

Abstract
Ginsenoside Rb3 is one of the major active components in protopanaxdiol type ginsenosides, and has demonstrated anti-diabetic activity. However, the mechanism of this action has yet to be elucidated. The present study investigated the effects of ginsenoside Rb3 on the AMP-activated protein kinase (AMPK) gluconeogenesis pathway. The present study involved the use of HepG2 cells and western blot analysis to systematically evaluate the effect of ginsenoside Rb3 on AMPK signaling proteins and key factors of gluconeogenesis [phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase, forkhead transcription factor 1 (FOXO1) and hepatic nuclear receptor 4α (HNF4α)]. The results indicated that 25 µM ginsenoside Rb3 significantly activated AMPK activity, increased the ratio of p-AMPK/total-AMPK, and had synergistic effects with the activator of AICAR on the activation of AMPK. Further analysis indicated that the expression of the transcription factor FOXO1 and HNF4α protein, two important factors in the pathway of HepG2 cell gluconeogenesis, was significantly suppressed by ginsenoside Rb3. PEPCK and G6Pase were subsequently inhibited, which led to the suppression of gluconeogenesis. These effects were partially blocked by the AMPK inhibitor, Compound C, which indicated that the inhibition effects of ginsenoside Rb3 on hepatic gluconeogenesis were predominantly due to the activation of the AMPK signaling pathway. These data suggested that ginsenoside Rb3 can suppress hepatic gluconeogenesis, at least partially through stimulation of AMPK activity.
Keywords: ginsenoside Rb3, diabetes, HepG2 cells, adenosine monophosphate-activated protein kinase, hepatic gluconeogenesis
Introduction
Diabetes affects patients worldwide, and is characterized by absolute or relative insulin insufficiency, causing high blood and urine glucose levels (1). Long-term high blood sugar levels can damage cardiovascular, kidney, eye and nervous tissues, and can cause great harm to the patient. Treatments for diabetes include insulin therapy, islet cell transplantation, gene therapy, oral hypoglycemic drugs, exercise and diet therapy (2). Furthermore, the current oral hypoglycemic drugs have three major mechanisms: i) Promoting insulin secretion, ii) increasing insulin sensitivity and iii) inhibiting α-glucosidase (3). Hypoglycemic drugs from traditional Chinese herbal remedies are continually emerging. These have multiple functions, including reducing blood sugar levels, reducing blood fat and improving blood viscosity (4–6).
Adenosine monophosphate-activated protein kinase (AMPK), which is activated by metabolic stressors including hypoxia, low glucose and nutrient deprivation, regulates cellular and systemic energy homeostasis in mammalian cells (7). Activation of AMPK in skeletal muscle, liver and adipose tissue enhances metabolism, improves insulin sensitivity, and may be favorable for the treatment of diabetes (8). AMPK is an attractive drug target that serves a key role in the regulation of whole-body energy homeostasis (9). Activation of hepatic AMPK leads to increased fatty acid oxidation and, simultaneously, inhibition of hepatic glucose production, as well as lipogenesis and cholesterol synthesis (9,10). A number of previous studies found that AMPK may adjust key enzymes [phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase)] involved in hepatic gluconeogenesis. The transcription factors cyclic-AMP response element binding protein (CREB), hepatocyte nuclear factor 4α (HNF4α) and forkhead box protein O1 (FOXO1) are the key regulatory factors of the AMPK channel. TORC2, a signaling protein that modulates CREB activity, is phosphorylated by AMPK. Through this mechanism, CREB expression is inhibited by AMPK. AMPK can adjust SHP, HNF4α and FOXO1 to achieve the inhibition of hepatic gluconeogenesis, which has the potential to reduce blood glucose (10).
Ginsenoside Rb3, one of the major active components of protopanaxdiol type ginsenosides, has received much attention due to its various bioactivities, including antioxidant activity and microcirculatory improvement (11), neuroprotection (12), cardiovascular protection (13,14), prevention of obesity (15) and prevention of diabetes (16,17). Previous research has indicated that ginsenoside Rb3 showed antidiabetic activity in alloxan-induced diabetic mice, and increased glucose consumption in C2C12 myotubes (16). Furthermore, our group previously demonstrated that ginsenoside Rb3 can decrease blood glucose, increase blood glucose tolerance and antioxidants, improve serum lipid disorders, and improve insulin sensitivity and resistance in diabetic mice (17). Numerous animal studies and clinical trials have ascertained that ginsenoside Rb3 has significant hypoglycemic effects (15–17). Although the hypoglycemic effects of ginsenoside Rb3 appear promising, it has not yet been used clinically as an antidiabetic drug, predominantly due to the fact that the molecular mechanism has yet to be fully elucidated. The current study assessed the effect of ginsenoside Rb3 on hepatic gluconeogenesis mediated by AMPK in HepG2 cells, and provided a theoretical basis for research on the molecular regulatory mechanism of gluconeogenesis inhibition.
