

Abstract
Cancer stem cells (CSC) are associated with tumor resistance and are characterized in gastric cancer (GC). Studies have indicated that Notch and wnt‐beta‐catenin pathways are crucial for CSC development. Using CD44+ CSCs, we investigated the role of these pathways in GC carcinogenesis. We performed cell proliferation, wound healing, invasion, tumorsphere, and apoptosis assays. Immunoblot analysis of downstream signaling targets of Notch and wnt‐beta‐catenin were tested after gamma‐secretase inhibitor IX (GSI) treatment. Immunohistochemistry, immunofluorescence, and Fluorescence activated cell sorting (FACS) were used to determine CD44 and Hairy enhancer of split‐1 (Hes1) expression in human GC tissues. CD44+ CSCs were subcutaneously injected into NMR‐nu/nu mice and treated with vehicle or GSI. GC patients with expression of CD44 and Hes1 showed overall reduced survival. CD44+ CSCs showed high expression of Hes1. GSI treatment showed effective inhibition of cell proliferation, migration, invasion, tumor sphere formation of CD44+ CSCs, and induced apoptosis. Importanly, Notch1 was found to be important in mediating a crosstalk between Notch and wnt‐beta‐catenin in CD44+ CSCs. Our study highlights a crosstalk between Notch and wnt‐beta‐catenin in gastric CD44+ CSCs. Expression of CD44 and Hes1 is associated with patient overall survival. GSI could be an alternative drug to treat GC. Stem Cells Translational Medicine 2017;6:819–829
Keywords: Cancer stem cells, CD44, Gastric cancer, Gamma‐secretase inhibitor IX, Notch, wnt‐beta‐catenin
Significance Statement.
Failure to eradicate cancer stem cells (CSC) have always been a major challenge in the field of cancer therapies. CSCs usually exihibit properties of chemo and radio resistance and are also associated with tumor relapse and metastases. Therefore, proving these CSCs to be critical targets for cancer cure. In our present study, we specifically target the CD44+ population of gastric CSCs both in vitro and in vivo using a gamma‐secretase inhbitor (GSI). GSI mediated concomitant inhibition of Notch and wnt‐beta‐catenin CSC associated pathways in CD44+ gastric cancer (GC). Targeting by GSI can evolve as an effective and potential therapeutic approach to treat patients with GC. Analysis of Notch signaling and CD44 might help to monitor treatment outcome of patients with GC. GSI can be considered as an effective drug targeting CSC in the field of GC.
Introduction
Gastric cancer (GC) is the third leading cause of cancer‐related death worldwide 1, 2. Only few patients can undergo curative surgery 1, 2, 3. The response rate to multiagent chemotherapy is 50% or greater, but median survival is still limited 3. Therefore, new targeted approaches are urgently needed.
Cancer stem cells (CSCs) are playing an important role in the occurence and development of GC 4, 5, 6, 7. CSCs are pluripotent cells with the function of self‐renewal, and are important for tumor proliferation, metastasis 8, 9. CSCs are important for drug resistancy and refractory radiation 10. For GC, it was shown that CD44 is the only CSC marker associated with tumor formation in immunodefiencient mice 11. CD44 is a class I transmembrane glycoprotein that can act as a receptor for extracellular matrix components and it has been shown that CD44+ CSCs are important for metastasis of gastrointestinal cancer 11, 12, 13, 14, 15. Therefore, direct targeting of CD44+ CSCs instead of treating unspecific bulk tumor cells would be of great interest.
The Notch signaling pathway has been implicated in the pathogenesis of a number of malignancies, including GC 16, 17. The interaction of Notch ligands with their Notch receptors, promotes a gamma‐secretase‐dependent cleavage of the receptor and release of the Notch intracellular domain (NICD), resulting in activation of the pathway 16, 17. NICD translocates to the nucleus and induces target genes like Hairy enhancer of split‐1 (Hes1). In some of T‐cell lymphoblastic leukemia, the Notch1 gene was discovered 18, 19. Over the last decades it was also well demonstrated that Notch signaling is activated in GC 20, 21, 22, 23, 24, 25.
The wnt‐beta‐catenin pathways are a group of signal transduction pathways characterized in GC 25. The best characterized wnt signaling pathways are the canonical wnt pathway or b‐catenin dependent 26. Activation of the wnt pathway was also shown to promote GC carcinogenesis 27, 28.
Interestingly, previous reports show that Notch and wnt‐beta‐catenin are activated in CSCs, associated with GC carcinogenesis, prognosis, and chemotherapy resistance 29, 30, 31. The Notch signaling and wnt‐beta‐catenin pathway regulation of GC may therefore depend primarily on the control of CSC. In the present study, we sought to further evaluate the potential benefit of targeting Notch signaling in GC. Interestingly, we identified a coexpression of CD44 and Hes1 positive cells in human GC material and correlated their concomitance with overall limited survival. We identified Notch1 to be important in mediating Notch and wnt‐beta‐catenin crosstalk in CD44+ CSCs. Finally, we studied in detail the biological impact of the gamma‐secretase inhibitor IX (GSI) in human gastric CD44+ CSCs and in a xenograft mouse model.
Materials and Methods
Cell Culture and In Vitro Treatment
Human GC cell lines were obtained from Derek Zieker (Wiesbaden, Germany), Kuang Hung Cheng (Kaohsiung, Taiwan) and human pancreatic cancer cells from Nabeel Bardeesy (Boston, USA). MKN45 was cultured in RPMI 1640 + Glutamax (Invitrogen, Germany) with 20% FCS (Biochrom, Germany) and 100 U/ml penicillin/streptomycin (Invitrogen, Germany) at 37°C in 5% CO2. KP3, were cultured under same conditions. Detailed information is provided in the Supporting Information.
