

Summary
Tumor development and the generation of induced pluripotent stem cells are highly comparable processes with striking similarities. Cellular plasticity is inherent to tumor evolution, rendering cells that acquire a stem cell‐like phenotype, for which Sox2 activation has proved instrumental for the plastic acquisition of stemness properties in tumor cells. Understanding the molecular mechanisms underlying both events might uncover novel approaches for the development of anticancer therapeutics and constitute model systems for understanding tumor generation and ensuring the biosafety of cell‐based therapies. Stem Cells Translational Medicine 2017;6:335–339
Significance Statement.
Defining the molecular drivers of cancer stem‐like cells will provide avenues for the rational design of cancer treatment regimens aimed at cancer stem cells, which combined with conventional treatments targeting the bulk tumor cells, will provide novel therapies for cancer patients. Screening methods based on pluripotency transcriptional reporters would help to identify new drugs targeting cancer stem cells and cellular plasticity.
Cancer Stem Cells and Pluripotency Reprogramming
The idea of a tumor being composed of nearly homogenous limitless replicating cells has been replaced by a concept in which tumors are composed of extremely heterogeneous cell populations. Subpopulations of cancer cells with enhanced tumorigenicity and bearing stemness features (defined as expression of stem cell genes), named cancer stem cells (CSCs) or tumor‐initiating cells (TICs), have been isolated from almost any type of tumor. TICs or CSCs share with pluripotent and adult stem cells gene networks that are essential for self‐renewal and pluripotency 1, 2. The reactivation of signaling pathways and genes essential for embryonic development during tumor progression and the ability of tumor cells to differentiate into many unrelated tissues and cell types, when placed in the right environment, highlight the link between pluripotency and cancer 3, 4, 5.
The generation of induced pluripotent stem cells (iPSCs) through genetic reprogramming of differentiated cells has created a wealth of knowledge on the mechanisms that control stemness properties 6. The similarities found between the process of pluripotency reprogramming and tumorigenesis are striking. Both processes require the expression or activation of oncogenes, the inactivation of tumor suppressor genes such as p53 or Rb 7, 8, 9, 10, overriding of senescence and apoptotic barriers, extensive epigenetic changes, and a metabolic switch toward a glycolytic metabolism 11, 12, 13. Historical similarities also exist between the method used to generate iPSCs and the in vitro oncogenic transformation protocols used to identify the first oncogenes: target cells are transduced with a cocktail of viruses expressing a variety of suspected oncogenes (OKSM for pluripotency—Oct4, Klf4, Sox2, and c‐Myc). After extensive morphological selection of the resulting cell clones (colonies), some will be separated as established transformed lines (capable of generating tumors in immunosuppressed mice). When fibroblasts of identical genetic background were reprogrammed to iPSCs or transformed, their transcriptome changes were found to overlap significantly 11, although differences specific to the pluripotent state were present. Some of these common genes are responsible for the programs that make iPSCs tumorigenic, including response to wounding, lysosomes, and inflammation. Furthermore, transient expression of OKSM factors in vivo was capable of inducing dedifferentiation and generating pluripotent cells in multiple tissues; however, teratomas 14 and multiple dysplastic lesions 15 were readily observed in a multitude of organs. If OKSM induction was sustained long enough (more than 7 days), the tumors became independent of OKSM expression. These results highlight the relationship between the acquisition of pluripotency and tumor generation.
Pluripotency reprogramming has been finely mapped and occurs in two transcriptional waves. The initial wave occurs even in cells refractive to full reprogramming, with the second wave preceding the changes in DNA methylation necessary for stable pluripotency to be achieved 16. Transformation imposes activation of certain roadblocks that prevent the cell from fully reprograming into the pluripotent state. For example, applying the typical four‐factor reprogramming protocol to the breast carcinoma MCF7 cell line rendered clones that were only partially reprogrammed, with activation of just one of the endogenous pluripotency genes: Sox2 17. These clones were enhanced in cancer stem cell‐related features, but none were fully reprogrammed iPSCs. It is possible that this is why natural tumor formation has been found, but not in vivo natural reprogramming.
Cellular Plasticity, Pluripotency, and Metastatic Invasion
The differentiated state tends to be a stable condition; however, cellular plasticity manifests under stress circumstances, such as tissue damage or infection. In such circumstances, short‐lived progenitors can dedifferentiate and acquire the ability to regenerate cells from other lineages, such as an entire mammary gland tree or intestinal crypts 18, 19, demonstrating that potency and plasticity are inherent cellular features.