Materials and methods
Materials
Ginsenoside Rb3 standards were isolated and purified in our laboratory (the Chinese Herbal Medicine Laboratory of Jilin Agricultural University, Changchun, China). HepG2 cells were purchased from the Shanghai Cell Institute of Chinese Academy of Science (Shanghai, China). AICAR (an AMPK activator), trypsin, and Dulbecco's modified Eagle's medium (DMEM) culture medium were purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Antibodies against AMPK (cat. no. sc-25792), P-AMPK (cat. no. sc-33524), PEPCK (cat. no. sc-32879), G6Pase (cat. no. sc-25840), FOXO1 (cat. no. sc-C29H4), HNF4α (cat. no. sc-8987) and goat anti-rabbit IgG-horseradish peroxidase (HRP) secondary antibodies (cat. no. sc-2004) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Dimethyl sulfoxide (DMSO) and the fetal bovine serum (FBS) and trypsin were purchased from Roche Applied Science (Penzberg, Germany). MTT was purchased from the Tianjin Lu Xin Chemical Technology Company (Tianjin, China). Compound C (an AMPK selective inhibitor) was purchased from North Kangtai Clinical Reagent Company (Beijing, China). The glucose uptake was measured using the Amplex Red Glucose/Glucose Oxidase Assay kit (cat. no. A22189; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The enhanced chemiluminescence (ECL) kit (cat. no. 32109) was purchased from Guangzhou Baotaike Biological Company (Guangzhou, China). X-ray film was from the Eastman Kodak Company (Rochester, NY, USA).
Cell culture
HepG2 cells, which were frozen with liquid nitrogen, were placed in a water bath at 37°C and then transferred to a sterile tube (10 ml). Culture medium (3 ml) was added to cells and evenly mixed. The pelleted cells were collected after centrifugation at 1,000 × g for 5 min and were cultured with DMEM (supplemented with 15% FBS). The culture medium was replaced the following day. The recovered cells were grown in DMEM (10% FBS), treated with 0.25% trypsin at an 80% confluency and then passaged to a 1:3 ratio. The cells used in subsequent experiments were in the logarithmic growth phase.
Cell experiments
HepG2 cells (1×105 cells/ml) were seeded into 6-well tissue culture plates in the logarithmic growth phase and cultured with DMEM (supplemented with 10% FBS). HepG2 cells were cultured for 12 h at 37°C with 5% CO2, then the medium was removed and replaced with medium containing 25 µM ginsenoside Rb3 and/or 1 mM AICAR and/or 10 µM Compound C was added. Total protein and nucleoprotein were extracted after a 24-h incubation by cell lysis. The total protein was immunoblotted with antibodies specific for AMPK, p-AMPK, PEPCK and G6Pase, while nucleoprotein was immunoblotted with FOXO1 and HNF4α. This Western blotting procedure is described in a subsequent section.
MTT assay
The cell viability of HepG2 cells was assessed using an MTT assay following treatment in the absence or presence of ginsenoside Rb3 (0, 3.125, 6.25, 12.5, 25 and 50 µM) for 24 h. The cells were treated with MTT in accordance with the manufacturer's protocol, and the supernatant was removed and DMSO added to dissolve the blue crystals. The optical density (OD) values were measured with a microplate reader at 490 nm, and then the cell viability rate was calculated [cell viability rate(%)=treatment group average OD/control group average ODx100]. Finally, the ginsenoside Rb3 concentration that resulted in no damage to HepG2 cells was determined to be the optimum concentration for glucose production and hepatic gluconeogenesis in the subsequent experimental analysis.
Glucose production assay
According to the method reported previously (14,15), the experiment was set up with three treatment groups [0 (control), 12.5 and 25 µM ginsenoside Rb3] in triplicate. The procedures were as follows: HepG2 cells were cultured to the exponential phase; 1-ml cell suspensions in 24-well plates were cultured with 10% FBS DMEM (5% CO2 concentration) at 37°C for 24 h; then, the medium was replaced with the glucose production buffer solution (5% CO2 concentration) at 37°C for 4 h. Finally, the medium culture was transferred into an Eppendorf tube, and glucose production from HepG2 cells was measured with the Amplex Red Glucose/Glucose Oxidase Assay kit.
Analysis of protein content
HepG2 cells were lysed with 4°C lysis buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with a protease inhibitor (1 mM phenylmethylsulfonyl fluoride) and phosphatase inhibitors (50 mM NaF, 0.1 mM sodium vanadate, 10 mM sodium molybdate, 20 mM 3-glycerol phosphate, 10 mM 4-nitropyrophosphate). Protein concentration was measured using the Bradford assay (cat. no. 5000002; Bio-Rad Laboratories Inc., Hercules, CA, USA). The immunoblotting analysis was performed by western blot. Total protein extracts (40 µg) were separated by 12% SDS-PAGE. The polyvinylidene difluoride (Merck Millipore, Darmstadt, Germany) protein hybridization membranes were first soaked with methanol for 15 sec to activate the membrane and then placed in the pre-cooled transmembrane buffer. The fibrin glue and transmembrane device were placed in the correct orientation, and then the proteins were transferred to membranes at 100 V at 4°C for 2 h. The membranes were then washed with Tris buffered saline-Tween (TBST) three times for 10 min each. Next, the membranes were blocked with 5% non-fat milk powder for 2 h. The primary and secondary antibody incubations were performed with an antibody hybridization process using standard hybridization techniques (18). The blocked membranes were incubated overnight in TBST with the primary antibodies against AMPK (1:500), p-AMPK (1:500), PEPCK (1:1,000), G6Pase (1:1,000), PGC-1α (1:500), HNF4α (1:1,000), FOXO1 (1:1,000), and β-actin (1:2,000) at 4°C. Membranes were then incubated and hybridized in TBST with anti-rabbit IgG-HRP secondary antibodies (1:1,000) at 37°C for 1 h. Subsequently, membranes were washed with TBST three times for 10 min each. Finally, the membranes were treated with ECL reagent and X-ray film was used to obtain the hybrid picture. X-ray films were scanned, and the results were analyzed with Quantity One software v. 4.6 (Bio-Rad Laboratories, Inc.). The ratio of the target protein expression to the reference protein expression was calculated as relative expression, and different treatments were compared with one-way analysis of variance.
