood subsets after HSPCT. At the time of neutrophil engraftment, the percentage of GFP+ (gene‐modified) blood granulocytes was 50% in all animals (Fig. 3, left). By day +50, 25% of GFP+ lymphocytes were detected. GFP+ lymphocytes stabilized (A11224, Z13018) or doubled between days +50 and +100 (A11208). These data are consistent with HSPCT in historic monkeys and patients, in that the initial marking was higher in granulocytes than in lymphocytes, as myeloid reconstitution occurs before lymphocyte recovery [17]. The number of GFP+ granulocytes and lymphocytes correlated positively with the CD34+ cell dose and was 1‐log higher for the recipient that received the highest cell dose (Fig. 3, middle; Z13018, 60 × 106 CD34+ cells per kilogram). Gene marking in lymphocytes >300 days after transplantation ranged from 11% to 34%, with up to 30% marking in CD3+ T lymphocytes (supplemental online Table 1). Marking in erythrocytes and platelets from A11224 and Z13018 were ∼5% and ∼25% GFP+ at days ∼700 and ∼450 after transplantation, respectively (Fig. 4A).
Figure 3.
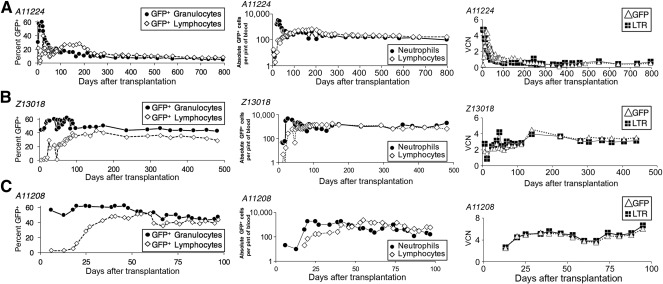
Long‐term engraftment and in vivo multilineage contribution to hematopoiesis by endothelial cell‐expanded gene‐modified autologous CD34+ cells. Detection of gene‐modified cells in animals A11224 (A), Z13018 (B), and A11208 (C). Left: Percentages of GFP+ cells (gene marking by flow cytometry) in peripheral blood lymphocytes and granulocytes. Middle: Absolute cell numbers of GFP+ cells in peripheral blood lymphocytes and neutrophils. Right: Gene marking as determined by the mean VCN per circulating leukocyte genome equivalent (normalized to β‐globin copy number) by Taqman qPCR to detect the lentiviral LTR and GFP transgene. Abbreviations: GFP, green fluorescent protein; LTR, long terminal repeat; VCN, vector copy number.
Figure 4.
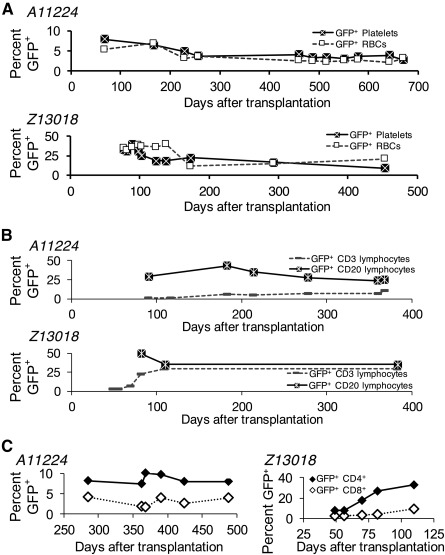
Detection of gene‐modified platelets, RBCs, and lymphoid subsets in vivo. (A): Detection of GFP+ platelets and RBCs in animal A11224 (top) and animal Z13018 (bottom). (B): Detection of GFP+ CD3+ T lymphocytes and CD20+ B lymphocytes in animal A11224 (top) and animal Z13018 (bottom). (C): Detection of GFP+ CD4 and CD8 T‐lymphocyte subsets in animal A11224 (left) and animal Z13018 (right). For determination of gene marking in lymphoid subsets, cells in the lymphoid subgate within the forward scatter by side scatter (FSC × SSC) gate were costained with fluorophore‐conjugated CD3, CD4, CD8, and CD20 antibodies. The percentages of GFP+ cells were then evaluated within the CD3, CD4, CD8, and CD20 subset gates. Abbreviations: GFP, green fluorescent protein; RBCs, red blood cells.
Analysis of marrow biopsies at day +120 revealed GFP marking in granulocytes (∼35%), monocytes (∼30%), and lymphocytes/blasts (∼20%), but no human ECs were detected (supplemental online Fig. 1). Six percent of LT‐HSPCs (CD34+CD38−CD90+CD49f+CD45RA−) in the marrow were GFP+. Gene marking in LT‐HSPCs at day +120 after transplantation was the same as the level of gene marking in the blood +800 days after transplantation (A11224). This finding is consistent with the canonical view of HSPC ontogeny, in which committed progenitors from HSPCs undergo proliferation on route to repopulation of the hematopoietic system with lineage‐committed mature blood cells [30].