CD44+ Cell Sorting and Flow Cytometry (FACS)
CD44+ population was sorted from human GC cell line MKN45. Cells were blocked in 100 μl of blocking buffer (5% bovine serum albumin (BSA)/PBS) for 30 minutes on iceand centrifuged at 800 ×g for 5 minutes at 4°C. The cells were then incubated with a primary antibody against CD44 (1:100; Cell Signaling Technology, Germany) or HES1 (1:100; Abcam, Germany) for 25 minutes on ice. Cells were washed with ice‐cold 1 mM EDTA/1%BSA/PBS and were stained with FITC conjugated goat antimouse IgG (1:200; Dianova, Jackson Immuno Research Laboratories, Inc., USA) for CD44 or Alexa fluor 594 goat anti rabbit seconday (1:100; Life technologies, Germany) for HES1, respectively, for 20 minutes in the dark. The stained cells were washed twice with 1 mM EDTA/1%BSA/PBS, centrifuged at 800 ×g for 5 minutes and the cell pellet was suspended in 200 μl of 1 mM EDTA/1%BSA/PBS prior to analysis. The stained cells were measured with FACS Calibur (Becton Dickinson Immunocytometry Systems, USA). KP3 cells was used as a positive control as previously published (33). Detailed information is provided in the Supporting Information.
Animals and In Vivo Treatment
NMRI‐nu/nu female nude mice were obtained from Charles River Laboratories International (Sulzfeld, Germany). In total, 10 mice were used for sorted MKN45 xenograft experiments. CD44+ MKN45 cells (1 × 106) were subcutaneously injected into each flank for each mouse. Mice were assigned into treatment groups (n = 5 mice per group). When tumor volume reached a size of 100 mm3, mice were treated intraperitoneally with GSI (10 mg/kg/body weight) or control phosphate buffered saline (PBS) for four times per week followed by 3 days rest. Health status and treatment side effects were inspected every day. The mice used in this study were maintained in the animal care of Medizinische Universitätsklinik Tübingen (UKT), Germany. All experimental protocols for animal care of UKT and Baden Württemberg (protocol no: M 11/12). Detailed information is provided in the Supporting Information.
Patient Samples and Tissue Microarray
Tissues were representing normal gastric mucosa (N), inflammation (G), and GC. Detailed information about stainings and dataanalysis are provided in the Supporting Information.
Statistical Analysis
All experiments were repeated three times. The results were analyzed using software Graphpad prism version 5.0 (USA) and SPSS Version 11.0 (USA). The tests include one way ANNOVA analysis of variance and Student's t test along with Bonferroni post‐test and paired and unpaired t tests. Differences were considered as statistically significant when the p value was < .05 and nonsignificant “n.s.” when the p value was > .05. For survival analysis, cases with missing date of death were censored. Univariate survival analysis was performed using the Kaplan‐Meier method comparing the survival curves with the log‐rank test. Differences were considered as statistically significant when the p‐value was less <.05 (*), <.005 (**) and <.001 (***).
Results
CD44 and Hes1 are Up‐Regulated in Human GC and are Associated with Patients Overall Survival
We screened a panel of five human GC cancer cell lines (MKN45, SNU, KATO III, 2313287, NCI‐N87) to analyze the level of CD44 expression and to validate the activation of Notch and wnt‐beta‐catenin signaling. FACS analysis and immunoblotting showed a relatively higher expression of targets (CD44, Hes1, wnt 5a/b) in MKN45 cells compared to the others (Fig. 1A, 1B). Furthermore, we analyzed the expression of the same markers in fresh human tissues (Normal‐N, Gastritis‐G, Cancer‐GC) (Fig. 1C). The highest expression of CD44, Hes1, and wnt 5a/b was observed in GC and the lowest in normal tissue (N). To further analyze the coexpression of Hes1 in CD44+, and CD44− MKN45 cells we performed FACS analysis. In the CD44+ cell population we found a significant (p < .001) expression of Hes1 (79.2% vs. 22%) (Fig. 1D). In addition, we investigated the CD44 and Hes1 expression by immunohistochemistry and immunfluorescence in GC tissues from 269 patients. We found positive expression of CD44 and Hes1 in 86% of the patients (Fig. 1E and Supporting Information Fig. 1A). We also found by immunoflurescent labeling coexpression of CD44 and Hes1 (Supporting Information Fig. 2). Next, we examined the correlation of CD44 and Hes1 expression with patients survival of 269 patients with GC for which survival data is available (Fig. 1F and Supporting Information Fig. 1C) 32. Interestingly, patients with positive expression of CD44 and Hes1 showed significant impaired (p = .004) overall survival. To determine the influence of coexpression we also examined the correlation of single CD44 versus Hes1 expression with patient's survival (Supporting Information Fig. 1D). We found that Hes1 expression has a higher influence on overall survival compared to CD44 expression (p = .004). Additional analysis showed that CD44 and Hes1 is highly expressed in older patients (60–70 years) and that there is no significant difference between the expression and gender (Supporting Information Fig. 1B, 1C). Our results show that both human GC tissues and MKN45 cellshave high levels of coexpression of CD44 and Hes1. GC patients with double positive expression (Hes1+ and CD44+) have the shortest survival. These data suggest that the CSC marker CD44 and the Notch signaling target gene Hes1 can act as a prognostic factor for patients survival.
Figure 1.