Statistical analysis
Data are expressed as the mean ± standard error of the mean. Significant differences from the control group were determined by Student's t-test. All statistics were calculated with SPSS statistical software (version 13.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of ginsenoside Rb3 on cell viability of HepG2 cells
Fig. 1. indicates that 3.125, 6.25, 12.5, and 25 µM ginsenoside Rb3 treatments caused limited damage to HepG2 cells after ginsenoside Rb3 incubation for 24 h; however, 50 µM ginsenoside Rb3 led to significant damage to HepG2 cells, as indicated by a significant reduction in cell viability (P<0.05). With use of the latter dose, the cells exhibited blebbing and apoptosis. Therefore, 12.5 and 25 µM ginsenoside Rb3 were determined to be suitable concentrations for further experiments.
Figure 1.
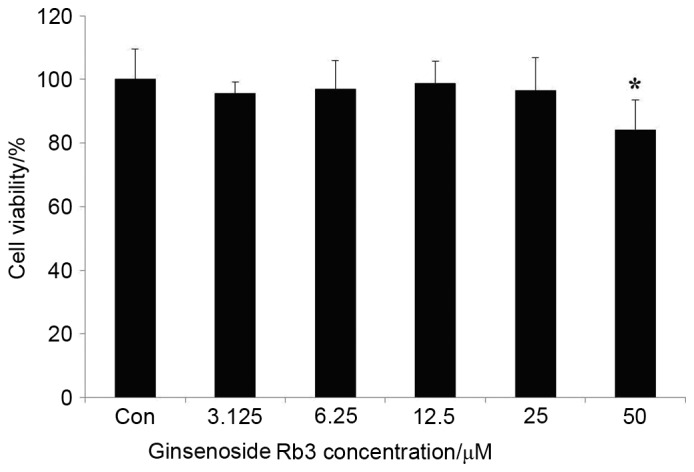
HepG2 cells were incubated with various concentrations of ginsenoside Rb3 (0, 3.125, 6.25, 12.5, 25 and 50 µM) for 24 h, and cell viability was determined by an MTT assay. Data are expressed as the mean ± standard error (n=6). *P<0.05 vs. control. Con, control.
Effect of ginsenoside Rb3 on glucose production of HepG2 cells
Subsequent to the determination of a suitable ginsenoside Rb3 dose, the HepG2 cells were rinsed with PBS, dissociated with protease, and then the protein was extracted. Finally, the measured glucose content was compared with the extracted total protein content on the basis of quantitative protein, so that the ratio could more accurately reflect the inhibitory effect of ginsenoside Rb3 on the glucose production ability of HepG2 cells. Fig. 2 indicates that both doses of ginsenoside Rb3 had an inhibitory effect on gluconeogenesis in HepG2 cells. However, only 25 µM ginsenoside Rb3 significantly inhibited gluconeogenesis (P<0.05).
Figure 2.
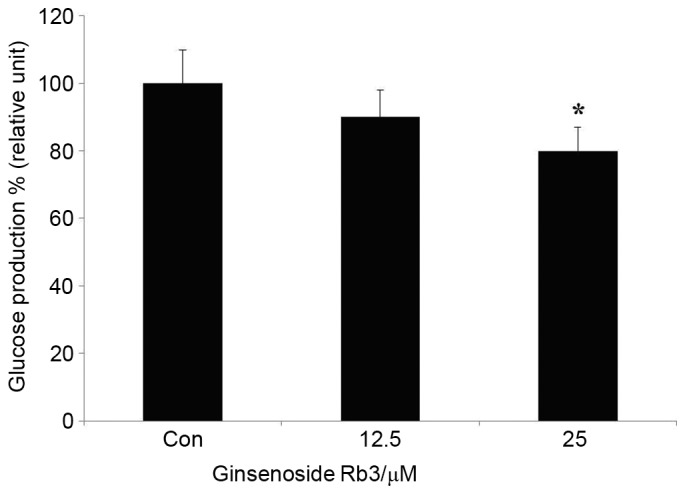
HepG2 cells were incubated with different concentrations of ginsenoside Rb3 (0, 12.5 and 25 µM), and glucose production from HepG2 cells was determined by a colorimetric glucose oxidase assay. Data are expressed as the mean ± standard error (n=6). *P<0.05 vs. control. Con, control.
Effect of ginsenoside Rb3 on PEPCK expression
To further investigate the molecular mechanism of ginsenoside Rb3 on gluconeogenesis, expression levels of PEPCK, a key enzyme of the gluconeogenesis pathway, were analyzed by western blot analysis. Fig. 3 indicates that treatment with ginsenoside Rb3 after 24 h inhibited the expression of PEPCK. Inhibition of PEPCK expression by 25 µM ginsenoside Rb3 treatment reached a significant level (P<0.05), which may be consistent with the inhibitory effect on glucose production of HepG2 cells.
Figure 3.
