

Abstract
To address concerns regarding the tumorigenic potential of undifferentiated human pluripotent stem cells (hPSC) that may remain after in vitro differentiation and ultimately limit the broad use of hPSC‐derivatives for therapeutics, we recently described a method to selectively eliminate tumorigenic hPSC from their progeny by inhibiting nicotinamide phosphoribosyltransferase (NAMPT). Limited exposure to NAMPT inhibitors selectively removes hPSC from hPSC‐derived cardiomyocytes (hPSC‐CM) and spares a wide range of differentiated cell types; yet, it remains unclear when and how cells acquire resistance to NAMPT inhibition during differentiation. In this study, we examined the effects of NAMPT inhibition among multiple time points of cardiomyocyte differentiation. Overall, these studies show that in vitro cardiomyogenic commitment and continued culturing provides resistance to NAMPT inhibition and cell survival is associated with the ability to maintain cellular ATP pools despite depletion of NAD levels. Unlike cells at earlier stages of differentiation, day 28 hPSC‐CM can survive longer periods of NAMPT inhibition and maintain ATP generation by glycolysis and/or mitochondrial respiration. This is distinct from terminally differentiated fibroblasts, which maintain mitochondrial respiration during NAMPT inhibition. Overall, these results provide new mechanistic insight into how regulation of cellular NAD and energy pools change with hPSC‐CM differentiation and further inform how NAMPT inhibition strategies could be implemented within the context of cardiomyocyte differentiation. Stem Cells Translational Medicine 2017;6:1191–1201
Keywords: Nicotinamide phosphoribosyltransferase, NAD, Pluripotent stem cells, Cardiomyocyte, Differentiation
Significance Statement.
This study provides new mechanistic insight into how regulation of cellular NAD and energy pools change with cardiomyocyte differentiation from human pluripotent stem cells (hPSC) and further informs how inhibition of nicotinamide phosphoribosyltransferase could be implemented within the context of cardiomyocyte differentiation to eliminate tumorigenic cells. Here we show how to refine approaches for eliminating tumorigenic cells that may be present in cardiomyogenic differentiation cultures, with shorter pulse treatments as a potential strategy to eliminate hPSC from early hPSC‐CM cultures, while longer treatments may be used for treating hPSC‐CM cultures later in differentiation.
Introduction
Human embryonic (hESC) and induced pluripotent stem cells (hiPSC), collectively termed human pluripotent stem cells (hPSC), are a renewable source for in vitro generation of a wide variety of cell types that are useful for the study of human development, disease modeling, and regenerative medicine. Significant progress has been made in developing efficient strategies for differentiating hPSC to many cell types, including cardiomyocytes (hPSC‐CM) that have proven useful for drug toxicity testing and human disease modeling 1, 2, 3, 4. However, the broad use of hPSC‐derived cells for human therapeutics has been limited by technical challenges regarding cell purity, subtype heterogeneity, maturation stage, mode of transplantation, and safety concerns including the potential for remnant hPSC to form teratomas at the site of transplantation 5, 6, 7, 8, 9.
To address the concern for teratoma tumor formation, several methods have been proposed to selectively eliminate undifferentiated hPSC from cultures of differentiated cells 10, 11, 12, 13, 14, 15, many of which target differences in cellular metabolism between hPSC and their differentiated progeny. We recently showed that inhibition of nicotinamide phosphoribosyltransferase (NAMPT) selectively eliminates hPSC and is advantageous for its efficacy in a broad range of culture conditions 16, 17. NAMPT, the rate limiting enzyme in the salvage pathway utilizing nicotinamide for NAD synthesis, is important for maintaining sufficient NAD levels to support pluripotency and for reprograming somatic cells to hiPSC 18. Ultimately, NAMPT inhibition in hPSC leads to a loss in ATP and cell death 17, 18. Early stage hPSC‐derivatives, including hPSC‐CM and neural progenitors, are resistant to short periods of NAMPT inhibition, whereas mature, differentiated cells, such as retinal epithelial cells and fibroblasts display increased resistance, suggesting that differentiation and maturation promote survival during NAMPT inhibition 17. In part, alterations in NAD or metabolic regulation during cellular differentiation may explain the differential sensitivity between hPSC and hPSC‐CM to NAMPT inhibition. hPSC are characterized by relatively immature mitochondria, increased reliance on glycolysis, and elevated flux through the pentose phosphate pathway 19, 20. As hPSC differentiate to hPSC‐CM, several changes in metabolism have been identified, including an increase in flux through the tricarboxylic acid cycle, use of alternative metabolic substrates, and increased mitochondrial maturation and respiration 6, 14, 18, 20, 21, 22, 23. Importantly, many of these metabolic changes occur in a time‐dependent manner during differentiation 24, 25 and a number of recent studies have exploited these changes to select for hPSC‐CM or promote maturation 14, 21, 26.
While hPSC‐CM show resistance to NAMPT inhibition 16, 17, it is not yet understood how in vitro differentiation influences susceptibility to NAMPT inhibition. Such information would inform how and when NAMPT inhibition based approaches could be used for the elimination of tumorigenic cells from hPSC‐derived progeny. Also, further understanding of metabolic flexibility, and how it changes during differentiation, could benefit strategies designed to drive maturation in vitro and select maturation stage specific cells. Therefore, the goals of this study were to determine when cells become resistant to NAMPT inhibition during hPSC‐CM differentiation and how cells maintain ATP despite a loss of NAD. Changes in cellular responses to NAMPT inhibition were examined across multiple timepoints of hPSC‐CM differentiation and in a terminally differentiated fibroblast cell line. We find that while day 10 hPSC‐CM are resistant to shorter exposure times, day 28 hPSC‐CM can survive 72 hours NAMPT inhibition and maintain the ability to spontaneously contract. We show that NAMPT inhibitor‐induced death in hiPSC is not associated with cell proliferation or poly (ADP‐ribose) polymerase (PARP) activation. Rather, we show cell survival during NAMPT inhibition correlates with an ability to maintain ATP levels, despite decreased NAD levels, through continued utilization of glycolysis or mitochondrial respiration in a cell type dependent manner.
Materials and Methods
Cell Culture and Reagents
hiPSC (DF6‐9‐9T), hESC (H9) and human dermal fibroblasts (ATCC CRL‐2097) were cultured as previously described 16, 17. Cardiomyocyte differentiation was performed using DF6‐9‐9T plated at 5.73‐6.25e4 cells per cm2 and H9 plated at 1.55e4 cells per cm2 with differentiation as described 27. For differentiation using the CDM3 protocol, DF6‐9‐9T plated at 2.95e4 cells/cm2 cultured for 96 hours and differentiation was performed using the CDM3 protocol as described 28, with the exception that lactate selection was initiated at day 12 of differentiation for 96 hours. Experiments were performed on cultures with cells in a beating monolayer that are routinely >80% troponin T2 (TNNT2) positive by day 10 (17, 27, Supporting Information Fig. 1). Day 28 hiPSC‐CM were passaged using a modified protocol from Burridge et al. 28. Cells were incubated in 0.5 U/ml Liberase TH and 50 U/ml DNase I in 1 ml RPMI for 20 minutes at 37°C, then 1 ml TrypLE express was added. Cells were dispersed with gentle trituration every 1–2 minutes and plated on Matrigel at 4.6e5 cells per cm2 unless otherwise specified. For all experiments, cells were given 72 hours to recover prior to further use. Glucose deprivation media was prepared using base media with no glucose and with addition of media components as previously reported 16, 27.