Epithelial to mesenchymal transition (EMT) is a genetic program active during embryonic development and responsible for tissue remodeling and cellular motility. It is a well‐known process associated with fate determination in cells of epithelial lineage and therefore a candidate for playing a role during tumor progression. This process is activated during tumor invasion and metastasis as cells detach from the primary tumor mass, acquire mesenchymal motility properties, and migrate to distal locations. EMT is driven by transcription factors such as SNAI1/2, ZEB1/2, or TWIST1/2, resulting in enhanced migration and invasive potential of epithelial cells and is critical for the metastatic spread of epithelial tumors (a review is provided in 20). This process was demonstrated to potentiate the stem cell features of cancer cells 21, ensuring the colonization of distal sites and the generation of new tumors. Several mechanisms have been proposed to explain the EMT‐induced stemness features: the EMT inducer Zeb1 inhibits the expression of the miRNA200 family, upregulating the polycomb protein Bmi1 and inducing stemness 22.
In contrast, the reverse process of EMT, mesenchymal to epithelial transition (MET), is required to fully reprogram fibroblasts to iPSCs 23, apparently contradicting the role of EMT in generating stem cell properties. Moreover, the four canonical Yamanaka pluripotency factors transcriptionally block the EMT process: Sox2/Oct4 suppresses the EMT mediator SNAIL; c‐Myc downregulates transforming growth factor (TGF)‐β1 and TGF‐β receptor 2; and Klf4 induces epithelial genes, including E‐cadherin 23, 24, 25. In order to reconcile these observations, a sequential model is necessary. EMT is required first for tumor cell motility and invasion, and then MET takes over to regain self‐renewal and therefore the ability to colonize new niches. This possibility was demonstrated in breast cancer stem cells 26 and through the isolation of cell clones enriched in TICs showing either one of these programs and their modulation through expression of mesenchymal or epithelial promoting genes (SNAIL or E‐cadherin) 27. Cells expressing the epithelial gene program that showed enhanced self‐renewal (i.e., CSC properties) also upregulated genes associated with pluripotency and self‐renewal, including KLF4, MYC, SOX2, KLF9, and LIN28A, thus linking these abilities to the pluripotency properties observed in iPSCs. Expression of the epithelial determinant E‐cadherin was shown in breast cancer cell lines to be required to uncover stem cell properties such as the formation of mammospheres in nonadherent cell culture 28. This sequential model to explain how tumor cells can migrate and home in distal tissues suggests that cancer cells have inherent cellular plasticity and are able to fluctuate between CSCs and more differentiated states.
Sox2 Expression Activation as a Tool
We, and others, have demonstrated the role of some of the major pluripotency regulators in the onset of cancer. Sox2 seems to have a prominent role. The Sox2 gene is amplified and/or its expression is increased in lung and esophageal squamous cell carcinoma (SSC), identifying it as an oncogene for these cancers 29. Also, Sox2 is expressed in both mouse and human preneoplastic skin lesions and SCC but not in normal epidermis. The deletion of Sox2 in melanoma or SCC caused regression of tumors, and a number of genes involved in proliferation, stemness, and cell survival are regulated by Sox2 in these tumors, providing further evidence of a role for Sox2 in carcinogenesis 30. Sox2 is also expressed in breast carcinoma mammospheres and is necessary for tumor formation in vivo 31. Using elegant lineage tracing experiments in mouse models of medulloblastoma 32, CSCs were mapped to rare therapy‐resistant quiescent cells that expressed Sox2. Thus, the aberrant activation of Sox2 within a group of transformed cells causes them to shift toward a CSC phenotype, highlighting the plastic nature of neoplastic cells and lining to the mechanisms used to control the pluripotent state. The observation that Sox2 expression is not required for late stages of tumor development suggests that, similar to pluripotency reprogramming, once reprogramming has occurred and the tumor fate has been started (or at least primed), the initiating genetic events might no longer be necessary for the later stages of malignant development 31, 33. Therefore, the initiating lesion could become a passenger mutation.