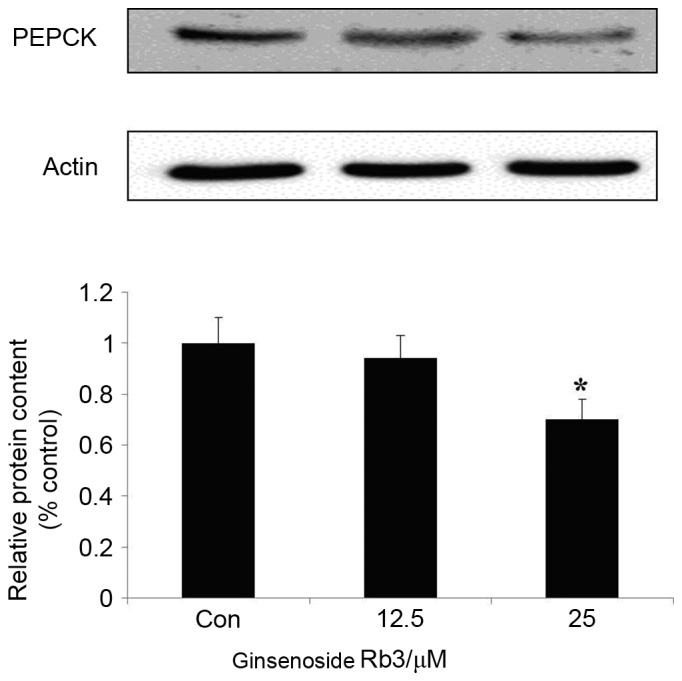
Effect of ginsenoside Rb3 on PEPCK in HepG2 hepatocytes. The cells were incubated with 0, 12.5, or 25 µM ginsenoside Rb3. Total protein was extracted after a 24-h incubation period, using the protein extraction kits. Western blotting was performed in the cells to check total protein levels of PEPCK. Data are expressed as the mean ± standard error of the mean (n=3). *P<0.05 vs. control group. PEPCK, phosphoenolpyruvate carboxykinase.
Effect of ginsenoside Rb3 on the expression levels of AMPK and p-AMPK in HepG2 cells
AMPK is an attractive target that serves a key role in the regulation of energy homeostasis at the whole body level (9). Activation of hepatic AMPK leads to increased fatty acid oxidation and simultaneously inhibits hepatic glucose production, in addition to lipogenesis and cholesterol synthesis (10). To further investigate the regulation of gluconeogenesis and AMPK expression by ginsenoside Rb3, analysis with AICAR (AMPK agonists) and Compound C (AMPK inhibitors) was performed. In the experiment, 6 groups were used: i) Control group, an untreated group, ii) 25 µM ginsenoside Rb3 group, iii) 1 mM AICAR group, iv) 10 µM Compound C group, v) 25 µM ginsenoside Rb3+1 mM AICAR group, and vi) 25 µM ginsenoside Rb3+10 µM Compound group.
The results show that ginsenoside Rb3 and AICAR activated AMPK, and that the expression levels of p-AMPK significantly increased (P<0.05; Fig. 4B). In addition, ginsenoside Rb3 and AICAR demonstrated a synergistic effect on p-AMPK expression levels compared with each group alone (P<0.05), whilst the p-AMPK expression levels following treatment with a combination of the two treatments was significantly higher than that of either alone (P<0.05).
Figure 4.

Effect of ginsenoside Rb3 on AMPK in HepG2 hepatocytes. (A) p-AMPK expression level, (B) AMPK expression level and (C) ratio of p-AMPK/AMPK in HepG2 hepatocytes under different treatment. The cells were incubated with 25 µM Rb3 and/or 1 mM AICAR and/or 10 µM Compound C. Total protein was extracted after a 24-h incubation using protein extraction kits. Western blotting was performed in the cells to assess the phosphorylation and total protein levels of AMPK. The ratio of pAMPK/AMPK was quantified in three independent experiments per condition. Data are expressed as the mean ± standard error of the mean (n=3). *P<0.05 vs. control group; #P<0.05 vs. ginsenoside Rb3 group. AMPK, adenosine monophosphate-activated protein kinase; Con, control; Rb3, ginsenoside Rb3.
Compound C, an AMPK inhibitor, was able to significantly inhibit expression of total AMPK and activated p-AMPK in HepG2 cells (P<0.05; Fig. 4A and B), and the ratio of p-AMPK to total AMPK was significantly reduced (Fig. 4C). The results indicated that Compound C had a marked inhibitory effect on p-AMPK expression. In the group in which ginsenoside Rb3 and Compound C were used at the same time, the expression of p-AMPK is higher than that of Compound C treatment alone (P<0.05). In particular, the ratio of p-AMPK to AMPK was significantly higher compared with that following the use of Compound C alone. Therefore, ginsenoside Rb3 significantly increased the expression and activation of AMPK (P<0.05), which was likely associated with the mechanism causing the partial reduction of the inhibitory effect by Compound C on AMPK expression. In addition, the combined use of ginsenoside Rb3 and Compound C caused the expression of p-AMPK and the ratio of p-AMPK/total AMPK to be significantly lower than that of the ginsenoside Rb3 single treatment (P<0.01), which indicated that Compound C could partly block the effects of ginsenoside Rb3 on AMPK activation.
Effect of ginsenoside Rb3 on the expression of PEPCK and G6Pase
The expression levels of PEPCK and G6Pase, which are the key gluconeogenic enzymes, were analyzed by western blot analysis in HepG2 cells. These data indicated that ginsenoside Rb3, AICAR (an AMPK activator) and a combinatorial treatment with these significantly inhibited the expression of G6Pase (Fig. 5) and PEPCK (Fig. 6) in comparison with the control group (P<0.05). The expression levels of PEPCK and G6Pase were significantly increased following the use of Compound C (P<0.05). In addition, the expression levels of PEPCK and G6Pase were significantly lower in the control group compared with the combination of ginsenoside Rb3 and Compound C (P<0.05); however, the expression levels of PEPCK and G6Pase following a single treatment with Compound C were significantly higher compared with those subsequent to treatment with ginsenoside Rb3 only (P<0.05), which indicates that the inhibitory effect of ginsenoside Rb3 on PEPCK and G6Pase is partially blocked by Compound C. Overall, the inhibition of gluconeogenesis by ginsenoside Rb3 was demonstrated to be associated with AMPK activation.