We tracked engraftment by quantitative PCR analysis for detection of GFP and the lentivirus LTR, the latter to rule out contribution of lentiviral transduced (LTR+GFP−) ECs. No difference was found between the GFP and LTR viral copy number per circulating leukocyte (VCN), which stabilized at 2 and 5 VCN, respectively, for A11224, Z13018, and A11208 (Fig. 3, right).
EC‐Expanded CD34+ Cells Reconstitute Polyclonal Hematopoiesis
To determine polyclonal repopulation, we performed retroviral integration site analysis on blood leukocytes. For the longest engrafted animal (A11224), reconstitution was polyclonal in bulk leukocytes. Two hundred different clones were detected at each time point (Fig. 5). We examined clonal diversity in sorted blood cell subsets. Between days +182 and +322, we observed no restriction of clonal diversity in CD3+ cells, and tracked multiple clones over time. At day 312, we detected shared clones in the CD3+, CD20+, and granulocyte lineages, demonstrating that EC‐expanded gene‐modified LT‐HSPCs were present at the time of infusion and were capable of sustaining hematopoiesis 1 year after transplantation. In Z13018, we also observed highly polyclonal reconstitution over time up to 107 days after transplantation, with nearly 2,000 individual clones identified at each time point (Fig. 5). Early reconstitution was so clonally diverse that not a single clone identified was found to contribute >1% of the genomes analyzed.
Figure 5.
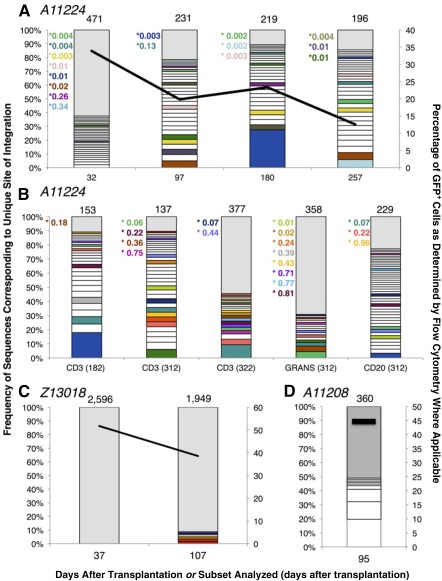
Gene‐modified peripheral blood leukocytes demonstrate polyclonal engraftment after endothelial cell (EC) expansion and myeloablative transplantation. Each graph represents the clonal diversity observed at each time point and/or in each blood cell subpopulation examined as a function of the frequency of genomic sequences identified in the pool (primary y‐axis) for each animal: A11224 (A, B), Z13018 (C), and A11208 (D). Each bar designates the relative frequency of each clonal integration sequence identified in the sample from the most abundant (bottom of bar) to the least abundant (top of bar). The total number of unique sites of integration identified in each sample is listed at the top of each bar. Unique integration sites (clones) sequenced at a frequency >1% of the pool are designated by open boxes. All clones contributing <1% sequence frequency to the pool are grouped in a single gray box. Colored boxes designate clones tracked in multiple samples over time in the same animal. White boxes indicate the clones that were not observed in any other sample from the same animal. Clones tracked across multiple samples that did not contribute >1% frequency in a time point are designated by colored text citing the percentage of sequences identified within or next to the corresponding gray box. Overlay lines designate the percentage of GFP+ cells in the sample analyzed as determined by flow cytometry (secondary y‐axis). For (B), the sorted lineage is indicated and the time point (days after transplantation) are indicated in parentheses. Abbreviation: GFP, green fluorescent protein.
For A11208, we analyzed one time point (+95 days) and found polyclonal reconstitution. Based on the level of lentivirus gene‐modified blood leukocytes (44.6%), the overall contribution of the most abundant clone to leukocytes was ∼8.5%. This proviral integration mapped within the sequence encoding mRNA JV727540 in the rhesus genome (BGI CR_1.0/rheMac3 assembly). The function of this mRNA is unknown, and integration within this mRNA sequence has not previously been reported in conjunction with insertional mutagenesis; however, we cannot rule out the disease state in this animal (fatal malaria infection) as a possible cause for the clonal skewing observed at this time point. These data indicate that ECs promote polyclonal CD34+ cell expansion ex vivo.