Characterization of Day 28 hPSC‐CM
Day 28 hPSC‐CM were collected and flow cytometry for TNNT2 was performed as previously described (Abcam, Cambridge, MA, www.abcam.com, ab8295‐1 µg primary antibody per 1e6 cells) 27. For qPCR, day 28 hESC‐CM were treated with dimethyl sulfoxide (DMSO) or STF‐31 (2.5 uM) for 72 hours. RNA was isolated using RNeasy Plus Mini kit (Qiagen, Germantown, MD, www.qiagen.com) and reverse transcribed with iScript Reverse Transcription Supermix (Biorad, Hercules CA, www.bio-rad.com). For qPCR, 10 ng of cDNA was loaded per assay with TaqMan universal mastermix and run with Quant Studio 6 Flex (Thermo Fisher Scientific, Waltham, MA, www.thermofisher.com). TaqMan primers were used for POU5F1 (Hs04260367_gH), NANOG (Hs04260366_g1), and TNNT2 (Hs00165960_m1). Samples were analyzed using the ΔΔCT method normalized to beta‐actin (ACTB) and hiPSC positive control for hPSC genes (POU5F1 and NANOG) and Day 28 hESC‐CM treated with DMSO for TNNT2.
Cell Viability Assays
Two different NAMPT inhibitors, FK866 and STF‐31 29, 30, 31 were used throughout the study. Cells were treated with 100 nM FK866, 2.5 µM STF‐31, or glucose deprivation continuously for 72 hours with daily medium replacement. Cell viability was assessed with neutral red assay and SYTOX Green nucleic acid stain as previously described 17 with the time of SYTOX incubation extended to 1 hour. To mitotically inactivate cells, 24 hours after plating, hPSC were treated with 8 µg/ml mitomycin c for 1.5 hours then washed four times with D‐PBS prior to treatment with 2.5 µM STF‐31 for 0–72 hours. For the fibroblast cell growth curve, STF‐31 treatment was initiated 24‐hour post plating and cell counts were performed daily up to 72 hours of continuous treatment. For mitotic inactivation and fibroblast growth curve measurements, cell viability was measured by trypan blue exclusion using a hemocytometer and viable cell count was reported for one well of a 24‐well plate.
Imaging
Immunofluorescence detection of TNNT2 was performed on day 28 hiPSC‐CM passaged onto 18 mm coverslips as previously described 17 using mouse anti‐troponin T2 (3 µg/coverslip; ThermoFisher) and anti‐mouse IgG1 Alexa 568 (2 µg/coverslip; Thermofisher). For detection of mitochondrial potential, cells were incubated with 50 nM tetramethylrhodamine ethyl ester (TMRE) for 15 minutes at 37°C in 5% CO2, rinsed in D‐PBS and imaged immediately. Immunofluorescent and TMRE staining were imaged on a Nikon Eclipse 90i confocal microscope. Brightfield imaging of cell contraction and morphology was performed on a Nikon Ti‐U inverted microscope.
Nucleotide Analysis
Nucleotides were collected from hPSC, hPSC‐CM, and fibroblasts using acidic lysis and the levels of nucleotides were assessed using high performance liquid chromatography (HPLC) as previously described 17, 32.
Lactate Assay
Lactate secretion into media was measured following 24–72 hours of treatment with NAMPT inhibitors or glucose deprivation in subconfluent hiPSC, day 28 hiPSC‐CM, and fibroblasts. Cells were washed twice with D‐PBS and placed in 500 µl of DMEM/F12 (hiPSC), DMEM (day 28 hiPSC‐CM and fibroblasts), DMEM minus glucose (glucose deprivation) without phenol red and were incubated for 2 hours at 37°C. Media were collected and placed on ice or stored at −80°C until use. Cells were lysed and total protein per well was quantified with Qubit Protein Assay (Thermofisher). Lactate levels in the medium were determined using lactate assay kit I or II (Sigma‐Aldrich, St. Louis, MO, www.sigmaaldrich.com) per manufacturer's instructions and normalized to total protein.
Mitochondrial Stress Test
Cells were plated in XF24 plates as follows: hiPSC (6e4 cells per well for 48 hours), day 28 hiPSC‐CM (1.2e5 cells per well for 72 hours) and fibroblasts (8.5e3 cells per well for 24 hours). Oxygen consumption rate was measured by a mitochondrial stress test with a Seahorse Bioscience XF24 Analyzer using subsequent additions of oligomycin (1.25 µM for hiPSC, and 3 µM for fibroblasts and day 28 hiPSC‐CM), carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP) (1 µM all cell types), and Antimycin A (10 µM). Components of respiration were calculated as recommended by the manufacturer (Agilent, Santa Clara, CA, www.agilent.com) and normalized to total protein (µg).
Statistics
Data are represented as mean with standard error of the mean. When appropriate, one‐way analysis of variance (ANOVA) or students two tailed t test was performed when comparing treatments within a cell type. For comparisons among time points and treatment groups, unpaired, two‐way ANOVA was performed. All ANOVA calculations were performed with multiple comparisons using Tukey post hoc test. All statistics were analyzed using GraphPad Prism version 6.07.
Results
Survival During NAMPT Inhibition Increases with Cardiomyocyte Differentiation and Maturation
To determine when cardiomyocyte differentiation alters susceptibility to NAMPT inhibition, cells were treated with NAMPT inhibitors, STF‐31 or FK866, continuously for 72 hours beginning on day 0 (confluent monolayer of hiPSC), day 5 (committed cardiac progenitors), day 10 (committed cardiomyocytes that spontaneously contract), and day 28 (time point by which cells show increased oxidative phosphorylation from alternative substrates 21 and adopt a more elongated mitochondrial morphology as compared to day 10 cells (Supporting Information Fig 2) and 18, 23, 33). Cell viability under NAMPT inhibition was assessed by neutral red uptake (an indirect assay of ATP levels) and SYTOX cell death assay (dependent on cell membrane permeability). Consistent with our previous studies 16, 17, continuous NAMPT inhibition is toxic to hiPSC (Fig. 1a, 1b). However, the number of cells that survive NAMPT inhibition increases with differentiation. Day 5 represents the first time in differentiation where a population of cells survive prolonged NAMPT inhibition (Fig. 1a, 1b and Supporting Information Fig. 3a, 3b). Although day 5 vehicle control treated hiPSC‐CM and hESC display increased cell death, possibly due to addition of IWR‐1 at this stage of differentiation, a population of cells remains viable after 72 hours of NAMPT inhibition. Moreover, a pulse treatment for 24 hours with 5 µM STF‐31 on day 5 avoids significant toxicity (Supporting Information Fig. 4A) and does not affect the ability of these cells to continue differentiating into contracting monolayers by day 15 (Supporting Information video 1 and 2). Day 10 hiPSC‐CM and hESC‐CM have increased cell survival with NAMPT inhibition; however, spontaneous contraction ceases by 72 hours of treatment and increased cell death is observed by 96 hours (data not shown). The toxicity resulting from continuous NAMPT inhibitor treatment at day 5 and 10 is consistent with our previous report 17, demonstrating that treatment with 2.5 µM STF‐31 for 24–48 hours did not produce adverse effects on hiPSC‐CM, although measurable toxicity was observed with 72 hours treatment.