Figure 5.
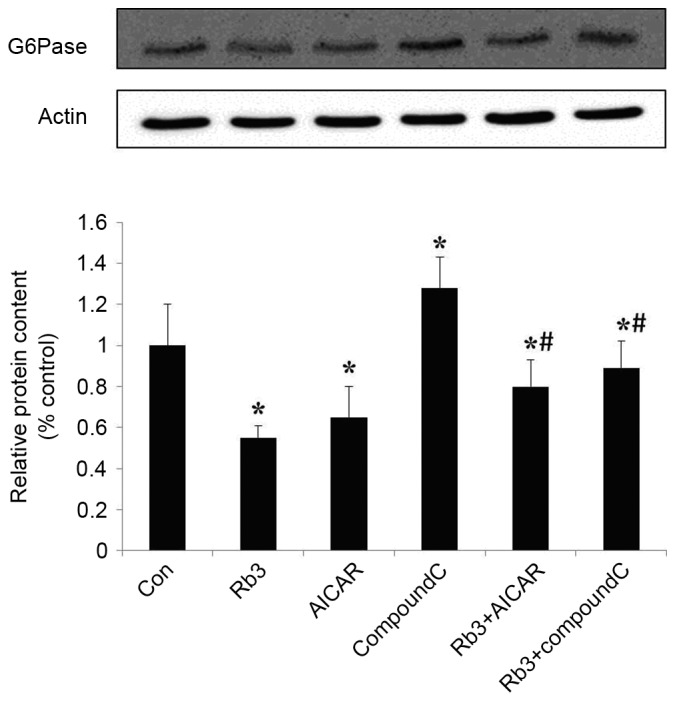
Effect of ginsenoside Rb3 on G6Pase in HepG2 hepatocytes. The cells were incubated with 25 µM Rb3 and/or 1 mM AICAR and/or 10 µM Compound C. Total protein was extracted after a 24-h incubation, using the protein extraction kits. Western blotting was performed in the cells to check total protein levels of G6Pase. Data are expressed as the mean ± standard error of the mean (n=3). *P<0.05 vs. control group; #P<0.05 vs. ginsenoside Rb3 group. G6Pase, glucose-6-phosphatase; Rb3, ginsenoside Rb3.
Figure 6.
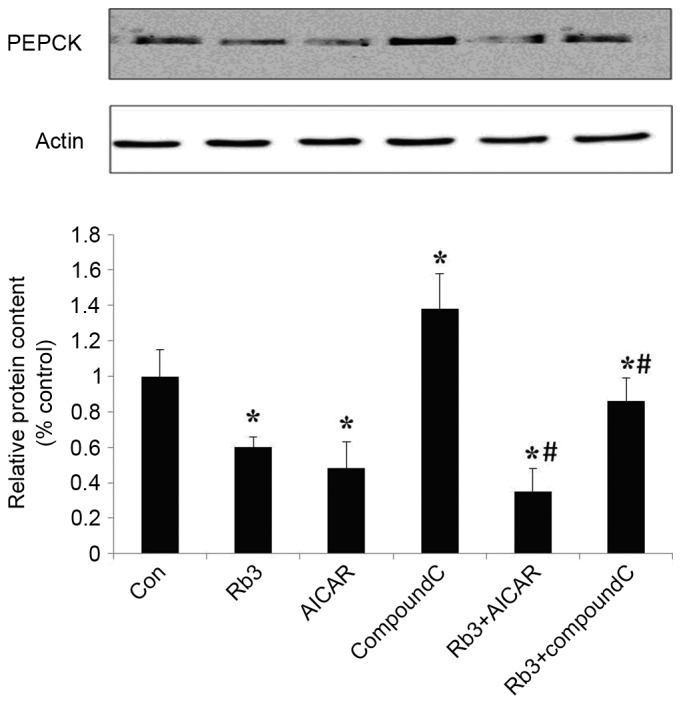
Effect of Rb3 on PEPCK in HepG2 hepatocytes. The cells were incubated with 25 µM Rb3 and/or 1 mM AICAR and/or 10 µM Compound C. Total protein was extracted after a 24-h incubation using protein extraction kits. Western blotting was performed in to determine total protein expression levels of PEPCK. Data are expressed as the mean ± standard error of the mean (n=3). *P<0.05 vs. control group; #P<0.05 vs. Rb3 group. PEPCK, phosphoenolpyruvate carboxykinase; Con, control; Rb3, ginsenoside Rb3.
Effect of ginsenoside Rb3 on gluconeogenesis nuclear transcription factors in HepG2 cells
Transcription factors are crucial in the regulation of eukaryotic gene expression (9). To further explore the molecular mechanism underlying the effect of ginsenoside Rb3 on gluconeogenesis, the expression levels of FOXO1 and HNF4α, two transcription factors, were analyzed by protein hybridization (Figs. 7 and 8). The expression levels of FOXO1 were assessed by western blot analysis (Fig. 7). Ginsenoside Rb3 and AICAR had inhibitory effects on the expression levels of FOXO1, and a combination of the two had a significant inhibitory effect on the expression levels of FOXO1 (P<0.05). The expression of transcription factor FOXO1 was significantly lower when both were combined, compared with the use of either alone (P<0.05). Notably, although AICAR did not enhance the inhibitory effects of ginsenoside Rb3 on the expression of FOXO1, the expression levels of FOXO1 were significantly higher when Compound C and ginsenoside Rb3 were combined, compared with the use of ginsenoside Rb3 alone (P<0.05). Compound C lessened the inhibitory effect of ginsenoside Rb3 on FOXO1 expression.
Figure 7.