Whole Transcriptome Analysis Reveals Differential Gene Expression in Expanded and Unexpanded CD34+ Cells
Given that EC‐expanded cells retain engraftment potential, we next compared the gene expression profiles of expanded and unexpanded CD34+ cells. CD34+ cells from three donors cultured on ECs for 7 days were FACS depleted of human ECs with Tra‐1‐85 antibody, which binds to the human CD147 surface antigen but does not bind to macaque cells [22], combined with immunomagnetic selection for CD34+ cells (Fig. 6). Transcriptome analysis of populations of CD34+ cells revealed that coculture with cytokines plus ECs or cytokines alone alters the HSPC transcriptional profile (Fig. 6). Of the 75 genes that were differentially expressed in EC‐expanded cells compared with the other populations, the most differentially expressed gene was FOS, which was identified as a putative downstream target of Notch signaling and also regulates hematopoietic stem cell (HSC) cycle entry and mitosis [31]. McKinney‐Freeman et al. found that highly proliferative fetal liver HSCs downregulate FOS [32]. Notably, CD86 is also upregulated in EC‐expanded CD34+ cells. CD86+ HSCs have prolonged self‐renewal activity and lymphopoietic potential, and loss of these cells resulted in impaired lymphopoiesis in vivo [33]. These findings are consistent with previous studies indicating that the vascular niche balances HSPC self‐renewal and differentiation, in part, through Notch signaling [20]. We have shown that ECs expand repopulating CD34+ cells and alter their transcriptional profile without compromising engraftment, as indicated by the long‐term engraftment and robust lymphoid reconstitution achieved for animals A11224 and Z13018.
Figure 6.

Differential gene expression in CD34+ cells unexpanded or expanded with cytokines or expanded with cytokines and ECs. Hierarchical clustering and transcriptional analysis of EC‐expanded CD34+ cells and cytokine‐expanded CD34+ cells from three donors (A11224, R11145, Z13018) and from unexpanded CD34+ cells (two donors). Heat maps display the relative abundance of genes that were differentially expressed across the three cell populations (unexpanded CD34+ cells, cytokine‐expanded CD34+ cells, and EC‐expanded CD34+ cells). EC‐expanded CD34+ cells were EC depleted and enriched for CD34+ cells before analysis. All other subsets were sorted according to expression of CD34 before analysis. Scaling is relative to the maximum FPKM for each gene across all samples. (For example, if the unexpanded CD34+ cells had the greatest mRNA abundance for a particular gene, all other values would be relative to the level in unexpanded CD34+ cells.) The numbers within parentheses next to each gene name provide that value of the maximum FPKM from all samples. All genes shown in this heat map are differentially expressed. The color scale to the left of the heat map is scaled to the FPKM values to the maximum. The genes are ordered according to the p value (from top to bottom, smallest to largest). For all genes shown, the following criterion was applied: FPKM cutoff of 20 (p ≤ .05, log twofold change). Abbreviations: ECs, endothelial cells; FPKM, fragments per kilobase of exon per million reads mapped; Max, maximum.
Discussion
Methods that support ex vivo maintenance and/or expansion of long‐term repopulating bone marrow‐derived CD34+ cells would improve engraftment and therapeutic benefit for gene‐modified HSPC gene therapy applications. Validation of long‐term engraftment of expanded cells in a clinically predictive animal model would enable clinical trials and translation. We [15, 16], and other groups [34, 35, 36], have shown that human CD34+ CD38− cells expand severalfold after coculture with endothelial cells and reconstitute hematopoiesis in vivo, with increased SCID mouse repopulation potential compared with either unexpanded or cytokine‐expanded CD34+ cells. In previously published studies describing EC‐mediated human BM CD34+ cell expansion from Chute et al., CD34+ CD38− cells cocultured with primary ECs plus cytokines expanded 5‐fold [34] and 13‐fold [36]. In our studies, the EC and cytokine coculture system supported up to 17‐fold expansion of CD34+CD38− cells (before PGE2 exposure).
In previously published studies of the nonhuman primate, expanded autologous BM CD34+ cells reconstituted hematopoiesis in and rescued lethally irradiated baboons after coculture with porcine microvascular endothelial cells (PMVECs) [37] or human brain microvascular endothelial cells [38]. These studies provided the first evidence that endothelial cells produce prohematopoietic factors capable of simultaneously increasing the CD34+ cell dose by severalfold while maintaining long‐term repopulating HSPCs. However, use of either xenogenic (porcine) or human brain ECs as an “off the shelf” HSPC expansion product is not clinically feasible, because sourcing ECs from xenogenic (pig) or adult human organs (brain tissue) for clinical use and expansion of primary unmodified ECs would present challenges.