Figure 1.
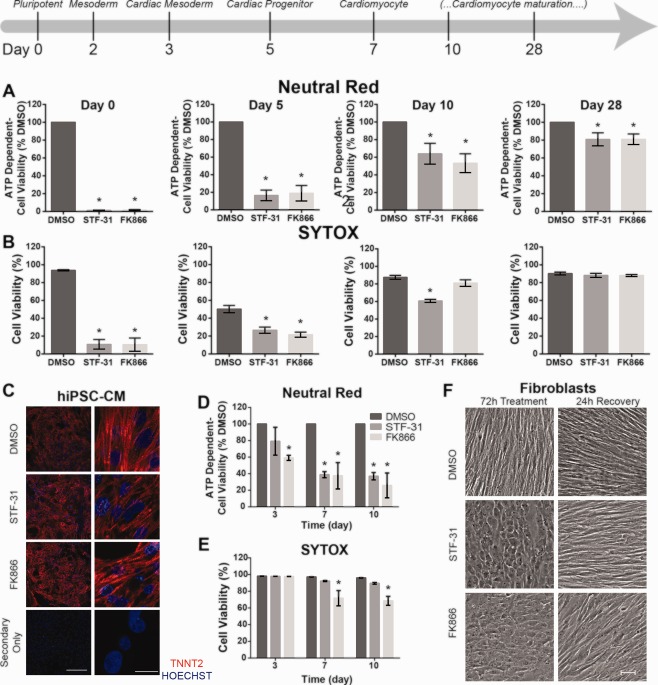
Nicotinamide phosphoribosyltransferase inhibition mediated toxicity decreases as human pluripotent stem cells differentiate and continue to mature. (A, B): Bar graphs of cell viability as measured by neutral red (A) or SYTOX cell death assay (B) in cultures at various stages of differentiation (day 0, 5, 10, 28) treated with 2.5 µM STF‐31 or 100 nM FK866 for 72 hours (C): Representative immunofluorescence staining for cardiac troponin T2 (red) and nuclei (Hoechst‐blue) in passaged day 28 hiPSC‐CM treated with 2.5 µM STF‐31 or 100 nM FK866 for 72 hours with imaging at ×20 (left) and ×100 (right). Bottom panel represents staining with secondary antibody only. Scale bar is 200 µm and 20 µm, respectively. (D, E): Bar graphs of cell viability as measured by neutral red (D) or SYTOX cell death assay (E) in human dermal fibroblasts following 3‐10 days of continuous treatment with 2.5 µM STF‐31 or 100 nM FK866. (F): Representative brightfield images showing fibroblast morphology at 10x following 72 hours continuous treatment with 2.5 µM STF‐31 or 100 nM FK866 and 24 hours recovery after washout of treatment at 72 hours. Scale bar is 50 µm. Data are represented as mean ± SEM for 3‐6 biological replicates in each group (N = 3 STF‐31 and FK866 treatment; N = 4–6 DMSO). *, p < .05. Abbreviations: DMSO, dimethyl sulfoxide; hiPSC‐CM, human induced pluripotent stem cells‐derived cardiomyocytes.
In contrast to cells from earlier time points of differentiation, 72 hours NAMPT inhibition in day 28 hiPSC‐CM and hESC‐CM does not result in significant cellular toxicity (Fig. 1a, 1b and Supporting Information Fig. 3a, 3b). To confirm that these observations are not dependent on the differentiation protocol, an alternative CDM3 cardiomyocyte differentiation protocol was used. Results are consistent between the two differentiation protocols used, as there is no significant difference in cell viability after 72 hours of STF‐31 treatment in Day 28 hiPSC‐CM generated with the CDM3 differentiation protocol (Supporting Information Fig. 3c). For all cell types and differentiation protocols, day 28 hPSC‐CM continue to spontaneously contract throughout 72 hours treatment (Supporting Information video 3 and 4) and remain positive for cardiac TNNT2 with structural organization comparable to vehicle control (Fig. 1c). Relative expression of TNNT2 and pluripotency markers POU5F1 and NANOG are not altered with STF‐31 treatment in day 28 hESC‐CM and expression of pluripotency markers is minimal when compared to hPSC (Supporting Information Fig. 5). These results indicate that cell survival during NAMPT inhibition increases with time in culture, where measurable viability (∼15%–30%) is first observed in committed cardiac progenitors and increased total viability (∼50%–80%) is observed in the early phases of cardiomyocyte commitment.
Fibroblasts Survive Prolonged Treatment With NAMPT Inhibitors
In addition to hiPSC‐CM, other hiPSC‐derived progeny and somatic cell lines are resistant to NAMPT inhibition 17. Here, the effects of prolonged NAMPT inhibition on human dermal fibroblasts were examined to determine if terminally differentiated cells are more resistant to longer treatment times as compared to immature cell types (i.e., day 5 or 10 hiPSC‐CM). Dermal fibroblasts were selected because they are a terminally differentiated cell type derived from the same mesoderm lineage as cardiomyocytes and are the precursor cells from which the hiPSC are derived. In fibroblasts treated for 72 hours, only FK866 treatment led to a significant decrease in viability as determined by neutral red assay. No toxicity was detected by SYTOX assay for either NAMPT inhibitor by 72 hours (Fig. 1d, 1e). Increasing the treatment time to 7–10 days similarly led to a decrease in viability as measured by neutral red uptake for both inhibitors. However, only FK866 led to significant, though incomplete, toxicity as measured by SYTOX (∼30%) (Fig. 1e). Although, fibroblast toxicity was not affected following 72 hours of STF‐31, altered cellular morphology was observed by brightfield microscopy (Fig. 1F). Between 48 and 72 hours of treatment, fibroblasts treated with both STF‐31 and FK866 display a rounded morphology, without alteration in cell attachment. These changes are reversible as cells allowed to recover for 24 hours following removal of NAMPT inhibitors are morphologically indistinguishable from control cells. Altogether, terminally differentiated fibroblasts are similar to day 28 hiPSC‐CM in their resistance to the toxic actions of prolonged periods of NAMPT inhibition as compared to hPSC or progenitor cells.