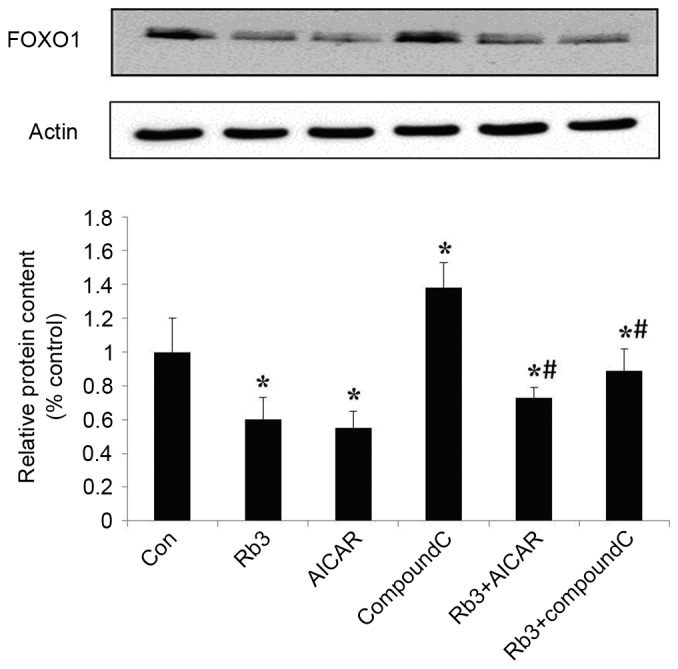
Effect of Rb3 on FOXO1 in HepG2 hepatocytes. The cells were incubated with 25 µM Rb3 and/or 1 mM AICAR and/or 10 µM Compound C. Total protein was extracted after a 24-h incubation using the protein extraction kits. Western blotting was performed to determine total protein expression levels of FOXO1. Data are expressed as the mean ± standard error of the mean (n=3). *P<0.05 vs. control group; #P<0.05 vs. Rb3 group. FOXO1, forkhead transcription factor 1; Con, control; Rb3, ginsenoside Rb3.
Figure 8.
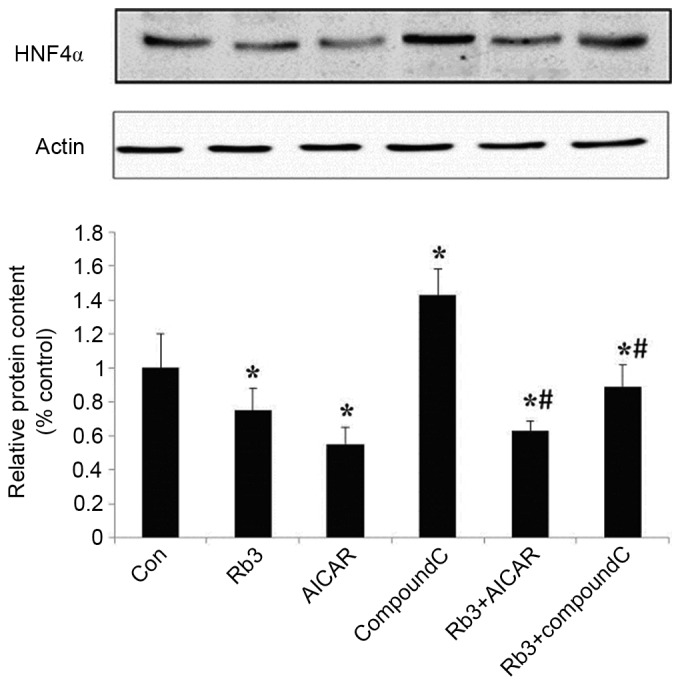
Effect of Rb3 on HNF4α in HepG2 hepatocytes. The cells were incubated with 25 µM Rb3 and/or 1 mM AICAR and/or 10 µM Compound C. Total protein was extracted after a 24-h incubation period using protein extraction kits. Western blotting was performed to determine total protein levels of HNF4α. Data are expressed as the mean ± standard error of the mean (n=3). *P<0.05 vs. control group; #P<0.05 vs. Rb3 group; Con, control; Rb3, ginsenoside Rb3.
The expression levels of HNF4α were analyzed (Fig. 8) and revealed that the expression of HNF4α was significantly inhibited by ginsenoside Rb3 and AICAR alone, or by their combination (P<0.05), and that the expression levels of HNF4α were significantly lower when both were combined, compared with the use of ginsenoside Rb3 alone (P<0.05). HNF4α was markedly (but not significantly) higher compared with the use of AICAR alone, and Compound C significantly reduced the inhibitory effect of ginsenoside Rb3 on HNF4α expression levels (P<0.05).
Discussion
Previous reports indicated that ginsenoside Rb3 exerted antidiabetic activity in alloxan-induced diabetic mice, and increased glucose consumption in C2C12 myotubes (16,19). In addition, our previous study demonstrated that ginsenoside Rb3 decreases blood glucose, increases blood glucose tolerance and antioxidants, improves serum lipid disorders and improves insulin sensitivity and resistance in diabetic mice (17). Numerous animal studies and clinical trials have ascertained that ginsenoside Rb3 has a significant hypoglycemic effect (15–17). To analyze the regulatory mechanism underlying the effect of ginsenoside Rb3 on AMPK, the present study used AMPK inhibitor Compound C (20–22) and AMPK activator AICAR (23–26).