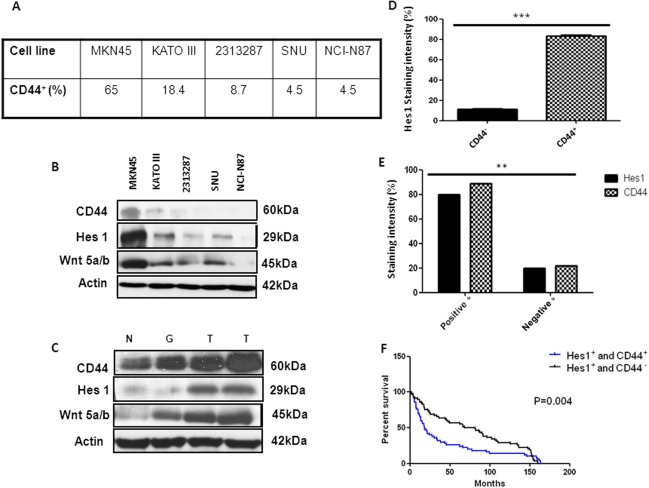
Expression of CD44 and hairy enhancer of split‐1 (Hes1) in human gastric cancer (GC) cell lines and tissues. (A): Table showing the % of CD44 expression from a panel of five human GC cell lines. (B): Immunoblot analysis of human GC cell lines for same targets with highest expression in MKN45 cells. (C): Immunoblot analysis of CD44, Hes1, and wnt 5a/b expression in human tissues (Normal‐N, Gastritis‐G, Cancer‐GC) with significant increase in the tumor samples. β‐actin was used as a loading control. (D): Activation of Notch pathway in CD44+ cancer stem cells was assesed by FACS showing a significant increase in Hes1 expressing cells compared to CD44− cells. (E): Bar graphs representing the different intensities of CD44 and Hes1 expression in a panel of 269 human GC tisssues. 80% of the samples showed positive CD44 and 89% positive HES1 expression. *GC tumor tissues showing both moderate and strong staining for Hes1 or CD44 were considered to be positive whereas tumor tissues showing both weak and negative staining were considered to be negative. (F): Kaplan‐Meier overall survival curve for GC patients stratified by Hes1 and CD44 expression, showing a significant diference. **p < .005. Abbreviation: Hes1, hairy enhancer of split‐1.
GSI Treatment Results in an Inhibition of Proliferation, Migration, Invasion and Induces Apoptosis in CD44+ CSCs
CD44+ cells were treated with three different concentrations of GSI (2.5, 5 & 15 µM). GSI treatment reduced the number of viable CD44+ cells in a dose and time dependent manner (Fig. 2A). In order to confirm that GSI specifically targets CD44+ population of MKN45 cells, we performed a cell proliferation assay on both CD44+ and CD44− sorted MKN45 cells. GSI specifically inhibited the proliferation of CD44+ cells as compared to the CD44− population. We compared the specificity of GSI for CD44 with another standard chemotherapy (5‐Fluouracil; 5‐FU) widely used for GC. 5‐FU treatment failed to show specific inhibition of CD44+ cells as compared to the CD44− cells (Supporting Information Fig. 3A, 3B). These results clearly demonstrate that GSI effectively targets CD44+ gastric CSCs for inhibition of MKN45 cells. Immunofluorescence stainings for DAPI, CD44, and Hes1 after treatment with Dimethylsulfoxid (DMSO) or GSI (2.5, 5 & 15 µM) are confirming these results (Supporting Information Fig. 3C). Significant (p < .001) inhibition of wound healing was observed with GSI in a concentration dependent manner (Fig. 2B). GSI treatment inhibited cell invasion in CD44+ cells in a significant (p < .001) concentration dependent manner (Fig. 2C). We noticed that GSI treatment showed relatively higher induction of apoptosis as compared to the DMSO group in a significant (p < .001) dose and time dependent manner (Fig. 2D). The highest induction of apoptosis was observed mostly after 96 hours under 15 µM GSI treatment in CD44+ cells. Thus, treatment with GSI can effectively inhibit cell proliferation, migration, invasion and induces apoptosis on human CD44+ CSCcells.
Figure 2.
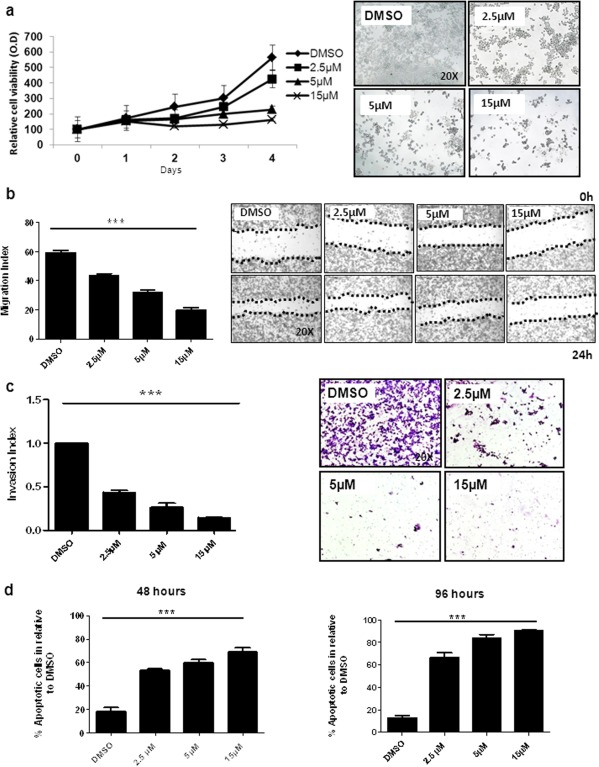
Gamma‐secretase inhibitor IX (GSI) inhibits proliferation, migration, invasion and induces apoptosis in CD44+ cancer stem cells (CSCs). (A): Cell proliferation assay representing GSI (2.5, 5 & 15 µM) or DMSO treated CD44+ CSCs and corresponding light microcope pictures showing treatment result on day 4. (B): Bar graphs representing the migration index of CD44+ CSCs after DMSO or GSI (2.5, 5 & 15 µM) treatment. Photographs were taken under light microscope (10× magnification) at 0 and 24 hours. (C): CD44+ cells were treated for 48 hours with control (DMSO) and GSI (2.5, 5 & 15 µM) to investigate the effect of GSI on invasiveness of CD44+ CSCs. Light microscope photographs were taken at (20× magnification) and invasion index was calculated and plotted. (D): GSI (2.5, 5 & 15 µM) or DMSO treated CD44+ CSCs. Apoptosis was quantified by staining with Annexin V and propidium iodide (PI) using flow cytometry for 48 hours and 96 hours. ***p < .001. Abbreviation: DMSO, Dimethylsulfoxid.
GSI Treatment Prevents Tumorsphere Formation in CD44+ CSCs
Cancer cells with stem cell properties harbors the potential to form tumor spheres under low attachment conditions 29, 31. GSI (2.5, 5 & 15 µM) inhibited significant (p < .001) the tumor sphere formation under treatement specifically under the highest dosage as compared tothe untreated DMSO control cells (Supporting Information Fig. 4A, 4B, 4C). Thus, the results show that GSI inhibits the tumorsphere forming potential of CD44+ GC cells.