Toward the clinical development of an HSPC expansion platform that could be an “off‐the‐shelf” product for use in clinical applications, we previously developed an ex vivo vascular niche from E4ORF1‐transduced, Akt‐activated endothelial cells from human cord blood, a source that is readily available and contains cells that are easily harvested [15, 16]. These Akt‐activated ECs produce additional prohematopoietic and angiocrine factors compared with unmodified primary ECs. In the present study, we demonstrated for the first time the ability of this ex vivo vascular niche to support CD34+ cell expansion, leading to long‐term engraftment of EC‐expanded BM CD34+ cells in a clinically relevant model. Transplant recipients showed long‐term (500–800 days after transplantation) multilineage polyclonal HSPC engraftment, robust gene‐modified lymphoid reconstitution, and gene marking in platelets and erythrocytes at levels not achievable previously after transplantation of mobilized/primed transduced CD34+ cells without in vivo selection. EC‐expanded CD34+ cells have a unique transcriptional profile, with upregulation of genes that are important for self‐renewal.
Although our data clearly demonstrate long‐term engraftment of EC‐expanded CD34+ cells, further studies focused on identifying the precise factors produced by ECs that maintain and expand LT‐HSPCs and/or enhance homing to and lodging in the marrow would facilitate clinical translation. Chute et al. [39], and others, demonstrated that transplantation of ECs alone rescues in vivo hematopoiesis of irradiated mice, highlighting the critical role of the vascular niche in hematopoietic regeneration. Regenerative hematopoiesis through contact with prohematopoietic factors is endothelial‐context dependent, as human HSPCs exposed to irradiation and then cultured with ECs recovered reconstitution potential while irradiated HSPCs cultured with cytokines alone did not rescue HSPC reconstitution [40]. Toward understanding the mechanism through which ECs support hemogenic regeneration after injury, subsequent studies in transgenic mice showed that conditional deletion of prohematopoietic factors in ECs, including vascular endothelial growth factor [41], SCF [42], chemokine C‐X‐C motif ligand 12 [43], Notch ligand Jagged‐1 [44], and pleiotrophin [45], impaired hematopoietic reconstitution. However, supplementing expansion cultures with prohematopoietic factors alone is not sufficient to maintain the HSPC reconstitution potential, suggesting that there are likely additional unidentified acellular paracrine or autocrine factors produced by the vascular niche that are required for LT‐HSPC maintenance.
Current CB HSPC expansion strategies have used small molecules [14] or BM mesenchymal cells [46] or have exploited the modulation of key signaling pathways [19] that balance HSPC homeostasis, self‐renewal, and differentiation. Csaszar et al. [47] demonstrated that Notch ligand delta‐1 (DL1)‐mediated HSPC expansion in the context of a fed‐batch system reduced IL‐6 cis‐ and trans‐signaling in CB HSPCs, increasing HSPC engraftment. Although such methods have been successful for expanding CB HSPCs, they have been almost exclusively evaluated with CB and have not translated to adult steady‐state BM. A few groups have applied these strategies to nonhuman primate mobilized CD34+ cells. Goessling et al. and others showed that mobilized CD34+ cells treated with 16,16 dimethyl PGE2 engrafted long‐term [7, 8, 9]. Although PGE2 increased homing, no difference was seen in the engraftment of PGE2‐treated mobilized CD34+ cells and untreated cells. In our study, gene‐modified BM CD34+ cells were expanded on ECs and then briefly pulsed with PGE2, which might have affected HSPC homing. However, the increase in CD34+CD38− cell numbers that occurred was attributed to EC plus cytokine cocultures and not to PGE2, because the cells were not transduced, cultured, or expanded in the presence of PGE2. We hypothesized that the combination of EC expansion, EC coinfusion, and PGE2 pulse may synergize to improve CD34+ cell homing to, and lodging in, the bone marrow, thereby enhancing long‐term hematopoietic reconstitution.
Additional transplantation studies of gene‐modified CD34+ cells in nonhuman primates that evaluate the effect of each independent variable on HSPC engraftment would clarify the mechanism through which EC expansion and EC infusion independently increase hematopoietic reconstitution with BM CD34+ cells. Controlled studies in nonhuman primates (i.e., comparative engraftment of unexpanded CD34+ cells, unexpanded CD34+ cells coinfused with ECs, EC‐expanded CD34+ cells in which the EC fraction is depleted before infusion, cytokine‐expanded CD34+ cells alone, and unexpanded PGE2‐pulsed CD34+ cells) would reveal the effect that each independent variable has on hematopoietic reconstitution. Although we acknowledge that transient exposure to PGE2 could improve HSPC homing, acute treatment alone is likely not sufficient to rescue/recover the engraftment potential of an EC‐expanded CD34+ product that had lost all multipotent repopulating cells during ex vivo culture.