The HepG2 cell line is a human hepatocellular carcinoma cell line, and has several of the biological characteristics of normal liver cells. It is commonly used to conduct experiments regarding hepatic gluconeogenesis (27,28). During these experiments, the level of glucose synthesis can be measured in HepG2 cells to reflect gluconeogenesis and its effect on the treatment of type 2 diabetes mellitus. Therefore, the HepG2 cell line is often used for drug-carrier screenings for diabetes treatment, and allows for thorough investigation of the hypoglycemic effect (27,28).
Previous results obtained using various animal models of type 2 diabetes confirm the physiological importance of hepatic AMPK in glucose homeostasis (9). The AMPK pathway has been reported to regulate the phosphorylation and nuclear exclusion of CREB-regulated transcription coactivator 2 (TORC2) (29). In response to fasting stimuli, TORC2 is dephosphorylated and transported from the cytoplasm to the nucleus, where it enhances the transcriptional activation of gluconeogenic genes. The transcriptional coactivator mediates CREB-dependent transcription (PGC-1α). Expression of the coactivator PGC-1α further induces the transcription of key gluconeogenic enzymes such as PEPCK and G6Pase in association with the factors HNF4α and FOXO1. AMPK is able to phosphorylate TORC2 and lead to the inhibition of gluconeogenesis in the cytoplasm (18,30).
The current results show that ginsenoside Rb3 and AICAR influence the activity of AMPK in HepG2 cells, in addition to PEPCK and G6Pase, which are key rate-limiting enzymes in the pathway of gluconeogenesis, and had significant inhibitory effects when used in combination. This may be due to the ability of ginsenoside Rb3 to inhibit gluconeogenesis. In addition, the present study also revealed that the combined use of ginsenoside Rb3 and AICAR dramatically enhanced the inhibition of PEPCK, and showed strong synergistic effects. Compound C could effectively block the inhibitory effect of ginsenoside Rb3 on PEPCK and G6Pase, which indicated that AMPK was an important target site of action of ginsenoside Rb3. In addition, FOXO1 and HNF4α, two key nuclear transcription factors in the gluconeogenic pathway, and analysis of the expression levels performed in the present study indicated that ginsenoside Rb3 had significant inhibitory effects on their expression, and that Compound C could partially block this inhibitory effect. The current study therefore suggests that the inhibitory effect of ginsenoside Rb3 on these two transcription factors may be associated with the activation of AMPK. In the regulation of HNF4α protein expression, the use of a combination of the two drugs also demonstrated a reduced inhibitory effect of AICAR on HNF4α expression. Therefore, ginsenoside Rb3 and AICAR both disrupt the regulation of certain transcription factors. These results suggest that ginsenoside Rb3 had a significant inhibitory effect on gluconeogenesis, which is associated, at least partly, with the activation of AMPK and its downstream signaling pathway in HepG2 cells.
Several antidiabetic drugs, such as metformin, regulate blood glucose by transcriptional inhibition of the gluconeogenic program (31,32). The glucose-lowering effect of metformin has been predominantly attributed to its ability to suppress hepatic gluconeogenesis through the AMPK signaling pathway (9). Additional studies are required to compare the use of ginsenoside Rb3 with that of metformin.
In conclusion, the current results provide further insight into the mechanism of hepatic gluconeogenesis and its modulation by ginsenoside Rb3 in HepG2 cells in vitro. Furthermore, ginsenoside Rb3 suppresses hepatic gluconeogenesis, at least in part, by activating AMPK. These data support the hypothesis that preparations of ginsenoside Rb3 have potential to prevent and treat type 2 diabetes.
Acknowledgements
The present study was supported by the Jilin Science and Technology Development Plan (grant no. 20060902).
Glossary
Abbreviations
- AMPK
adenosine monophosphate-activated protein kinase
- PEPCK
phosphoenolpyruvate carboxykinase
- G6Pase
glucose-6-phosphatase
- FOXO1
forkhead transcription factor 1
- HNF4α
hepatic nuclear receptor 4α
References
- 1.Biadgo B, Melku M, Abebe SM, Abebe M. Hematological indices and their correlation with fasting blood glucose level and anthropometric measurements in type 2 diabetes mellitus patients in Gondar, Northwest Ethiopia. Diabetes Metab Syndr Obes. 2016;9:91–99. doi: 10.2147/DMSO.S97563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Budinsky A, Wolfram R, Oguogho A, Efthimiou Y, Stamatopoulos Y, Sinzinger H. Regular ingestion of opuntia robusta lowers oxidation injury. Prostaglandins Leukot Essent Fatty Acids. 2001;65:45–50. doi: 10.1054/plef.2001.0287. [DOI] [PubMed] [Google Scholar]
- 3.Li Q, Zhou JP, Zhang HB. Research progresses in anti-diabetic drugs. Progress in Pharmaceutical Sciences. 2013;37:417–427. [Google Scholar]
- 4.Lai Yu, Chai Dandan, Niu Rui, Xiaodong S. Study the mechanism of potentilla discolor Bunge extract on blood sugar in diabetic rats. Aisa-Pacific Traditional Medicine. 2016;12:17–19. [Google Scholar]
- 5.Wan Y, Wu J, Wu Q. A review of the hypoglycemic activity of Siraitia Grosvenorii. Food Research and Development. 2016;37:188–191. [Google Scholar]