GSI Treatment Concomitantly Inhibits Notch and Wnt‐Beta‐Catenin Pathway in CD44+ CSCs
Hes1 was activated in CD44+ cells we like to further understandwhich Notch receptor is mainly important in activating the Notch pathway in gastric CD44+ CSCs. GSI treatment showed a relative down‐regulation of Notch1 in a dose and time‐dependent manner whereas Notch2 and Notch3 levels remained relatively unchanged (Fig. 3A). NICD showed a down‐regulation under GSI therapy compared to DMSO treated cells. c‐Myc is a direct downstream of Notch1 and also mediates the expression of other common downstreams like Rac1, Akt, Erk, Stat3 33, 34, 35. Immunoblots clearly showed a relative down‐regulation of CD44, c‐Myc, Rac1 and phospho forms of Akt, Erk, and Stat3 after GSI treatment (Fig. 3B). The full forms of Erk, Aktand Stat3 remained unchanged after GSI treatment. Next, we wanted to analyze if GSI treatment inhibits also the wnt‐beta‐catenin pathway in CD44+ CSCs. Immunoblotting clearly showed a relative inhibition of wnt 5a/b, beta‐catenin and other common wnt pathway downstreams like Axin2, APC, LPR6 after GSI treatment (Fig. 3C). These data clearly support a coactivation of both the Notch and wnt‐beta‐catenin pathways in CD44+ CSCs and suggest that, GSI inhibits the activation of theseprimary important pathway involved in the maintenance of GC CSCs. We observed that GSI had at least a partial effect on the regulation of EMT in CD44+ cells. N‐cadherin was partially effected specifically under 15‐µM GSI treatment for 96 hours (Fig. 3D). In contrast, E‐cadherin showed no significant change under GSI therapy. We also examined the expression of PARP and found an increase under highest GSI dosage (Fig. 3D).
Figure 3.
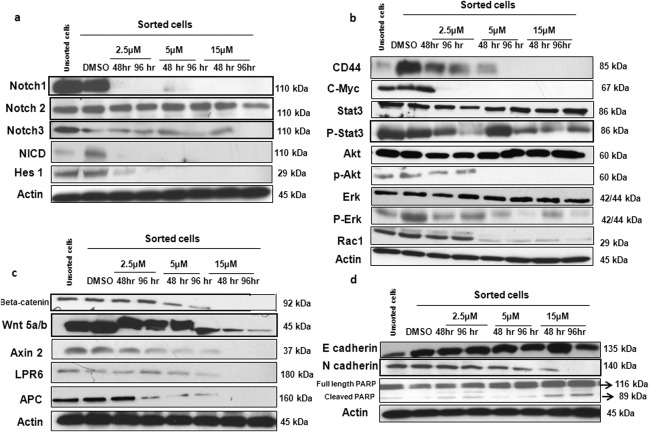
Gamma‐secretase inhibitor IX (GSI) concomitantly inhibits wnt‐beta‐catenin and Notch pathways crosstalk in CD44+ cancer stem cells (CSCs). CD44+ CSCs were treated with GSI (2.5, 5 & 15 µM) or DMSO for 48 hours and 96 hand compared to unsorted cells. (A): GSI treatment was evaluated on Notch signaling pathway components (Notch1, Notch2, Notch3, Notch intracellular domain, hairy enhancer of split‐1) by immunoblotting. (B): Drug treatment on downstream targets involved in the crosstalk between Notch and wnt‐beta‐catenin pathway (CD44, c‐Myc, Stat3, p‐Stat3, Akt, p‐Akt, Erk, p‐Erk, Rac‐1) evaluated by immunoblotting. (C): Inhibition of wnt‐beta‐catenin pathway and its downstreams (Beta‐catenin, wnt 5a/b, Axin2, LPR6, APC) by GSI evaluated by immunoblotting. (D): Immunoblots showing the expression of EMT markers: E‐cadherin, N‐cadherin, and cleavage of PARP after GSI treatment. β‐actin was used as a loading control. Abbreviations: DMSO, Dimethylsulfoxid; Hes1, hairy enhancer of split‐1; NICD, Notch intracellular domain.
GSI Concomitantly Inhibits Notch and Wnt‐Beta‐Catenin Crosstalk via Down‐Regulation of Notch1 in CD44+ CSCs
To reconfirm our proposed hypothesis of GSI for inhibition of CD44+ CSCs, siRNA mediated silencing of CD44 and Notch1was performed. CD44 silencing lead to the down‐regulation of all the important common downstream effector proteins involved in mediating the Notch and wnt‐beta‐catenin crosstalk. CD44, Hes1, P‐Erk, P‐Akt, Rac1, P‐Stat3, c‐Myc, Notch1, wnt 5a/b, and beta‐catenin were down‐regulated under CD44 siRNA treatment as observed by immunoblotting (Fig. 4A). These obeserved CD44 siRNA effect was similar to the effects achieved under GSI treatment. Notch1 was also silencedvia siRNA to see whether Notch1 is specifically important for mediating the crosstalk and also for GSI mediated inhibition of the Notch pathway. Notch1 siRNA treatment showedbyimmunoblottingsimilar down‐regulation of CD44, Hes1, P‐Erk, P‐Akt, Rac1, P‐Stat3, c‐Myc, Notch1, wnt 5a/b, and beta‐catenin (Fig. 4B). These findings were also very similar to GSI treatment results. Therefore, we confirm our hypothesis that GSI inhibits CD44+ CSCs specifically via down‐regulation of Notch1, which in turn leads to the inhibiton of Notch and wnt‐beta‐catenin pathway crosstalks involved in the maintenance of CSCs.
Figure 4.