Survival Correlates With the Maintenance of ATP levels
NAMPT inhibition in hPSC leads to a loss of NAD, decrease in mitochondrial respiration and glycolysis, and ultimately ATP depletion 17. Therefore, cellular NAD+ and ATP levels were measured in the presence or absence of NAMPT inhibition throughout hPSC‐CM differentiation and in fibroblasts to determine whether survival from the toxic actions of STF‐31 and FK866 associates with altered nucleotide levels in these cell types in comparison to hPSC. In hPSC, NAMPT inhibition causes a rapid depletion in NAD+ followed by a decrease in ATP levels by 24 hours (Fig. 2a and Supporting Information Fig. 6). In contrast, hPSC‐CM and fibroblasts maintain ATP levels, despite loss of NAD+ (Fig. 2a and Supporting Information Fig. 6a). In day 10 hPSC‐CM, NAD+ levels decrease within 24 hours of NAMPT inhibition while ATP levels are maintained until 48 hours. In day 28 hPSC‐CM, NAMPT inhibition results in the depletion of NAD+ by 48 hours of treatment without significant loss of ATP at 72 hours (Fig. 2a and Supporting Information Fig. 6). These findings suggest that differentiation of hPSC to hPSC‐CM is associated with an increased ability to maintain ATP levels under conditions in which cellular NAD+ is depleted. Consistent with cell viability data described above, as hPSC‐CM differentiate they become partially resistant to a loss in ATP during NAD+ depletion resulting from NAMPT inhibition by day 10 and are fully resistant by day 28. Terminally differentiated fibroblasts are similar to day 28 hPSC‐CM as they maintain ATP throughout treatment with STF‐31 (Fig. 2a).
Figure 2.
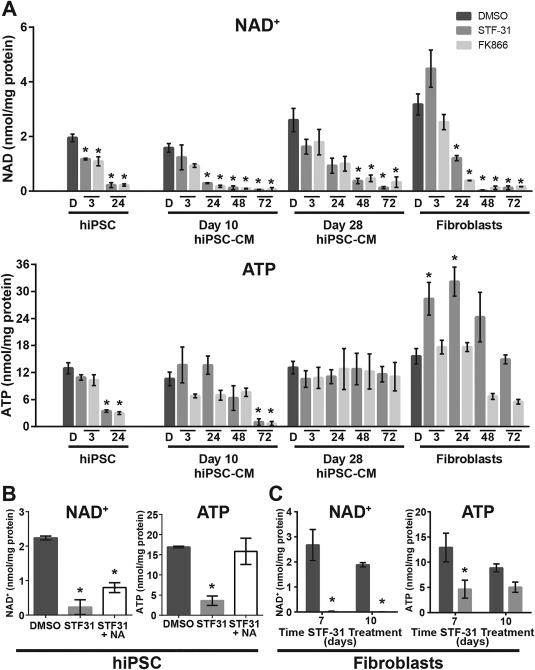
ATP levels are maintained in differentiated cardiomyocytes despite NAD+ depletion. (A): Bar graphs representing cellular nucleotide pools of NAD+ and ATP following 3‐72 hours treatment with 2.5 µM STF‐31, and 100 nM FK866 in hiPSC (only 3‐24 hours), day 10 and 28 hiPSC‐CM, and fibroblasts (3‐72 hours). (B): Bar graphs depicting NAD+ and ATP levels in hiPSC with 24 hours 2.5 µM STF‐31 or combined treatment with 10 µM NA and 2.5 µM STF‐31. (C): Bar graphs depicting NAD+ and ATP levels in fibroblasts after 7 and 10 days of continuous treatment with 2.5 µM STF‐31. Data are represented as mean ± SEM for 3‐7 biological replicates in each group (N = 3–4 STF‐31 and FK866 treatment; N = 4–7 DMSO). *, p < .05. Abbreviations: DMSO, dimethyl sulfoxide; hiPSC‐CM, human induced pluripotent stem cell‐derived cardiomyocytes.
To further determine the relationship between NAD+ levels, ATP maintenance and cell survival, hiPSC were cotreated with STF‐31 and nicotinic acid (NA, 10 µM), which provides an alternative substrate for NAD synthesis to bypass the inhibition of NAMPT. NAD+ levels are restored to ∼35% of DMSO control and prevent ATP depletion (Fig. 2b). Thus, we observe that a partial rescue of NAD+ levels is sufficient to completely prevent depletion of ATP (Fig. 2b) and cell viability 17 in hiPSC during NAMPT inhibition. In the case of fibroblasts, exposure to extended periods of NAMPT inhibition (2.5 µM STF‐31 for 7 and 10 days) is associated with the maintenance of ATP despite almost complete loss of cellular NAD+ (Fig. 2c). Taken together, these data suggest that the ability of differentiated cells to survive NAMPT inhibition depends on their ability to maintain a small pool of NAD+ that supports the cells' ability to preserve ATP levels.
Cell Death due to NAMPT Inhibition—The Role of Cell Proliferation and PARP Activation
During cell growth or proliferation there may be an increased demand for NAD or ATP. To evaluate if the selective toxicity mediated by NAMPT inhibition in hPSC is dependent on the highly proliferative nature of these cells, hiPSC were mitotically inactivated with a short pulse treatment of mitomycin c (8 µg/ml for 1.5 hours). Control and mitotically inactivated hiPSC were treated with 2.5 µM STF‐31 for 0–72 hours and the total viable cell number was determined (Fig. 3A). As expected, DMSO control hiPSC proliferate until 48 hours after which they reach confluence. Mitomycin c treatment prevented proliferation in hiPSC. Comparable to proliferating hiPSC, STF‐31 treatment in mitotically inactive hiPSC was toxic, leading to the absence of live cells by 72 hours. While these findings show that cell proliferation does not modify the ability of NAMPT inhibitors to induce hiPSC death, they suggest that NAMPT inhibition may attenuate hiPSC proliferation, as hiPSC treated with STF‐31 for 24 hours do not increase in cell number. However, the decrease in cell proliferation appears to be cell type selective as NAMPT inhibition does not alter the fibroblast growth curve (Fig. 3b).
Figure 3.
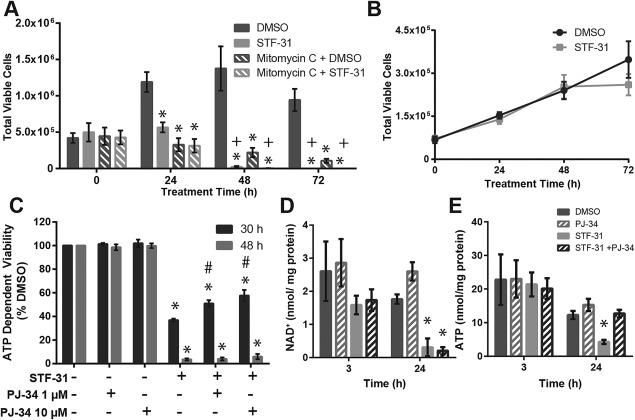
Nicotinamide phosphoribosyltransferase Inhibition mediated toxicity is independent of proliferation and proliferation or poly (ADP‐ribose) polymerase activity. (A): Bar graph of total viable cells as measured by trypan blue exclusion in proliferating and mitotically inactivated DF6‐9‐9T (mitomycin c for 3 hours) treated with 2.5 µM STF‐31 for 24 and 72 hours. (B): Growth curve of fibroblasts treated with 2.5 µM STF‐31 and 100 nM FK866 for 0‐72 hours. (C): Cell viability measured by neutral red in DF6‐9‐9T treated with 2.5 µM STF‐31 and 1 or 10 µM PJ‐34 for 30 and 48 hours. (D, E): NAD+ (D) and ATP (E) levels measured by HPLC after treatment with 2.5 µM STF‐31 and 10 µM PJ‐34 for 3 and 24 hours. Data are represented as mean ± SEM for N = 3 biological replicates in each group. *, p <.05 compared to DMSO +, p < .05 compared to 0 hours time point in treatment group #, p < .05 compared to STF‐31. Abbreviation: DMSO, dimethyl sulfoxide.