In addition to increasing our understanding of tumor biology and the roots of cancer, Sox2 activation could be used as a tool to accelerate drug discovery for cancer treatment. After the initial flurry of activity for anticancer stem cell drug development a decade ago, the biology of cancer stem cells has proved complex and difficult to translate into effective therapeutic strategies. Conventional drug screening relies on validated targets for which fast and automated assays are developed. However, to monitor the effect of test compounds on cancer stem cells, targets must be defined. This is, however, cumbersome, because for many tumor indications, cancer stem cell targets have not been clearly defined, with often nonoverlapping combinations of markers defining cell populations with cancer stem cell activities or tumor initiation ability. This most likely reflects the changing nature of the stemness capacity in tumor cells. The evolution of tumor cell populations and the existence of competing programs promoting invasiveness and pluripotency points to a dynamic state in tumor cells characterized by the interconversion of cells with and without stem cell properties, regardless of the actual nature of the driver promoter for plasticity. Aberrant activation of Sox2 in tumor cells might provide this specificity for cancer stem cell‐targeted drug screening. We, and others, have proved that the use of fluorescence protein expression‐based reporters for Sox2 enhancer element activation identifies cells with tumor initiating activity 34, 35, 36, 37. Compounds that are capable of blocking activation of Sox2 in the relevant cell systems might be good candidates for anticancer drug development. These reporter systems could also be used to track CSCs in tumors, study CSC niches, and study the interactions between CSCs and tumor stroma.
Modeling of Tumor Formation
Few models of human cancer progression are currently available. Primary human cells can undergo cancer progression in mouse xenografts models but require previous transformation with oncogenes different from the endogenous genetic changes found in natural tumors. Patient‐derived human tumor cells are also not a good model, because oncogenic transformation has already occurred, making it difficult to recapitulate the events leading to the generation of CSCs in the first place. Reprogramming primary cancer cells to pluripotent states could be a useful tool to “normalize” tumor cells and then capture the early stages of tumor progression, regardless of the controversy regarding whether the neoplastic state is itself reversible, as only certain cancer genomes or cell types are amenable to this manipulation 38, 39, 40, 41. Small compound screening for drugs that can substitute the expression of pluripotency factors 42 might be used to reveal new drugs able to reprogram cancer cells to a pluripotent state, preventing tumor spread from rapid proliferating nonstem cells. However, methods to control this stem cell population must be set in place in order for this therapy to be successful. Nevertheless, partially reprogrammed tumor cells can be induced to differentiate into disease‐affected lineages, resulting in an unprecedented tool to model tumorigenesis and understand the role of specific oncogenic mutations in that precise tumor type and allowing patient‐specific tumor models. This is very relevant for cancers for which no cellular model is available, such as pancreatic ductal adenocarcinoma. Thus, combining pluripotency reprogramming and animal models demonstrated the continuum from a premalignant phenotype to malignant progression 41, although just one of nine patient samples could be fully reprogrammed, evidencing the intrinsic barriers that transformation imposes on reprogramming. The democratization of genome‐editing technologies within the past few years has made it viable to generate human disease cellular models with greater efficiency. Correction of specific genetic alterations in the genomes of reprogrammed tumor cells would allow one to directly connect genotype and phenotype to establish causality (e.g., for validation of new cancer gene candidates as markers of premalignancy) in an undifferentiated cancer background during its evolution toward more differentiated states. Novel biomarkers for the early detection of cancers might be discovered using this approach, maximizing the possibilities for curative interventions.
The design of new animal models that consider the reprogramming capacities of the oncogenes (e.g., the ones referred to in 43) is necessary to mimic human disease and generate models useful for the development of new therapeutic approaches. Built‐in CSC transcriptional reporters would help to track the evolution of CSC populations during the in vivo evaluation of the next generation of antitumor drugs and contribute to the development of more effective chemotherapy regimens targeting both the bulk of the tumor and the CSC compartment.
Tools for Ensuring Biosafety of Cell‐Based Therapies
As mentioned, some of the multifactorial events involved in the reprogramming and acquisition of stemness properties in pluripotent stem cells can also lead to the generation of malignant tumor cells (Fig. 1). Currently, the major limitation for the use of pluripotent stem cells, and cells derived from pluripotent stem cells, in the clinic is the safety concern, because tumorigenicity remains an unsolved concern for clinical application 44 (Fig. 2). Therefore, a greater understanding of the parallelisms between these two processes might lead to novel approaches to prevent the transformation of cells used as cell therapies, ensuring the biosafety of these treatments. The advances in regenerative medicine and the generation of induced pluripotent stem cells are providing cancer scientists with a new set of tools that will be very useful for the characterization of cancer stem cells.
Figure 1.

Schematic representation of the parallel processes of pluripotency reprogramming and tumorigenesis from a mechanistic viewpoint. Primary cells of diverse origins are transduced with pluripotency factors or oncogenes that, upon inhibition of tumor suppressors, extensive epigenetic remodeling, and a switch to a glycolytic metabolism, render clones or colonies with embryonic stem cell‐like morphology that are selected manually until fully pluripotent (self‐renewing and undifferentiated) colonies are established. In the case of tumor formation, selection for self‐renewing and undifferentiated (in terms of tumor cell potential) occurs naturally in the organism.