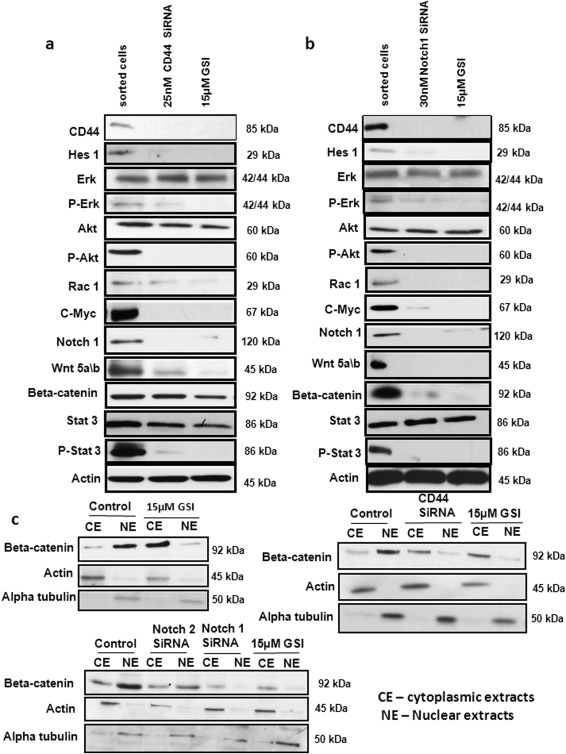
Gamma‐secretase inhibitor IX (GSI) targets CD44+ cancer stem cells (CSCs) via Notch1. (A): We silenced CD44 using siRNA and confirmed the effect of GSI on hairy enhancer of split‐1, p‐Erk, p‐Akt, RAC1, Notch 1, c‐Myc, Stat3, P‐Stat3, wnt 5a/b, beta‐catenin by immunoblot. (B): We also silenced Notch1 using siRNA and confirmed the effect of GSI on same targets. β‐actin was used as a loading control. (C): Complete inhibition of nuclear translocation of beta‐catenin was also observed after GSI treatment in CD44+ CSCs using immuno blot. Alpha‐tubulin was used as loading control. Abbreviations: CE, cytoplasmic extracts; GSI, gamma‐secretase inhibitor IX; Hes1, hairy enhancer of split‐1; NE, nuclear extracts.
A major hallmark of the activation of the wnt pathway is the accumulation of the beta‐catenin protein in the cytoplasm 36, 37. Our immunoblot results from the nuclear and cytoplasmic extractsof the CD44+ CSCs treated with DMSO or GSI (2.5, 5 & 15 µM) showed that GSI effectively inhibited translocation of beta‐catenin to the nucleus (Fig. 4C). Our results clearly show that GSI inhibits CD44+ CSC via Notch1 mediated inhibiton of the concomitant activation of the Notch and wnt‐beta‐catenin pathway.
GSI Treatment Effectively Inhibits the Growth of CD44+ CSC Xenograft Tumors
GSI treatment reduced CD44+ xenograft growth significantly (p < .001) to control group (Fig. 5A, 5C, Supporting Information Fig. 5A). All mice were sacrificed after 4 weeks of treatment and tolerated the treatments equally well and without significant weight loss (Fig. 5D). Histological analysis of resected xenograft tumors from GSI showed more necrosis as compared to thecontrol (Fig. 5B). The tumor weight was significant (p < .005) lower in the GSI group (Fig. 5E, Supporting Information Fig. 5A). GSI treatment showed a significant decrease in Ki67 staining (Fig. 6F and Supporting Information Fig. S5B). We analyzed by immunoflourescence/immunohistochemistrythe expression of CD44 and HES1 after treatments (Supporting Information Fig. 6A, 6B, 6C). We observed a considerable decrease in expression of both CD44 and Hes1 in GSI treated tumors. Analysis by immunoblot after GSI treatment showeda significant down‐regulation of CD44, Hes1, Notch1, c‐Myc, wnt 5a/b, beta‐catenin and also the common downstream effector proteins for both the pathways (p‐Erk, p‐Stat3, Notch1, Rac1) (Fig. 6A, 6B). Full forms of Erk, Akt, and Stat3 remained unchanged. Furthermore, common wnt pathway downstreams like Axin 2, APC, and LPR6 were also down‐regulated under GSI treatment (Fig. 6B). In vivo, GSI application influenced the epithelial marker E‐cadherin and mesenchymal marker N‐cadherin. Immunoblots also showed cleavage of PARP in GSI treated tumors compared to the control tumors (Fig. 6B). These data are in accordance with our in vitro results and support apoptosis as one of the main cell death mechanisms involved in GSI treatment. FACS analysis of blood obtained from untreated GC patients (n = 3) compared to healthy patients (n = 4) showed a significant (p < .001) upregulation of double positive cells (Fig. 6C). To determine whether in vivo GSI treatment targets CD44 and Hes1 positive CSCs, we performed a FACS analysis of the mononuclear cells isolated from the blood of both GSI treated and control group xenograft mice and compared the results to healthy mice (Fig. 6D). Control mice showed significantly (p < .005) higher double positive cells and GSI treated mice showed expression levels similar to normal mice. Thus, GSI treatment is not reducing the double positive cells more then the average count in mice without cancer.
Figure 5.
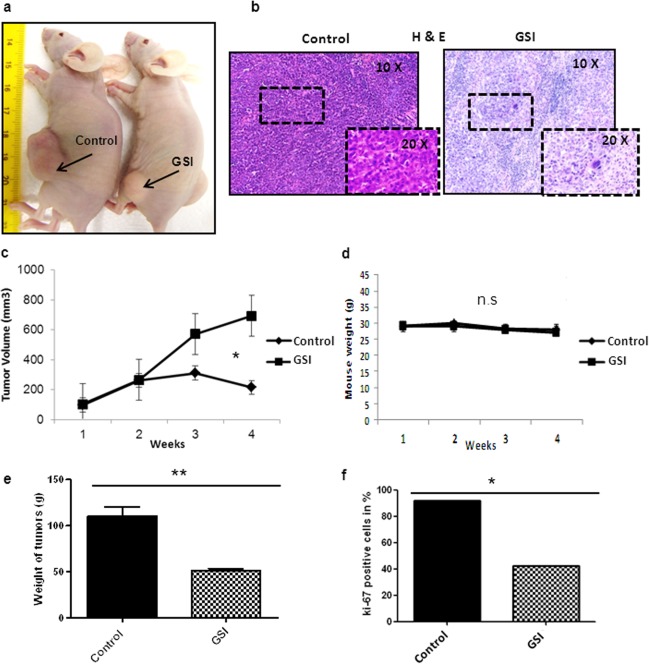
Gamma‐secretase inhibitor IX (GSI) inhibits CD44+ cancer stem cells xenograft tumor growth. (A): Pictures of mice taken after 4 weeks of treatment with indicated therapies. The arrows showing the xenograft tumors (n = 5/group). (B): H&E stainings of corresponding tumors (10× & 20× magnification), showing necrosis under GSI therapy. (C): Tumor growth curves of all right and left flanks tumors under indicated treatments. (D): Mouse weight curves for indicated therapies. Quantification of (E): tumor weight (F): Ki67 staining of the xenograft tumors according to different therapy groups. *p < .05, **p < .005. Abbreviation: GSI, gamma‐secretase inhibitor IX.