We have also tested other reagents that, like PGE2, showed promising results for CB CD34+ cell expansion and hematopoietic regeneration and reconstitution in mouse models. These methods included coculture of the Notch ligand DL1, mesenchymal stromal cells (MSCs), and forced expression of HOXB4. Watts et al. [48] expanded HOXB4‐transduced autologous nonhuman primate CB CD34+ cells, which had superior engraftment compared with unexpanded CB CD34+ cells. A follow‐up study to investigate whether activation of Notch signaling (through coculture with MSCs expressing Notch ligand DL) synergized with HOXB4 overexpression to further improve CB expansion and engraftment was tested in the autologous nonhuman primate setting [49]. That study showed that this combined expansion strategy accelerated neutrophil and platelet recovery compared with unexpanded and HOXB4‐expanded HSPC recipient historic controls. Although these methods, and others (PGE2, UM171, SR1), expanded CB CD34+ cells, no studies have been reported on the expansion of steady‐state BM CD34+ cells, until now.
Our findings showed that ECs support expansion of BM‐HSPCs (supplemental online Table 1 ), thus marking an important advance for HSPC gene therapy for patients who cannot tolerate mobilization but who would otherwise be candidates for gene therapy (e.g., sickle cell disease, myelofibrosis). Without mobilization, the yield from BM harvest results in a lower CD34+ cell dose. Because the success of CD34+ cell reconstitution is proportional to the cell dose, expansion of BM CD34+ cells would likely improve gene‐corrected HSPC reconstitution. ECs could thus be used to expand steady‐state BM for such patients.
In contrast to these expansion platforms, we provide the first evidence of a CB expansion strategy [16] that effectively translates to expansion of adult HSPCs to support hematopoietic reconstitution and long‐term engraftment of steady‐state adult BM CD34+ cells. We have shown for the first time that our ex vivo vascular niche expands steady‐state BM CD34+ cells while maintaining their stem cell properties (i.e., self‐renewal, multipotency, engraftment potential), leading to sustained long‐term engraftment (800 days), and enhances lymphoid and erythroid gene‐modified cell reconstitution. We have also shown that EC‐mediated expansion of HSPCs is safe, as the animals did not exhibit signs of infusional toxicity or any other health problems, such as unexpected thromboembolic or hemorrhagic complications that could be attributed to cotransplantation of ECs. As such, our studies set the stage for translation of endothelial cell‐mediated expansion of steady‐state BM CD34+ cells.
For the recipients of EC‐expanded CD34+ cells, hematopoietic recovery and engraftment occurred within the time frame observed in clinical BM HSPC transplantation (ANC >500 by days 14–20). Hematopoietic recovery and engraftment was achieved earlier compared with a previous study in which baboons were transplanted with similar BM CD34+ cell doses after expansion on PMVECs (ANC >500 by day 25 ± 1) [37]. The CD34+ cell dose per kilogram and the frequency of mixed hematopoietic colony‐forming units (CFU‐GM, CFU‐GEMM) have been shown to correlate with improved long‐term engraftment in patients [1, 25, 26, 27]. In our studies, the EC coculture supported up to 17‐fold expansion of CD34+CD38− cells that substantially increased the CD34+ cell dose per kilogram with a concomitant increase in CFU‐GEMM. The expansion of CD34+ cells and CFCs achieved in the present study was substantially higher than the levels achieved in earlier reports evaluating EC‐ plus cytokine‐expanded CD34+ cells in baboons [37, 38]. However, similar to those previous reports, expansion of macaque CD34+ cells was lower compared with human CD34+ cell expansion with human cytokines and ECs [16], which can be attributed to the use of human ECs and human cytokines, as nonhuman primate reagents were not available. In addition to the increased cell dose and positive safety profile associated with vascular niche‐expanded marrow CD34+ cells, we also achieved unprecedented levels of gene marking in CD3+CD4+ T lymphocytes, erythrocytes, and platelets without the requirement for in vivo selection.
Our previous studies in the nonhuman primate indicated that myeloid cells typically have the highest efficiency of gene marking in vivo after HSPC transplantation. In those studies, achieving high levels of transgene expression in lymphoid, erythroid, and megakaryocyte progeny was more challenging despite polyclonal repopulation [17, 18]. In addition, gene marking in the lymphocyte pool was predominantly in B cells, with <20% in GFP+ CD20+ B lymphocytes [17]. In contrast, marking in CD3+ T lymphocytes was <5% [50] without in vivo selection. In the study by Trobridge et al. [50], in vivo selection with O6‐benzylguanine combined with alkylating chemotherapy (temozolomide) increased marking in CD3+ cells to 7%. In contrast, here we show that reconstitution of nonhuman primates with endothelial cell‐expanded BM CD34+ cells led to an unprecedented 30% gene marking in CD3+CD4+ lymphocytes (range, 10%–30%) without in vivo selection. This high level marking in gene‐modified T cells would likely improve the efficacy of HSPC and gene therapies for the treatment of HIV and genetic immunodeficiencies.