Figure 2.
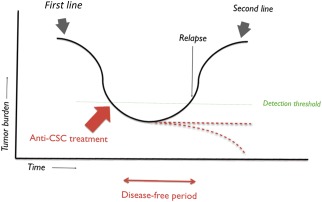
Proposed clinical positioning of anticancer stem cell‐directed treatments. Most oncologic patients will show a response to first‐line treatment; however, if the cancer stem cell compartment is left untouched, resilient cancer stem cells could fuel new tumor growth later on. Therefore, if the cancer stem cell compartment can be controlled after the first line of treatment, relapses and metastases might be prevented. Abbreviation: CSC, cancer stem cell.
Conclusion
The concept of cellular plasticity is challenging how we treat tumors and could explain why tumors relapse. The links found between the acquisition of pluripotency properties and tumor cell plasticity have uncovered novel targets that might be suitable for anticancer therapeutic development, attacking the molecular roots behind the generation of relapses and tumor metastases. More efficient chemotherapy regimens are needed that target the cancer stem cells and the bulk of the tumor composed of more differentiated cancer cells. Conventional cancer therapy has focused on attacking the proliferating cells, with little attention given to tumor cell structure and heterogeneity. Defining the molecular drivers of cancer stem‐like cells will provide avenues for the rational design of cancer treatment regimens aimed at CSCs that, combined with conventional treatments targeting the bulk tumor cells, will provide novel therapies for cancer patients. As complicated as targeted drug design might seem for discovering novel anticancer stem cell‐directed treatments, screening methods based on pluripotency transcriptional reporters could help to identify new drugs targeting cancer stem cells and cellular plasticity. Given the inherent plasticity of cancer cells, it is envisioned that these novel therapeutics would not likely be used as the first line of treatment but would be positioned as maintenance therapy for subsequent relapse prevention after the first line of presumably aggressive therapy has been completed. The arsenal of cytotoxic drugs targeting fast dividing tumor cells is well developed; therefore, combinations with novel drugs that prevent acquisition of stemness properties in cancer cells would ensure a durable anticancer response.
Author Contributions
J.M.I., J.G., and A.G.M.: manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
J.M.I. is an employee of Synpromics Ltd. J.G. is an employee of StemTek Therapeutics. A.G.M. is an employee of, and has stock options in, StemTek Therapeutics.
References
- 1. Ben‐Porath I, Thomson MW, Carey VJ et al. An embryonic stem cell‐like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong DJ, Liu H, Ridky TW et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008;2:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bussard KM, Boulanger CA, Booth BW et al. Reprogramming human cancer cells in the mouse mammary gland. Cancer Res 2010;70:6336–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pezzolo A, Parodi F, Marimpietri D et al. Oct‐4+/Tenascin C+ neuroblastoma cells serve as progenitors of tumor‐derived endothelial cells. Cell Res 2011;21:1470–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci USA 1976;73:549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 7. Kawamura T, Suzuki J, Wang YV et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009;460:1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong H, Takahashi K, Ichisaka T et al. Suppression of induced pluripotent stem cell generation by the p53‐p21 pathway. Nature 2009;460:1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tapia N, Schöler HR. p53 connects tumorigenesis and reprogramming to pluripotency. J Exp Med 2010;207:2045–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li H, Collado M, Villasante A et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009;460:1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Riggs JW, Barrilleaux BL, Varlakhanova N et al. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev 2013;22:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vazquez‐Martin A, Vellon L, Quirós PM et al. Activation of AMP‐activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012;11:974–989. [DOI] [PubMed] [Google Scholar]
- 13. Menendez JA, Corominas‐faja B, Cuyàs E. Metabostemness: Metaboloepigenetic reprogramming of cancer stem‐cell functions. Oncoscience 2014;1:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abad M, Mosteiro L, Pantoja C et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013;502:340–345. [DOI] [PubMed] [Google Scholar]
- 15. Ohnishi K, Semi K, Yamamoto T et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014;156:663–677. [DOI] [PubMed] [Google Scholar]