Figure 6.
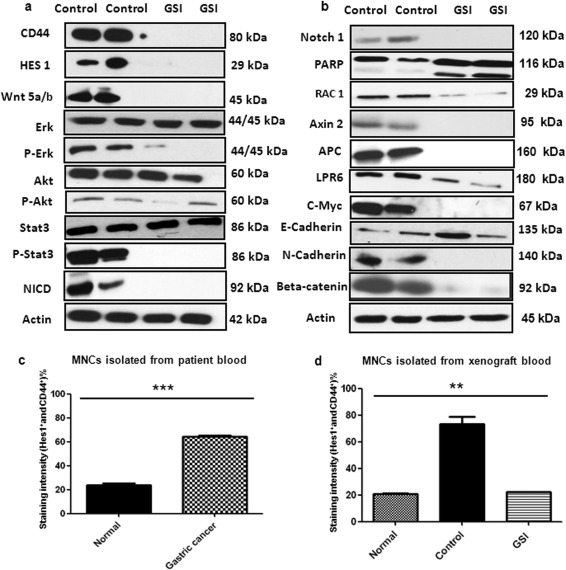
Gamma‐secretase inhibitor IX inhibits the growth of CD44+ cancer stem cells xenograft tumors via attenuation of Notch and wnt‐beta‐catenin signaling. (A): Immunoblot showing the expression of CD44, hairy enhancer of split‐1 (Hes1), wnt 5a/b, Erk, p‐Erk, Akt, p‐Akt, Stat3, p‐stat3, and beta‐catenin of treated xenograft tumors like indicated. (B): Immunoblots showing the expression of Notch1, PARP, Rac1, Axin 2, APC, LPR6, c‐Myc, E‐cadherin, and N‐cadherin of treated xenograft tumors like indicated. Actin was used as a loading control. (C): Bar graph showing the double positive cells (Hes1+ and CD44+) from the blood of normal compared to gastric cancer patients like indicated. (D): Bar graph showing the double positive cells (Hes1+ and CD44+) from the blood of mice like indicated. **p < .005, ***p < .001. Abbreviations: Hes1, hairy enhancer of split‐1; GC, gastric cancer, MNC, mononuclear cells, NICD, Notch intracellular domain.
Discussion
CSCs are known to be the most important candidate cells involved in resistance to chemotherapies and also involved in relapse. In this study, we demonstrate the advantage of GSI treatment in a subset of GC cells with CD44+ expression and in a xenograft mouse model. For the first time our data also highlight the relevance of Notch1 as a propable mediator for Notch and wnt‐beta‐catenin crosstalk involved in maintenance of gastric CD44+ CSCs.From five tested human GC cell lines, MKN45 showed the highest expression of CD44+ CSCs. Notch and wnt‐beta‐catenin signaling has been reported to be two of the main important pathways for CSC maintenance 29, 30, 31. We have previously published that CD44+/EpCAM+ pancreatic cancer cells are inhibited by GSI and that the decrease of CD44+ cells correlates with response to chemotherapy in a panel of gastrointestinal cancer 32, 38. However, the involvement of CD44 and Notch signaling as a new target in GC and the treatment of these specifc CD44+ CSCs by GSI was not validated in detail. We also show that the presence of CD44 and Hes1 acts as an effective prognostic factor for GC patient survival. Our present study mainly involves and focusses on the CD44+ GC stem cell population as an effective target in comparison to the CD44− population mainly due to two major reasons: First, CD44− population reflects to be easier targets for chemotherapy as these population do not posses the unique property of chemoresistance unlike the CD44+ CSC population. Second, it is true that we cannot rule out the possibilty of expression of other CSC markers (at a minimal amount) in CD44− population like CD133, and so on, but as our result shows that 5FU effectively kills these CD44− population thus, we can determine that CD44− population might not be the resistant reminanat population leading to tumor relapse in GC. Importantly, CD44+ CSC being majorly expressed in GC might be the main CSC involved in chemoresistance and effective target for GSI mediated therapy to overcome resistency associated with GC treatment.
First, in a subset of human GC cell lines, tissues, blood samples, tissue microarray (TMA)s and xenograft tumors we were able to demonstrate by western blots, immunohistochemistry, immunofluorescence, and FACS considerable expression of both targets (CD44, Hes1). Second, we analyzed 269 human GC tissues and found an interesting correlation between expression of CD44 and Hes1 with poor overall survival. Third, we showed in detail that inhibition of Notch signaling by GSI blocked cell proliferation, wound healing, invasion, and spheroid formation. Fourth, treatment of GC xenografts by GSI resulted in a significant impairment of tumor growth and thus, prooved to be an effective drug in vivo. Finally, our data also showed a crosstalk between Notch and wnt‐beta‐catenin signaling in CD44+ CSCs and that inhibition of these two pathways by GSI targets CD44+ CSC population in GC.