NAMPT inhibitor‐mediated toxicity of hiPSC correlates with NAD and ATP depletion. Thus, the potential role of PARP over‐activation was examined because it can cause NAD+ and ATP depletion 34. As shown in Figure 3c, the PARP inhibitor PJ‐34 does not modify STF‐31 mediated toxicity of hiPSC cells following 48 hours incubation with both inhibitors. However, a partial (∼50%), but significant protection is observed after 30 hours of cotreatment with STF‐31 and PJ‐34 compared to STF‐31 alone (Fig. 3c). These results suggest that PARP inhibition may delay hiPSC death; however, PARP over‐activation is ultimately not responsible for toxicity mediated by NAMPT inhibition. To assess whether the delay in toxicity could be explained by maintenance of NAD+ or ATP levels, nucleotides were measured when NAD+ is partially depleted (3 hours) and ATP levels are decreased (24 hours). While NAD+ levels were not altered by PARP inhibition (Fig. 3c), ATP levels were maintained at 24 hours (Fig. 3d). Consequently, the delay in NAMPT‐mediated toxicity with PARP inhibition may be attributed to maintenance of ATP levels at 24 hours; however, this occurs in a temporally limited manner.
Cardiomyocytes Maintain Glycolytic Flux During NAMPT Inhibition
In human cancer cells and C. elegans the depletion of NAD levels during NAMPT inhibition is associated with decreased glycolytic flux through attenuation of GAPDH activity 35, 36, 37, 38. This effect impairs both the generation of ATP and cycling of NAD(H). Here, the formation of lactate, a downstream product of glycolysis, was used to examine how NAD+ depletion affects glycolytic flux. Consistent with our previous observation that basal extracellular acidification rate decreases following 16 hours of treatment with STF‐31 in hiPSC 17, lactate secretion in hiPSC decreases following 24 hours of NAMPT inhibition (Fig. 4a). However, day 28 hiPSC‐CM continue to secrete lactate throughout 72 hours of NAMPT inhibition (Fig. 4a) (conditions in which there is a significant depletion of NAD+ (Fig. 2a)). Although lactate secretion is attenuated following 72 hours treatment with NAMPT inhibitors in day 28 hiPSC‐CM, as compared to vehicle control, the levels are well above those from hiPSC‐CM that have been deprived of glucose. In contrast to these observations, fibroblasts do not maintain lactate secretion after 48–72 hours of NAMPT inhibition. The decrease in lactate secretion correlates with the loss of NAD+ (Figs. 2a, 4a) and by 72 hours, lactate secretion is comparable to the amount produced following glucose deprivation. Overall, these data show glycolysis is differentially inhibited by NAD+ depletion in a cell type selective manner: hPSC‐CM preserve glycolytic flux despite loss of NAD+ whereas fibroblasts do not.
Figure 4.
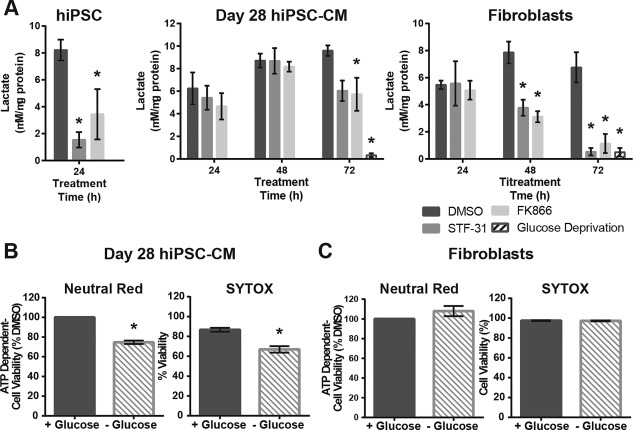
Nicotinamide phosphoribosyltransferase inhibition affects glycolytic flux in a cell type dependent manner. (A): Lactate secretion as measured by colorimetric assay following treatment with 2.5 µM STF‐31 or 100 nM FK866 for 24 hours in hiPSC and 24–72 hours in day 28 hiPSC‐CM and fibroblasts. (B, C): Bar graphs depicting cell viability following 72 hours of glucose deprivation as measured by neutral red and SYTOX cell death assay in day 28 hiPSC‐CM (B) and fibroblasts (C). Data are represented as mean ± SEM for 3–6 biological replicates in each group (N = 3 STF‐31 and FK866; N = 6 DMSO for hiPSC and fibroblasts, N = 3 cardiomyocytes). *, p < .05. Abbreviations: DMSO, dimethyl sulfoxide; hiPSC‐CM, human induced pluripotent stem cells‐derived cardiomyocytes.
To follow up on these observations, day 28 hPSC‐CM and fibroblast survival was examined under conditions of glucose deprivation to determine if a decreased glycolytic flux is toxic in either of these cell types. Cell viability was measured after 72 hours of glucose deprivation using ATP‐dependent neutral red and SYTOX cell death assays (Fig. 4b, 4c). While day 28 hiPSC‐CM continue to spontaneously contract throughout 72 hours of glucose deprivation (Supporting Information videos 6 and 7), approximately 20% of cells die under these conditions as determined by brightfield microscopy (Supporting Information video 6) and viability assays (Fig. 4b). Fibroblast viability was not significantly affected by glucose deprivation (Fig. 4c). Overall, these data suggest that the majority of day 28 hiPSC‐CM and fibroblasts can survive under conditions of limited glycolytic flux that is present during NAMPT inhibition or glucose deprivation. However, while both glucose deprivation and NAMPT inhibition are not toxic to day 28 hiPSC‐CM or fibroblasts, these conditions differentially affect glycolytic flux in these cell types.