The level of marking achieved in erythrocytes and platelets after transplantation of endothelial cell‐expanded HSPCs is 2‐ to 10‐fold greater than the level previously reported by our group and others for erythroid‐specific marking after transplantation with lentivirus transduced mobilized HSPC transplantation [51, 52]. Therefore, the gene marking observed in the erythroid and megakaryocyte progeny of endothelial cell‐expanded CD34+ cell progeny in vivo is unprecedented, as neither mobilization before transplantation nor in vivo selection with chemotherapy after transplantation were required to achieve these milestones in our study.
In summary, vascular niche‐mediated expansion of steady‐state BM CD34+ cells has the potential to increase the accessibility and improve the outcome of success of HSPC gene therapy for patient populations with varied hematopoietic disease indications, from frequent immunodeficiencies (infectious or genetic), hemoglobinopathies (sickle cell disease) and anemias (Fanconi anemia) to rare orphan diseases.
Conclusion
An ex vivo vascular niche sustains and/or expands bone marrow‐derived LT‐HSPCs that retain multilineage blood reconstitution of the clinically relevant nonhuman primate. These findings demonstrate safety and effectiveness of a bone marrow HSPC expansion platform that could be clinically translated to HSPC gene therapy applications.
Author Contributions
J.L.G.: conception and design, performance of all experiments with technical support where indicated, manuscript writing and editing, final approval of manuscript; J.M.B.: generation of ECs, contribution to experimental design, manuscript editing, final approval of manuscript; B.K. and Z.K.N.: performance of RNA sequencing analysis and related bioinformatics, final approval of manuscript; M.G.P.: isolation of RNA and performance of RNA sequencing experiments, final approval of manuscript; M.G. and D.J.N.: generation of ECs, feedback on experiments, manuscript editing, final approval of manuscript; J.E.A.: performance of RIS analysis and related bioinformatics, final approval of manuscript; S.R. and H.‐P.K.: general guidance, manuscript editing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
M.G. is an employee of Angiocrine Bioscience. D.J.N. is an employee of, and equity holder in, Angiocrine Bioscience. S.R. is a founder of Angiocrine Bioscience and an unpaid consultant. The other authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Acknowledgments
We thank Grace Choi and Jacob Isenberg for help in preparing this report and Krystin Norman, Devikha Chandrasekaran, Yan Yi Chan, Emily Menard, and Christina McAllister for technical assistance at Fred Hutchinson Cancer Research Center (FHCRC). We also thank Veronica Nelson, Erica Curry, Kelvin Sze, and the University of Washington National Primate Research Center staff, members of the H.P. Kiem Laboratory, and the Flow Cytometry Shared Resource Facility at FHCRC. We thank Grant Trobridge at Washington State University for work on retroviral integration site data processing. We thank Olivier Elemento and the Genomics Resources Core Facility at Weill Cornell Medical College for performing the RNA sequencing. This work was supported in part by NIH Grants HL115128, HL098489, HL084345, and HL053750. H.‐P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E.D. Thomas Endowed Chair for Cancer Research. S.R. is a Howard Hughes Medical Institute Investigator.