Notch1 as well as Notch2 are critical for the metaplastic transition of gastric epithelial cells 21. Kang et al. described the expression of Notch3 and Jagged2 in association with GC development 39. Recently, Li et al. also tested GSI to treat CD44+ MKN45 cells in vitro and in vivo, but they did not use more than one drug concentration and there study lacked human data 40. Our results clearly show less interactions with Notch2‐3 under GSI treatment compared with Notch1. Thus, pAKT which plays a key role in multiple cellular processes 41. Mitogen activated protein/extracellular signal‐regulated kinase (MAP/ERK) pathway, which plays an important role in the control of cellular processes like proliferation, and so on, and STAT3 (signal transducer and activator of transcription 3) are effectively impaired 42, 43. Furthermore, RAC1 (Ras‐related C3 botulinum toxin substrate), a protein important for cell motility is down‐regulated under GSI therapy 44. As a new aspect, we additionaly show a cross talk between Notch and wnt‐beta‐catenin pathway via Notch1 in gastric CD44+ CSCs and show these two pathways as attractive targets for GSI mediated therapy (Supporting Information Fig. S7). CD44 and specifically Notch1 silencing showed us an overall effective, similar outcome as achieved by GSI. This strengthens that GSI targets CD44+ CSCs via inhibition of Notch1 which in turn helps in the concomitant inhibition of Notch/wnt‐beta‐catenin pathway. CSC targeted therapy for GC by GSI also reduces the chances of toxic side effects to normal cells as it is site or target specific in action and also diminishes the chances of any recurrance or. In order to confirm the target specificity of GSI to CD44+ CSCs for MKN45, we showed an effective inhibition of mostly the CD44+ population CSCs as compared to the CD44− population. Moreover, on comparison to 5‐Fluoruracil treatment the same population of sorted cells (CD44+ and CD44−) GSI showed effective inhibition of CD44+ whereas 5‐Fluoruracil failed to show not only specific targeting of CD44+ CSCs but also effectiveness to impair proliferation of CD44+ MKN45 cells as compared to GSI.
In summary, we showed a coexpression of CD44 and Hes1 and a crosstalk between Notch and wnt‐beta‐catenin signaling in human GC. GSI treatment can inhibit GC growth in vitro and in vivo by targeting CD44+ CSCs. Coexpression of CD44 and Hes1 acts as a potential prognostic factor, but additional experiments are needed to determine our findings in a prospective study of patients with GC. Future therapeutic approaches of GSI in clinical trials of GC are of definite interest.
Conclusion
Our study highlights the potential role of CD44+ CSCs in GC. We for the first time validate the prognostic role of CD44+ and Hes1+ double positive cells as a probable potential futuristic prognostic marker for GC patients. Moreover, our data clearly demonstrates the concept of dual inhibition of major Notch and wnt‐beta‐catenin pathways by targeting the CD44+ gastric CSCs as a new treatment by GSI. We focuss on the concomitant inhibition of two major pathways involved in CSC maintenance (Notch and wnt‐beta‐catenin) using GSI in these CD44+ gastric CSC, therefore reducing all the chances of tumor relapse or metastasis associated via activation of feed back loop due to other pathways in gastric CSC. Current chemotherapeutic strategies available for GC treatment are mostly limited to the inhibition of cancer cells present within the tumor. The failure to eradicate the CSC strongly limits or hinders the ultimate effectiveness of conventional anti cancer strategies. Therefore, our results for the first time show that GSI mediated targeted therapy towards CD44+ gastric CSC can evolve to be an effective, novel and specific therapeutic approach to treat GC.
Author Contributions
S.B.: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; X.C.: Collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; K.C.B.: Collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; P.B.: Data analysis and interpretation, final approval of manuscript; J.G.: Provision of study material or patients, final approval of manuscript; M.G.: Provision of study material or patients, final approval of manuscript; T.K.: Provision of study material or patients, final approval of manuscript; N.P.M.: Financial support, final approval of manuscript; R.R.P.: Conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgment
R.R.P. is supported by Deutsche Krebshilfe, Grant #110870.
References
- 1. Jemal A, Bray F, Center MM et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- 2. de Martel C, Forman D, Plummer M. Gastric cancer: Epidemiology and risk factors. Gastroenterol Clin North Am 2013;42:219–240. [DOI] [PubMed] [Google Scholar]
- 3. Fontana E, Smyth EC, Cunnningham D et al. Improved survival in resected oesophageal and gastric adenocarcinoma over a decade: The Royal Marsden experience 2001‐2010. Gastric Cancer; 2016;19:1114–1124. [DOI] [PubMed] [Google Scholar]
- 4. Myoung‐Eun H, Sae‐Ock O. Gastric stem cells and gastric cancer stem cells. Anat Cell Biol 2013;46:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li K, Dan Z, Nie Y‐Q. Gastric cancer stem cells in gastric carcinogenesis, progression, prevention and treatment. World J Gastroenterol 2014;20:5420–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quante M, Wang TC. Stem cells in gastroenterology and hepatology. Nat Rev 2009;6:724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonnet D, Dick JE. Human acute myleoid leukemia is organized as a hierarchy that orginates from a primitive hematopoetic cell. Nat Med 1997;3:730–737. [DOI] [PubMed] [Google Scholar]
- 8. Han L, Shi S, Gong T et al. Cancer stem cells: Therapeutic implications and perspectives in cancer therapy. Acta Pharm Sin 2013;3:65–75. [Google Scholar]
- 9. Massard C, Deutsch E, Soria JC. Tumour stem cell‐targeted treatment: Elimination or differentiation. Ann Oncol 2006;17:1620–1624. [DOI] [PubMed] [Google Scholar]