Mitochondrial Respiration Continues During NAMPT Inhibition in Cardiomyocytes and Fibroblasts
To determine how NAD+ depletion affects mitochondrial oxidative phosphorylation, mitochondrial stress tests were performed under conditions in which NAD+ levels are significantly depleted in hiPSC (18 and 24 hours treatment) as well as day 28 hiPSC‐CM and fibroblasts (48 and 72 hours treatment). NAMPT inhibition in hiPSC results in an attenuation of all aspects of respiration measured by the mitochondrial stress test (Fig. 5a–5c and Supporting Information Fig. 4b). Despite a decrease in respiration, 24 hours of exposure to NAMPT inhibition is required to attenuate the coupling efficiency of hiPSC (Fig. 5b). At this time point, cell death has been initiated as evidenced by enhanced cleavage of caspases 17. Unlike hiPSC, day 28 hiPSC‐CM (Fig. 5c) and fibroblasts (Fig. 5d) maintain basal respiration. Only after 72 hours incubation with NAMPT inhibitors is basal respiration decreased in day 28 hiPSC‐CM, while fibroblasts maintain basal respiration throughout 72 hours of treatment with NAMPT inhibitors. The decrease in basal respiration coupling efficiency in day 28 hiPSC‐CM incubated for 72 hours correlates with a decrease in ATP linked respiration (Fig. 5c and Supporting Information Fig. 4c). Following shorter exposures to NAMPT inhibition, basal respiration is maintained yet maximal respiratory capacity is decreased (Fig. 5c, 5d). These results indicate that later stage hiPSC‐CM and fibroblasts have the capacity to maintain mitochondrial respiration under conditions of NAD+ depletion, although these cells have decreased total mitochondrial capacity.
Figure 5.
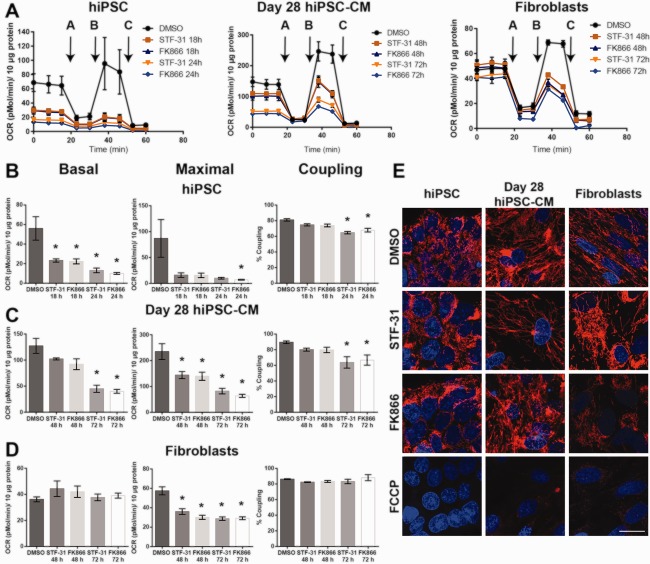
Mitochondrial respiration is maintained in differentiated cells following nicotinamide phosphoribosyltransferase inhibition. (A): Mitochondrial stress test was performed on hiPSC following 18 and 24 hours 2.5 µM STF‐31 or 100 nM FK866 and on day 28 hiPSC‐CM and fibroblasts following 48 and 72 hours treatment. Addition of oligomycin, FCCP, and antimycin for the mitochondrial stress test are denoted with arrows and letters A‐C within the figure, respectively. (B–D): Analysis of basal respiration, maximal respiratory capacity, and coupling efficiency in hiPSC (B), day 28 hiPSC‐CM (C), and fibroblasts (D). (E): Representative images from three biological replicates for tetramethylrhodamine ethyl ester staining after 24 hours of 2.5 µM STF‐31 or 100 nM FK866 in hiPSC and 72 hours in day 28 hiPSC‐CM and fibroblasts imaged at ×100. 10 µM FCCP for 45–60 minutes was used to confirm mitochondrial potential specific staining. Scale bar is 20 µm. Data are represented as mean ± SEM for three biological replicates in each group. *, p < .05. Abbreviations: DMSO, dimethyl sulfoxide; hiPSC‐CM, human induced pluripotent stem cells‐derived cardiomyocytes.
Basal respiration was significantly decreased in hiPSC and day 28 hiPSC‐CM following NAMPT inhibition for 24 and 72 hours, respectively. Therefore, we examined TMRE accumulation to evaluate whether mitochondrial membrane potential is still present in these cells after the same treatment strategy. TMRE accumulation is present in hiPSC and day 28 hiPSC‐CM at a time when NAMPT inhibition decreases basal respiration (Fig. 5e). In hiPSC, day 28 hiPSC‐CM, and fibroblasts, mitochondria remain TMRE positive in the presence of NAMPT inhibitors indicating a mitochondrial membrane potential is present despite a decrease in NAD+ or basal respiration associated with NAMPT inhibition. As a control, cells were treated with 10 µM FCCP (uncoupling agent) for 1 hours to confirm TMRE staining is present only in mitochondria with intact membrane potential (Fig. 5e). These findings indicate that day 28 hiPSC‐CM and fibroblasts have an ability to maintain mitochondrial respiration during NAMPT inhibition, and this respiration can support mitochondrial ATP synthesis.
Discussion
In this study, we provide evidence that cell survival under conditions of NAMPT inhibition increases during hPSC‐CM differentiation and maturation in vitro. Cell survival correlates with an ability to maintain ATP despite NAD+ depletion, and the mechanisms by which ATP pools are maintained appears to be the maintenance of glycolysis and/or mitochondrial respiration (Fig. 6). Overall, there are several factors that are important for determining cell survival following NAMPT inhibition, each of which appear to be cell type selective and maturation state dependent. These factors include the length of exposure to NAMPT inhibitors, the rate of NAD depletion, and the thresholds of NAD required to maintain NAD‐dependent metabolism.
Figure 6.
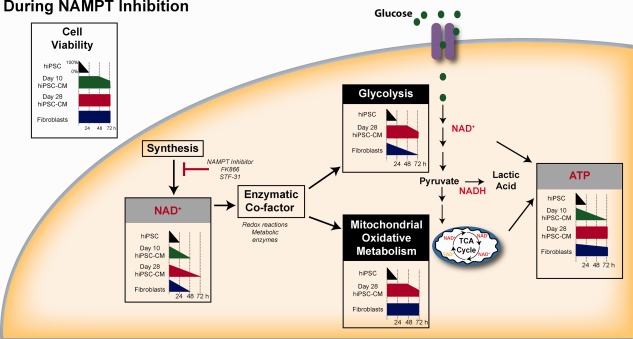
Schematic overview depicting changes in cell viability, nucleotide pools, and metabolic pathways following NAMPT inhibition in hiPSC, hiPSC‐CM, and fibroblasts. Abbreviations: hiPSC‐CM, human induced pluripotent stem cells‐derived cardiomyocytes; NAMPT, nicotinamide phosphoribosyltransferase; TCA, tricarboxylic acid. See www.StemCellsTM.com for supporting information available online.
Toxicity in hiPSC resulting from NAMPT inhibition does not correlate with cell proliferation or activation of the NAD degrading enzyme PARP. In contrast, it is the ability to maintain ATP despite the loss of cellular NAD+ pools that is an important factor in determining cell survival during NAMPT inhibition. The timing and mechanisms by which cells preserve ATP despite loss of NAD+ vary among the cell types tested here. NAMPT inhibition in hPSC results in a rapid loss of NAD that is followed by a decrease in glycolytic and mitochondrial metabolism. By day 10 of differentiation, ATP depletion is delayed and cell toxicity is minimal when NAMPT inhibition is limited to ≤48 hours of treatment. However, prolonged treatment leads to loss of cardiomyocyte contractility by 72 hours followed by near complete cell death by 96 hours (data not shown). Once hiPSC‐CM have reached 28 days of differentiation, they are resistant to the toxic actions of NAMPT inhibition, displaying both a delay in NAD+ depletion and increased maintenance of glycolysis and mitochondrial respiration. Strikingly, in day 28 hiPSC‐CM, glycolysis remains intact despite having NAD+ levels at the limits of detection by HPLC. This finding was unexpected as glycolytic flux and lactate production are both dependent on NAD as a redox cofactor. Similar observations were found with mitochondrial respiration in both dermal fibroblasts and day 28 hiPSC‐CM.