- 6.Zhang M, Shen Y. Research advances in pharmacological effects of oleanolic acid in hypoglycemia and antidiabetic complications. Anti-infection Pharmacy. 2015;12:801–806. [Google Scholar]
- 7.Ryu GR, Lee MK, Lee E, Ko SH, Ahn YB, Kim JW, Yoon KH, Song KH. Activation of AMP-activated protein kinase mediates acute and severe hypoxic injury to pancreatic beta cells. Biochem Biophys Res Commun. 2009;386:356–362. doi: 10.1016/j.bbrc.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 8.Magnoni LJ, Vraskou Y, Palstra AP, Planas JV. AMP-activated protein kinase plays an important evolutionary conserved role in the regulation of glucose metabolism in fish skeletal muscle cells. PLoS One. 2012;7:e31219. doi: 10.1371/journal.pone.0031219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viollet B, Guigas B, Leclerc J, Hébrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Han B, Yu X, Qu S, Sui D. Ginsenoside Rb3 ameliorates myocardial ischemia-reperfusion injury in rats. Pharm Biol. 2011;49:900–906. doi: 10.3109/13880209.2011.554845. [DOI] [PubMed] [Google Scholar]
- 12.Cui J, Jiang L, Xiang H. Ginsenoside Rb3 exerts antidepressant-like effects in several animal models. J Psychopharmacol. 2012;26:697–713. doi: 10.1177/0269881111415735. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Jiang Y, Yu X, Fu W, Zhang H, Sui D. Ginsenoside-Rb3 protects the myocardium from ischemia-reperfusion injury via the inhibition of apoptosis in rats. Exp Ther Med. 2014;8:1751–1756. doi: 10.3892/etm.2014.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T, Yu X, Qu S, Xu H, Han B, Sui D. Effect of ginsenoside Rb3 on myocardial injury and heart function impairment induced by isoproterenol in rats. Eur J Pharmacol. 2010;636:121–125. doi: 10.1016/j.ejphar.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 15.Rui L, Yi-Nan Z, Wen-Cong L. Inhibitory activity of ginsenoside Rb3 on pancreatic lipase. J Xidian Univ. 2011;23:522–525. [Google Scholar]
- 16.Bu QT, Zhang WY, Chen QC, Zhang CZ, Gong XJ, Liu WC, Li W, Zheng YN. Anti-diabetic effect of ginsenoside Rb(3) in alloxan-induced diabetic mice. Med Chem. 2012;8:934–941. doi: 10.2174/157340612802084199. [DOI] [PubMed] [Google Scholar]
- 17.Meng FL, Su XT, Zheng YN. Effects of Ginsenoside Rb3 on antihyperglycemia and antioxidation in diabetic mice. J South Chin Agr Univ. 2013;34:553–557. [Google Scholar]
- 18.Wei S, Li W, Yu Y, Yao F, A L, Lan X, Guan F, Zhang M, Chen L. Ginsenoside Compound K suppresses the hepatic gluconeogenesis via activating adenosine-5′monophosphate kinase: A study in vitro and in vivo. Life Sci. 2015;139:8–15. doi: 10.1016/j.lfs.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 19.Lee MS, Hwang JT, Kim SH, Yoon S, Kim MS, Yang HJ, Kwon DY. Gimenoside Rc, an active component of Panax ginseng, stimulates glucose uptake in C2C12 myotubes through an AMPK-dependent mechanism. J Ethnopharmacol. 2010;127:771–776. doi: 10.1016/j.jep.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Jin J, Mullen TD, Hou Q, Bielawski J, Bielawska A, Zhang X, Obeid LM, Hannun YA, Hsu YT. AMPK inhibitor Compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J Lipid Res. 2009;50:2389–2397. doi: 10.1194/jlr.M900119-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YM, Kim MY, Kim HJ, Roh GS, Ko GH, Seo HG, Lee JH, Chang KC. Compound C independent of AMPK inhibits ICAM-1 and VCAM-1 expression in inflammatory stimulants-activated endothelial cells in vitro and in vivo. Atherosclerosis. 2011;219:57–64. doi: 10.1016/j.atherosclerosis.2011.06.043. [DOI] [PubMed] [Google Scholar]
- 22.Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40–50. doi: 10.4161/auto.7.1.13883. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Martin N, Blas-García A, Morales JM, Marti-Cabrera M, Monleón D, Apostolova N. Metabolomics of the effect of AMPK activation by AICAR on human umbilical vein endothelial cells. Int J Mol Med. 2012;29:88–94. doi: 10.3892/ijmm.2011.802. [DOI] [PubMed] [Google Scholar]
- 24.Lee H, Kang R, Bae S, Yoon Y. AICAR, an activator of AMPK, inhibits adipogenesis via the WNT/β-catenin pathway in 3T3-L1 adipocytes. Int J Mol Med. 2011;28:65–71. doi: 10.3892/ijmm.2011.674. [DOI] [PubMed] [Google Scholar]
- 25.Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci USA. 2009;106:12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langelueddecke C, Jakab M, Ketterl N, Lehner L, Hufnagl C, Schmidt S, Geibel JP, Fuerst J, Ritter M. Effect of the AMP-kinase modulators AICAR, metformin and compound C on insulin secretion of INS-1E rat insulinoma cells under standard cell culture conditions. Cell Physiol Biochem. 2012;29:75–86. doi: 10.1159/000337589. [DOI] [PubMed] [Google Scholar]
- 27.Fei F, Xin-rong W, Ming L, Huan L. Effect of bioactive components in mulberry leaves on glucose metabolism in insulinresistant HepG2 cells. J Guang Pharm Col. 2011;27:637–639. [Google Scholar]
- 28.Li XL, He SM, Zhu Y, Feng B, Huang XQ, Chen T, Zheng GJ. Establishment and identify of HepG2 cells model of insulin resistance. Chin J Exp Trad Med Formul. 2013;19:203–207. [Google Scholar]
- 29.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 31.He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, Radovick S. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137:635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes Metab. 2011;13:1097–1104. doi: 10.1111/j.1463-1326.2011.01466.x. [DOI] [PubMed] [Google Scholar]