- 16. Polo JM, Anderssen E, Walsh RM et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012;151:1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corominas‐Faja B, Cufí S, Oliveras‐Ferraros C et al. Nuclear reprogramming of luminal‐like breast cancer cells generates Sox2‐overexpressing cancer stem‐like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle 2013;12:3109–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buczacki SJA, Zecchini HI, Nicholson AM et al. Intestinal label‐retaining cells are secretory precursors expressing Lgr5. Nature 2013;495:65–69. [DOI] [PubMed] [Google Scholar]
- 19. Van Keymeulen A, Rocha AS, Ousset M et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011;479:189–193. [DOI] [PubMed] [Google Scholar]
- 20. Yang J, Weinberg RA. Epithelial‐mesenchymal transition: At the crossroads of development and tumor metastasis. Dev Cell 2008;14:818–829. [DOI] [PubMed] [Google Scholar]
- 21. Mani SA, Guo W, Liao M‐J et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wellner U, Schubert J, Burk UC et al. The EMT‐activator ZEB1 promotes tumorigenicity by repressing stemness‐inhibiting microRNAs. Nat Cell Biol 2009;11:1487–1495. [DOI] [PubMed] [Google Scholar]
- 23. Li R, Liang J, Ni S et al. A mesenchymal‐to‐epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010;7:51–63. [DOI] [PubMed] [Google Scholar]
- 24. Samavarchi‐Tehrani P, Golipour A, David L et al. Functional genomics reveals a BMP‐driven mesenchymal‐to‐epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010;7:64–77. [DOI] [PubMed] [Google Scholar]
- 25. Redmer T, Diecke S, Grigoryan T et al. E‐cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep 2011;12:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu S, Cong Y, Wang D et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep 2014;2:78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Celià‐Terrassa T, Meca‐Cortés O, Mateo F et al. Epithelial‐mesenchymal transition can suppress major attributes of human epithelial tumor‐initiating cells. J Clin Invest 2012;122:1849–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manuel Iglesias J, Beloqui I, Garcia‐Garcia F et al. Mammosphere formation in breast carcinoma cell lines depends upon expression of E‐cadherin. PLoS One 2013;8:e77281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bass AJ, Watanabe H, Mermel CH et al. SOX2 is an amplified lineage‐survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 2009;41:1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boumahdi S, Driessens G, Lapouge G et al. SOX2 controls tumour initiation and cancer stem‐cell functions in squamous‐cell carcinoma. Nature 2014;511:246–250. [DOI] [PubMed] [Google Scholar]
- 31. Leis O, Eguiara A, Lopez‐Arribillaga E et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012;31:1354–1365. [DOI] [PubMed] [Google Scholar]
- 32. Vanner RJ, Remke M, Gallo M et al. Quiescent Sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014;26:33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lengerke C, Fehm T, Kurth R et al. Expression of the embryonic stem cell marker SOX2 in early‐stage breast carcinoma. BMC Cancer 2011;11:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iglesias JM, Leis O, Pérez‐Ruiz E et al. The activation of the Sox2 RR2 pluripotency transcriptional reporter in human breast cancer cell lines is dynamic and labels cells with higher tumorigenic potential. Front Oncol 2014;4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jung K, Gupta N, Wang P et al. Triple negative breast cancers comprise a highly tumorigenic cell subpopulation detectable by its high responsiveness to a Sox2 regulatory region 2 (SRR2) reporter. Oncotarget 2015;6:10366–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang S, Furuhashi M, Nakane R et al. Isolation and characterization of human breast cancer cells with SOX2 promoter activity. Biochem Biophys Res Commun 2013;437:205–211. [DOI] [PubMed] [Google Scholar]
- 37. Tang B, Raviv A, Esposito D et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Rep 2015;4:155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hochedlinger K, Blelloch R, Brennan C et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev 2004;18:1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carette JE, Pruszak J, Varadarajan M et al. Generation of iPSCs from cultured human malignant cells. Blood 2010;115:4039–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang X, Cruz FD, Terry M et al. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency based reprogramming. Oncogene 2013;32:2249–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim J, Hoffman JP, Alpaugh RK et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Reports 2013;3:2088–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shan J, Schwartz RE, Ross NT et al. Identification of small molecules for human hepatocyte expansion and iPS differentiation. Nat Chem Biol 2013;9:514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romero‐Camarero I, Barajas‐Diego M, Castellanos‐Martín A et al. New models towards assessing anti‐cancer therapeutics. Histol Histopathol 2012;27:157–170. [DOI] [PubMed] [Google Scholar]
- 44. Okano H, Nakamura M, Yoshida K et al. Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res 2013;112:523–533. [DOI] [PubMed] [Google Scholar]