The survival of day 28 hiPSC‐CM under conditions of glucose deprivation and NAMPT inhibition suggests that these cells are capable of maintaining ATP through multiple pathways. In response to NAMPT inhibition, mitochondrial respiration and glycolysis remain intact. However, glycolysis is dispensable in day 28 hiPSC‐CM, as cells continue to spontaneously contract and minimal cell death is observed following 72 hours of glucose deprivation. These results are consistent with previous studies showing that hPSC‐CM can survive in cultures deprived of glucose with lactate supplementation 14. Extending these observations, we show that by day 28, lactate supplementation is not required for hiPSC‐CM survival during glucose deprivation, as cells can utilize alternative pathways and substrates to generate ATP. In summary, by determining susceptibility to NAMPT inhibition at defined timepoints in differentiation, these data will inform how to refine approaches for eliminating tumorigenic cells that may be present in hPSC‐CM. While a 24–48 hours pulse treatment may be an effective strategy to eliminate hPSC from early hPSC‐CM cultures, longer treatments (72 hours) may be used for treating hPSC‐CM cultures later in differentiation. Moreover, these data add to our knowledge of cellular metabolism during differentiation, which is increasingly being considered in strategies to drive maturation in vitro and for the selection of maturation stage specific cells 6, 22).
These data also suggest that in some respects, day 28 hiPSC‐CM begin to resemble rat neonatal cardiomyocytes during NAMPT inhibition, as both cell types maintain ATP and remain viable in the presence of FK866 39. While explanted rat neonatal cardiomyocytes treated with NAMPT inhibitors have increased susceptibility to H2O2‐mediated cell death and have altered cellular signaling during NAMPT inhibition, this was observed at higher concentrations of FK866 (1–10 µM) than used in the current studies. It is not yet known if similar effects occur on hPSC‐CM at the concentrations necessary to eliminate undifferentiated hPSC, but this will be important to assess in future studies. Finally, it is not yet clear how hPSC‐CM and fibroblasts maintain ATP and cell viability despite decreased or undetectable levels of cellular NAD+. We speculate that one possibility could be the presence of a NAMPT‐inhibitor resistant pool of NAD, similar to what has been reported in HeLa cells 40. It is also possible that differential utilization of NAD synthesis pathways 41, 42, localization of NAD pools 42, 43, 44, or competition with other NAD requiring processes 45, 46 play a role in determining the quantity of NAD that is sufficient to support NAD‐dependent processes, such as glycolysis and lactate production in hPSC‐CM.
Conclusion
This study further supports our previous reports that NAMPT inhibition can be used to selectively eliminate undifferentiated hiPSC 16, 17 by providing more details concerning optimal timepoints and treatment duration with NAMPT inhibition. By assessing survival during NAMPT inhibition at various stages of differentiation (undifferentiated, cardiac progenitors, committed cardiomyocytes, and later stage cardiomyocytes), we show that resistance to NAMPT inhibition increases with cardiomyogenic commitment. However, differentiation alone is not sufficient to protect from prolonged NAMPT inhibition, as day 10 cells can only survive 48 hours of treatment. With continued culturing, possibly due to maturation, cells continue to develop resistance such that by day 28 cells can survive 72 hours treatment. Furthermore, this increased resistance to NAMPT inhibition correlates with an ability to maintain cellular energy pools through continued glycolytic flux or mitochondrial respiration despite a significant loss in NAD levels. Overall, we show a shift in susceptibility to NAMPT inhibition with in vitro cardiomyogenic differentiation and continued culturing, which provides new insight into how NAD regulation and differential maintenance of energy stores mediate survival during NAMPT inhibition during hiPSC‐CM differentiation.
Author Contributions
E.M.K. and R.L.G.: conceived and coordinated the study and wrote the manuscript; E.M.K.: designed, performed and analyzed all experiments; K.A.B.: assisted in designing, performing and analyzing the experiments shown in Figures 2 and 5. M.W.: assisted in performing and analyzing the experiments shown in Figure 1. A.N.: assisted in performing and analyzing the experiments in Supporting Information Figure 3, 5, and 6. JAC contributed to overall conception and design and interpretation of data. E.M.K., K.A.B., M.W., A.N., J.A.C., and R.L.G.: reviewed the results and approved the final version of the manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Supporting information
Supporting Information Figures.
Supporting Information Video 1.
Supporting Information Video 2.
Supporting Information Video 3.
Supporting Information Video 4.
Supporting Information Video 5.
Supporting Information Video 6.
Supporting Information Video 7.
Acknowledgments
This research was supported by NIH R00HL094708, NIH R01HL126785, and Institutional Research Grant 86‐004‐26 from the American Cancer Society (R.L.G.); NIH R01DK52194 and NIH R01DK44458 (J.A.C.); and NIH P30DK020595 to (K.A.B.). E.M.K. is a member of the MCW‐MSTP which is partially supported by a T32 grant from NIGMS, GM080202. We would like to thank Dr. Nathan Schuld for his careful review of the manuscript and the MCW Bioenergetics Shared Resource Core for performing mitochondrial stress test experiments.