References
- 1. Anguita‐Compagnon AT, Dibarrart MT, Palma J et al. Mobilization and collection of peripheral blood stem cells: Guidelines for blood volume to process, based on CD34‐positive blood cell count in adults and children. Transplant Proc 2010;42:339–344. [DOI] [PubMed] [Google Scholar]
- 2. Petrecca K, Atanasiu R, Akhavan A et al. N‐linked glycosylation sites determine HERG channel surface membrane expression. J Physiol 1999;515:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perseghin P, Terruzzi E, Dassi M et al. Management of poor peripheral blood stem cell mobilization: Incidence, predictive factors, alternative strategies and outcome. A retrospective analysis on 2177 patients from three major Italian institutions. Transfus Apheresis Sci 2009;41:33–37. [DOI] [PubMed] [Google Scholar]
- 4. Sun CL, Francisco L, Kawashima T et al. Prevalence and predictors of chronic health conditions after hematopoietic cell transplantation: A report from the Bone Marrow Transplant Survivor Study. Blood 2010;116:3129–3139; quiz 3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delaney C, Heimfeld S, Brashem‐Stein C et al. Notch‐mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med 2010;16:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dahlberg A, Brashem‐Stein C, Delaney C et al. Enhanced generation of cord blood hematopoietic stem and progenitor cells by culture with StemRegenin1 and Delta1(Ext‐IgG.). Leukemia 2014;28:2097–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoggatt J, Mohammad KS, Singh P et al. Prostaglandin E2 enhances long‐term repopulation but does not permanently alter inherent stem cell competitiveness. Blood 2013;122:2997–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cutler C, Multani P, Robbins D et al. Prostaglandin‐modulated umbilical cord blood hematopoietic stem cell transplantation. Blood 2013;122:3074–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goessling W, Allen RS, Guan X et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long‐term safety in preclinical nonhuman primate transplant models. Cell Stem Cell 2011;8:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan X, Gay FP, Lim FW et al. Low‐dose insulin‐like growth factor binding proteins 1 and 2 and angiopoietin‐like protein 3 coordinately stimulate ex vivo expansion of human umbilical cord blood hematopoietic stem cells as assayed in NOD/SCID gamma null mice. Stem Cell Res Ther 2014;5:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blank U, Ehrnström B, Heinz N et al. Angptl4 maintains in vivo repopulation capacity of CD34+ human cord blood cells. Eur J Haematol 2012;89:198–205. [DOI] [PubMed] [Google Scholar]
- 12. Lam AC, Li K, Zhang XB et al. Preclinical ex vivo expansion of cord blood hematopoietic stem and progenitor cells: Duration of culture; the media, serum supplements, and growth factors used and engraftment in NOD/SCID mice. Transfusion 2001;41:1567–1576. [DOI] [PubMed] [Google Scholar]
- 13. Boitano AE, Wang J, Romeo R et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010;329:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fares I, Chagraoui J, Gareau Y et al. Cord blood expansion: Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self‐renewal. Science 2014;345:1509–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kobayashi H, Butler JM, O’Donnell R et al. Angiocrine factors from Akt‐activated endothelial cells balance self‐renewal and differentiation of haematopoietic stem cells. Nat Cell Biol 2010;12:1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Butler JM, Gars EJ, James DJ et al. Development of a vascular niche platform for expansion of repopulating human cord blood stem and progenitor cells. Blood 2012;120:1344–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beard BC, Trobridge GD, Ironside C et al. Efficient and stable MGMT‐mediated selection of long‐term repopulating stem cells in nonhuman primates. J Clin Invest 2010;120:2345–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trobridge GD, Beard BC, Gooch C et al. Efficient transduction of pigtailed macaque hematopoietic repopulating cells with HIV‐based lentiviral vectors. Blood 2008;111:5537–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watts KL, Delaney C, Humphries RK et al. Combination of HOXB4 and Delta‐1 ligand improves expansion of cord blood cells. Blood 2010;116:5859–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Butler JM, Nolan DJ, Vertes EL et al. Endothelial cells are essential for the self‐renewal and repopulation of Notch‐dependent hematopoietic stem cells. Cell Stem Cell 2010;6:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernstein SH, Nademanee AP, Vose JM et al. A multicenter study of platelet recovery and utilization in patients after myeloablative therapy and hematopoietic stem cell transplantation. Blood 1998;91:3509–3517. [PubMed] [Google Scholar]
- 22. Gori JL, Butler JM, Chan YY et al. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J Clin Invest 2015;125:1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beard BC, Adair JE, Trobridge GD et al. High‐throughput genomic mapping of vector integration sites in gene therapy studies. Methods Mol Biol 2014;1185:321–344. [DOI] [PubMed] [Google Scholar]
- 24. Ginsberg M, James D, Ding BS et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFβ suppression. Cell 2012;151:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoo KH, Lee SH, Kim HJ et al. The impact of post‐thaw colony‐forming units‐granulocyte/macrophage on engraftment following unrelated cord blood transplantation in pediatric recipients. Bone Marrow Transplant 2007;39:515–521. [DOI] [PubMed] [Google Scholar]