- 10. Mimeault M, Hauke R, Mehta PP et al. Recent advances in cancer stem/progenitor cell research: Therapeutic implications for overcoming resistance to the most aggressive cancers. J Cell Mol Med 2007;11:981–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takaishi S, Okumura T, Tu S et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009;27:1006–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W, Dong LP, Zhang N et al. Role of cancer stem cell marker CD44 in gastric cancer: A meta‐anaylsis. Int J Clin Exp Med 2014;7:5059–5066. [PMC free article] [PubMed] [Google Scholar]
- 13. Takaishi S, Okumura T, Wang TC. Gastric cancer stem cells. J Clin Oncol 2008;26:2876–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lau WM, Teng E, Chong HS et al. CD44v8‐10 is a cancer–specific marker for gastric cancer stem cells. Cancer Res 2014;74:2630–2641. [DOI] [PubMed] [Google Scholar]
- 15. Palagani V, El Khatib M, Krech T et al. Decrease of CD44‐positive cells correlates with tumor response to chemotherapy in patients with gastrointestinal cancer. Anticancer Res 2012;32:1747–1756. [PubMed] [Google Scholar]
- 16. Koch U, Radtke F. Notch and cancer: A double‐edged sword. Cell Mol Life Sci 2007;64:2746–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Artavanis‐Tsakonas S, Rand MD, Lake RJ. Notch signaling: Cell fate control and signal integration in development. Science 1999;284:770–776. [DOI] [PubMed] [Google Scholar]
- 18. Radtke F, Raj K. The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat Rev Cancer 2003;3:756–767. [DOI] [PubMed] [Google Scholar]
- 19. Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006;107:2223–2233. [DOI] [PubMed] [Google Scholar]
- 20. Katoh M, Katoh M. Notch signaling in gastrointestinal tract. Int J Oncol 2007;30:247–251. [PubMed] [Google Scholar]
- 21. Yeh TS, Wu CW, Hsu KW et al. Differential Notch1 and Notch2 expresion and frequent activation of Notch signaling in gastric cancers. Arch Pathol Lab Med 2011;135:451–458. [DOI] [PubMed] [Google Scholar]
- 22. Tseng YC, Tseng YC, Tsai YH et al. Notch2‐induced COX‐2 expression enhancing gastric cancer progression. Mol Carcinog 2012;51:939–951. [DOI] [PubMed] [Google Scholar]
- 23. Hsu KW, Hsieh RH, Huang KH et al. Activation oft he Notch1/STAT3/Twist signaling axis promotes gastric cancer progression. Carcinogenesis 2012;33:1459–1467. [DOI] [PubMed] [Google Scholar]
- 24. Du X, Chng Z, Wang YH et al. Role of Notch signaling pathway in gastric cancer: A meta‐anaysis of the literature. World J Gastroenterol 2014;20:9191–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ooi CH, Ivanova T, Wu J et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet 2009;5:e1000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Semenov MV, Habas R, Macdonald BT. Wnt/beta‐catenin signaling: Components, mechanisms, and diseases. Dev Cell 2009;17:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lowy AM, Clements WM, Bishop J et al. β‐Catenin/Wnt signaling regulates expression of the membrane type 3 matrix metalloproteinase in gastric cancer. Cancer Res 2006;66:4734–4741. [DOI] [PubMed] [Google Scholar]
- 28. Chiurillo MA. Role of the Wnt/beta‐catenin pathway in gastric cancer: Ab in‐depth literature review. World J Exp Med 2015;5:84–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gupta R, Vyjas P, Enver T. Molecular targeting of cancer stem cells. Cell Stem Cell 2009;5:125–126. [DOI] [PubMed] [Google Scholar]
- 30. Takebe N, Miele L, Harris PJ et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat Rev Clin Oncol 2015;8:445−4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stojnev S, Krstic M, Ristic‐Petrovic et al. Gastric cancer stem cells: Therapeutic targets. Gastric Cancer 2014;1:13–25. [DOI] [PubMed] [Google Scholar]
- 32. Palagani V, El Khatib M, Kossatz U et al. Epithelial mesenchymal transition and pancreatic tumor initiating CD44+/EpCAM+ cells are inhibited by γ‐secretase inhibitor IX. PLoS One 2012;7:e46514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wenig AP, Millholland JM, Ohtani YY et al. c‐Myc is an important direct target ofNotch1 in T‐cell acute lymphoblasticleukemia/lymphoma. Genes Dev 2006;20:2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sahlberg SH, Spiegelberg D, Glimelius B et al. Evaluation of cancer stem cell markers CD133, CD44, CD24: Association with AKT isoforms and radiation resistance in colon cancer cells. PLoS One 2014;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chappell WH, Green TD, Spengeman JD et al. Increased protein expression of the PTEN tumor suppressor in the presence of constitutively active Notch‐1. Cell Cycle 2005; 4:1389–1395. [DOI] [PubMed] [Google Scholar]
- 36. Lu HH, Zhang R, Haydon RC. Wnt/β‐Catenin signaling pathway as novel cancer drug targets. Curr Cancer Drug Targets 2004;4:653–671. [DOI] [PubMed] [Google Scholar]
- 37. JI Han, KJ Na. Wnt/β‐Catenin Signaling Pathway in Canine Skin Melanoma and a Possibility as a Cancer Model for Human Skin Melanoma. In: Prof. Mandi Murph, ed. Melanoma in the Clinic ‐ Diagnosis, Management and Complications of Malignancy. ISBN: 978‐953‐307‐571‐6, InTech, DOI: 10.5772/21221.
- 38. Palagani V, El Khatib M, Kossatz U et al. Epithelial mesenchymal transition and pancreatic tumor iniating CD44+/EpCAM+ cells are inhibited by y‐secretase inhibitor IX. PLoS One 2012;7:e46514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kang H, An HJ, Song JY et al. Notch3 and Jagged2 contribute to gastric cancer development and to glandular differentiation associated with MUC2 and MUC5AC expression. Histopathology 2012;61:576–586. [DOI] [PubMed] [Google Scholar]
- 40. Li LC, Wang DL, Wu YZ et al. Gastric tumor‐iniating CD44+ cells and epithelial‐mesenchymal transiation are inhibited by gamma‐secreatse inhibitor DAPT. Oncol Lett 2015;10:3293–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sasaki T, Kuniyasu H. Signficance of AKT in gastric cancer. Int J Oncol 2014;45:2187–2192. [DOI] [PubMed] [Google Scholar]
- 42. Kawada I, Hasina R, Arif Q et al. Dramatic antitumor effects of the dual MET/RON small‐molecule inhibitor LY2801653 in non‐small cell lung cancer. Cancer Res 2014;74:884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiong A, Yang Z, Shen Y et al. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancer 2014;6:926–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bid HK1, Roberts RD, Manchanda PK et al. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther 2013;12:1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information