References
- 1. Takahashi K, Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development 2013;140:2457–2461. [DOI] [PubMed] [Google Scholar]
- 2. Chong JJ, Yang X, Don CW et al. Human embryonic‐stem‐cell‐derived cardiomyocytes regenerate non‐human primate hearts. Nature 2014;510:273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grskovic M, Javaherian A, Strulovici B et al. Induced pluripotent stem cells‐‐opportunities for disease modelling and drug discovery. Nat Rev Drug Discov 2011;10:915–929. [DOI] [PubMed] [Google Scholar]
- 4. Ebert AD, Liang P, Wu JC. Induced pluripotent stem cells as a disease modeling and drug screening platform. J Cardiovasc Pharmacol 2012;60:408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keung W, Boheler KR, Li RA. Developmental cues for the maturation of metabolic, electrophysiological and calcium handling properties of human pluripotent stem cell‐derived cardiomyocytes. Stem Cell Res Ther 2014;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu R, Blazeski A, Poon E et al. Physical developmental cues for the maturation of human pluripotent stem cell‐derived cardiomyocytes. Stem Cell Res Ther 2014;5:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee AS, Tang C, Rao MS et al. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med 2013;19:998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okano H, Nakamura M, Yoshida K et al. Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res 2013;112:523–533. [DOI] [PubMed] [Google Scholar]
- 9. Chong JJ, Murry CE. Cardiac regeneration using pluripotent stem cells‐‐progression to large animal models. Stem Cell Res 2014;13:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ben‐David U, Benvenisty N. Chemical ablation of tumor‐initiating human pluripotent stem cells. Nat Protoc 2014;9:729–740. [DOI] [PubMed] [Google Scholar]
- 11. Ben‐David U, Gan QF, Golan‐Lev T et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high‐throughput screen. Cell Stem Cell 2013;12:167–179. [DOI] [PubMed] [Google Scholar]
- 12. Rong Z, Fu X, Wang M et al. A scalable approach to prevent teratoma formation of human embryonic stem cells. J Biol Chem 2012;287:32338–32345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang C, Lee AS, Volkmer JP et al. An antibody against SSEA‐5 glycan on human pluripotent stem cells enables removal of teratoma‐forming cells. Nat Biotechnol 2011;29:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tohyama S, Hattori F, Sano M et al. Distinct metabolic flow enables large‐scale purification of mouse and human pluripotent stem cell‐derived cardiomyocytes. Cell Stem Cell 2013;12:127–137. [DOI] [PubMed] [Google Scholar]
- 15. Cao F, Drukker M, Lin S et al. Molecular imaging of embryonic stem cell misbehavior and suicide gene ablation. Cloning Stem Cells 2007;9:107–117. [DOI] [PubMed] [Google Scholar]
- 16. Boheler KR, Bhattacharya S, Kropp EM et al. A human pluripotent stem cell surface N‐glycoproteome resource reveals markers, extracellular epitopes, and drug targets. Stem Cell Reports 2014;3:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kropp EM, Oleson BJ, Broniowska KA et al. Inhibition of an NAD(+) salvage pathway provides efficient and selective toxicity to human pluripotent stem cells. Stem Cells Transl Med 2015;4:483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robertson C, Tran DD, George SC. Concise review: Maturation phases of human pluripotent stem cell‐derived cardiomyocytes. Stem Cells 2013;31:829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Folmes CD, Nelson TJ, Dzeja PP et al. Energy metabolism plasticity enables stemness programs. Ann N Y Acad Sci 2012;1254:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaspar JA, Doss MX, Hengstler JG et al. Unique metabolic features of stem cells, cardiomyocytes, and their progenitors. Circ Res 2014;114:1346–1360. [DOI] [PubMed] [Google Scholar]
- 21. Rana P, Anson B, Engle S et al. Characterization of human‐induced pluripotent stem cell‐derived cardiomyocytes: Bioenergetics and utilization in safety screening. Toxicol Sci 2012;130:117–131. [DOI] [PubMed] [Google Scholar]
- 22. Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell‐derived cardiomyocytes. Circ Res 2014;114:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prowse AB, Chong F, Elliott DA et al. Analysis of mitochondrial function and localisation during human embryonic stem cell differentiation in vitro. PLoS One 2012;7:e52214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ivashchenko CY, Pipes GC, Lozinskaya IM et al. Human‐induced pluripotent stem cell‐derived cardiomyocytes exhibit temporal changes in phenotype. Am J Physiol Heart Circ Physiol 2013;305:H913–922. [DOI] [PubMed] [Google Scholar]
- 25. Sartiani L, Bettiol E, Stillitano F et al. Developmental changes in cardiomyocytes differentiated from human embryonic stem cells: A molecular and electrophysiological approach. Stem Cells 2007;25:1136–1144. [DOI] [PubMed] [Google Scholar]
- 26. Yang X, Rodriguez M, Pabon L et al. Tri‐iodo‐l‐thyronine promotes the maturation of human cardiomyocytes‐derived from induced pluripotent stem cells. J Mol Cell Cardiol 2014;72:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhattacharya S, Burridge PW, Kropp EM et al. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J Vis Exp 2014;91:52010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burridge PW, Holmstrom A, Wu JC. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr Protoc Hum Genet 2015;87: 21.3.1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams DJ, Ito D, Rees MG et al. NAMPT is the cellular target of STF‐31‐like small‐molecule probes. ACS Chem Biol 2014;9:2247–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dragovich PS, Zhao G, Baumeister T et al. Fragment‐based design of 3‐aminopyridine‐derived amides as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). Bioorg Med Chem Lett 2014;24:954–962. [DOI] [PubMed] [Google Scholar]
- 31. Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res 2003;63:7436–7442. [PubMed] [Google Scholar]
- 32. Broniowska KA, Diers AR, Corbett JA et al. Effect of nitric oxide on naphthoquinone toxicity in endothelial cells: Role of bioenergetic dysfunction and poly (ADP‐ribose) polymerase activation. Biochemistry 2013;52:4364–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chung S, Dzeja PP, Faustino RS et al. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med 2007;4(suppl 1):S60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zong WX, Ditsworth D, Bauer DE et al. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 2004;18:1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang W, McReynolds MR, Goncalves JF et al. Comparative metabolomic profiling reveals that dysregulated glycolysis stemming from lack of salvage NAD+ biosynthesis impairs reproductive development in Caenorhabditis elegans . J Biol Chem 2015;290:26163–26179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan B, Dong S, Shepard RL et al. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, leads to altered carbohydrate metabolism in cancer cells. J Biol Chem 2015;290:15812–15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan B, Young DA, Lu ZH et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: Metabolic basis and potential clinical implications. J Biol Chem 2013;288:3500–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tolstikov V, Nikolayev A, Dong S et al. Metabolomics analysis of metabolic effects of nicotinamide phosphoribosyltransferase (NAMPT) inhibition on human cancer cells. PLoS One 2014;9:e114019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oyarzun AP, Westermeier F, Pennanen C et al. FK866 compromises mitochondrial metabolism and adaptive stress responses in cultured cardiomyocytes. Biochem Pharmacol 2015;98:92–101. [DOI] [PubMed] [Google Scholar]
- 40. Pittelli M, Formentini L, Faraco G et al. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem 2010;285:34106–34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zamporlini F, Ruggieri S, Mazzola F et al. Novel assay for simultaneous measurement of pyridine mononucleotides synthesizing activities allows dissection of the NAD(+) biosynthetic machinery in mammalian cells. FEBS J 2014;281:5104–5119. [DOI] [PubMed] [Google Scholar]
- 42. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: Functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol 2015;50:284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. VanLinden MR, Dolle C, Pettersen IK et al. Subcellular distribution of NAD+ between cytosol and mitochondria determines the metabolic profile of human cells. J Biol Chem 2015;290:27644–27659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nikiforov A, Dolle C, Niere M et al. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 2011;286:21767–21778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruggieri S, Orsomando G, Sorci L et al. Regulation of NAD biosynthetic enzymes modulates NAD‐sensing processes to shape mammalian cell physiology under varying biological cues. Biochim Biophys Acta 2015;1854:1138–1149. [DOI] [PubMed] [Google Scholar]
- 46. Dolle C, Rack JG, Ziegler M. NAD and ADP‐ribose metabolism in mitochondria. FEBS J 2013;280:3530–3541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figures.
Supporting Information Video 1.
Supporting Information Video 2.
Supporting Information Video 3.
Supporting Information Video 4.
Supporting Information Video 5.
Supporting Information Video 6.
Supporting Information Video 7.