- 26. Page KM, Zhang L, Mendizabal A et al. Total colony‐forming units are a strong, independent predictor of neutrophil and platelet engraftment after unrelated umbilical cord blood transplantation: A single‐center analysis of 435 cord blood transplants. Biol Blood Marrow Transplant 2011;17:1362–1374. [DOI] [PubMed] [Google Scholar]
- 27. Bedi R, Kumar L, Kochupillai V. Autologous peripheral blood stem cell transplantation: Predictors for haematopoietic reconstitution. Natl Med J India 2003;16:255–259. [PubMed] [Google Scholar]
- 28. Schmitz N, Linch DC, Dreger P et al. Randomised trial of filgrastim‐mobilised peripheral blood progenitor cell transplantation versus autologous bone‐marrow transplantation in lymphoma patients. Lancet 1996;347:353–357. [DOI] [PubMed] [Google Scholar]
- 29. Gonçalves TL, Benvegnú DM, Bonfanti G. Specific factors influence the success of autologous and allogeneic hematopoietic stem cell transplantation. Oxid Med Cell Longev 2009;2:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Notta F, Doulatov S, Laurenti E et al. Isolation of single human hematopoietic stem cells capable of long‐term multilineage engraftment. Science 2011;333:218–221. [DOI] [PubMed] [Google Scholar]
- 31. Okada S, Fukuda T, Inada K et al. Prolonged expression of c‐fos suppresses cell cycle entry of dormant hematopoietic stem cells. Blood 1999;93:816–825. [PubMed] [Google Scholar]
- 32. McKinney‐Freeman S, Cahan P, Li H et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell 2012;11:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimazu T, Iida R, Zhang Q et al. CD86 is expressed on murine hematopoietic stem cells and denotes lymphopoietic potential. Blood 2012;119:4889–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chute JP, Saini AA, Chute DJ et al. Ex vivo culture with human brain endothelial cells increases the SCID‐repopulating capacity of adult human bone marrow. Blood 2002;100:4433–4439. [DOI] [PubMed] [Google Scholar]
- 35. Chute JP, Muramoto G, Fung J et al. Quantitative analysis demonstrates expansion of SCID‐repopulating cells and increased engraftment capacity in human cord blood following ex vivo culture with human brain endothelial cells. Stem Cells 2004;22:202–215. [DOI] [PubMed] [Google Scholar]
- 36. Chute JP, Muramoto GG, Fung J et al. Soluble factors elaborated by human brain endothelial cells induce the concomitant expansion of purified human BM CD34+CD38− cells and SCID‐repopulating cells. Blood 2005;105:576–583. [DOI] [PubMed] [Google Scholar]
- 37. Brandt JE, Bartholomew AM, Fortman JD et al. Ex vivo expansion of autologous bone marrow CD34(+) cells with porcine microvascular endothelial cells results in a graft capable of rescuing lethally irradiated baboons. Blood 1999;94:106–113. [PubMed] [Google Scholar]
- 38. Araki H, Chute JP, Petro B et al. Bone marrow CD34+ cells expanded on human brain endothelial cells reconstitute lethally irradiated baboons in a variable manner. Leuk Lymphoma 2010;51:1121–1127. [DOI] [PubMed] [Google Scholar]
- 39. Chute JP, Muramoto GG, Salter AB et al. Transplantation of vascular endothelial cells mediates the hematopoietic recovery and survival of lethally irradiated mice. Blood 2007;109:2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muramoto GG, Chen B, Cui X et al. Vascular endothelial cells produce soluble factors that mediate the recovery of human hematopoietic stem cells after radiation injury. Biol Blood Marrow Transplant 2006;12:530–540. [DOI] [PubMed] [Google Scholar]
- 41. Hooper AT, Butler JM, Nolan DJ et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2‐mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009;4:263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding L, Saunders TL, Enikolopov G et al. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012;481:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013;495:231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poulos MG, Crowley MJ, Gutkin MC et al. Vascular platform to define hematopoietic stem cell factors and enhance regenerative hematopoiesis. Stem Cell Rep 2015;5:881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Himburg HA, Harris JR, Ito T et al. Pleiotrophin regulates the retention and self‐renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Reports 2012;2:964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Parekh C, Crooks GM. Critical differences in hematopoiesis and lymphoid development between humans and mice. J Clin Immunol 2013;33:711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Csaszar E, Wang W, Usenko T et al. Blood stem cell fate regulation by delta‐1‐mediated rewiring of IL‐6 paracrine signaling. Blood 2014;123:650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watts KL, Nelson V, Wood BL et al. Hematopoietic stem cell expansion facilitates multilineage engraftment in a nonhuman primate cord blood transplantation model. Exp Hematol 2012;40:187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watts KL, Delaney C, Nelson V et al. CD34(+) expansion with delta‐1 and HOXB4 promotes rapid engraftment and transfusion independence in a Macaca nemestrina cord blood transplant model. Mol Ther 2013;21:1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trobridge GD, Wu RA, Beard BC et al. Protection of stem cell‐derived lymphocytes in a primate AIDS gene therapy model after in vivo selection. PLoS One 2009;4:e7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kiem HP, Arumugam PI, Burtner CR et al. Pigtailed macaques as a model to study long‐term safety of lentivirus vector‐mediated gene therapy for hemoglobinopathies. Mol Ther Methods Clin Dev 2014;1:14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hayakawa J, Ueda T, Lisowski L et al. Transient in vivo beta‐globin production after lentiviral gene transfer to hematopoietic stem cells in the nonhuman primate. Hum Gene Ther 2009;20:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information