

This study highlights the significant role of the genetic repertoire of a microorganism in the similarity between Novosphingobium strains. The results suggest that the phylogenetic relationships were mostly influenced by metabolic trait enrichment, which is possibly governed by the microenvironment of each microbe’s respective niche. Using core genome analysis, the enrichment of a certain set of genes specific to a particular habitat was determined, which provided insights on the influence of habitat on the distribution of metabolic traits for Novosphingobium strains. We also identified habitat-specific protein hubs, which suggested delineation of Novosphingobium strains based on their habitat. Examining the available genomes of ecologically diverse bacterial species and analyzing the habitat-specific genes are useful for understanding the distribution and evolution of functional and phylogenetic diversity in the genus Novosphingobium.
KEYWORDS: Novosphingobium, core genome, habitat-specific genes, pangenome, regulatory hubs
ABSTRACT
Species belonging to the genus Novosphingobium are found in many different habitats and have been identified as metabolically versatile. Through comparative genomic analysis, we identified habitat-specific genes and regulatory hubs that could determine habitat selection for Novosphingobium spp. Genomes from 27 Novosphingobium strains isolated from diverse habitats such as rhizosphere soil, plant surfaces, heavily contaminated soils, and marine and freshwater environments were analyzed. Genome size and coding potential were widely variable, differing significantly between habitats. Phylogenetic relationships between strains were less likely to describe functional genotype similarity than the habitat from which they were isolated. In this study, strains (19 out of 27) with a recorded habitat of isolation, and at least 3 representative strains per habitat, comprised four ecological groups—rhizosphere, contaminated soil, marine, and freshwater. Sulfur acquisition and metabolism were the only core genomic traits to differ significantly in proportion between these ecological groups; for example, alkane sulfonate (ssuABCD) assimilation was found exclusively in all of the rhizospheric isolates. When we examined osmolytic regulation in Novosphingobium spp. through ectoine biosynthesis, which was assumed to be marine habitat specific, we found that it was also present in isolates from contaminated soil, suggesting its relevance beyond the marine system. Novosphingobium strains were also found to harbor a wide variety of mono- and dioxygenases, responsible for the metabolism of several aromatic compounds, suggesting their potential to act as degraders of a variety of xenobiotic compounds. Protein-protein interaction analysis revealed β-barrel outer membrane proteins as habitat-specific hubs in each of the four habitats—freshwater (Saro_1868), marine water (PP1Y_AT17644), rhizosphere (PMI02_00367), and soil (V474_17210). These outer membrane proteins could play a key role in habitat demarcation and extend our understanding of the metabolic versatility of the Novosphingobium species.
IMPORTANCE This study highlights the significant role of a microorganism’s genetic repertoire in structuring the similarity between Novosphingobium strains. The results suggest that the phylogenetic relationships were mostly influenced by metabolic trait enrichment, which is possibly governed by the microenvironment of each microbe’s respective niche. Using core genome analysis, the enrichment of a certain set of genes specific to a particular habitat was determined, which provided insights on the influence of habitat on the distribution of metabolic traits in Novosphingobium strains. We also identified habitat-specific protein hubs, which suggested delineation of Novosphingobium strains based on their habitat. Examining the available genomes of ecologically diverse bacterial species and analyzing the habitat-specific genes are useful for understanding the distribution and evolution of functional and phylogenetic diversity in the genus Novosphingobium.
INTRODUCTION
The genus Novosphingobium represents metabolically versatile members that belong to the class Alphaproteobacteria and family Sphingomonadaceae (1). Novosphingobium species have been isolated from a wide range of ecological habitats such as agricultural soil (2), pesticide-contaminated soil (3, 4), plant surfaces (5), and aquatic environments (6) (see Table 1). Previous studies have investigated Novosphingobium strains for their bioremediation capacity (7–10), nutrient cycling (11, 12), taxonomic characterization (3, 13), analysis of extracellular products (7), mutagenesis experiments on certain genes or gene clusters (14), disease conditions (15, 16), and application in nanoparticle formation for antibacterial activity (17).
TABLE 1 .
General genome characteristic features of the genus Novosphingobium
| Strain | Source of isolation | Genome size (bp) | No. of contigs/repliconsa | GC content (%) | No. of genes | No. of essential marker genes | % completeness | Genomic island size (bp) | Accession no. | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Novosphingobium sp. AAP1 | Freshwater lake | 4,750,579 | 50 | 65.6 | 4,304 | 106 | 99.07 | 501,818 | LJHO00000000 | Unpublished data |
| Novosphingobium sp. AAP83 | Freshwater lake | 4,232,088 | 84 | 59.4 | 4,074 | 106 | 99.07 | 286,348 | LJHY00000000 | Unpublished data |
| Novosphingobium sp. AAP93 | Freshwater lake | 4,267,112 | 149 | 65.5 | 3,948 | 104 | 97.20 | 219,491 | LJHZ00000000 | Unpublished data |
| N. acidiphillum DSM19966 | Acidic lake water | 3,708,535 | 55 | 64.3 | 3,496 | 104 | 97.20 | 248,334 | AUBA00000000.1 | Unpublished data |
| N. aromaticivorans DSM 12444 | The sample obtained at a depth of 410 m from a borehole sample that was drilled at the Savannah River Site | 4,233,314 | One Chr and two plasmids | 65.1 | 4,124 | 106 | 99.07 | 64,422 | CP000248.1, CP000676.1, CP000677.1 | Aylward et al. (12) |
| N. fuchskuhlense FNE08-7 | Isolated from a surface water sample of the southwest basin of Lake Grosse Fuchskuhle | 3,963,850 | 14 | 65.4 | 3,721 | 105 | 98.13 | 172,938 | LLZS00000000.1 | Unpublished data |
| Novosphingobium sp. MBES04 | Sunken wood from Suruga Bay | 5,361,448 | 33 | 65.4 | 5,202 | 103 | 96.26 | 528,404 | BBNP00000000 | Ohta et al. (5) |
| N. malaysiense Musc273 | Mangrove sediment | 5,027,021 | 85 | 63.4 | 4,887 | 106 | 99.07 | 135,248 | JTDI00000000 | Unpublished data |
| N. pentaromaticivorans US6-1 | Muddy sediment of Ulsan Bay | 5,457,578 | One Chr and five plasmids | 63.1 | 5,087 | 106 | 99.07 | 203,560 | CP009291, CP009292, CP009293, CP009294, CP009295, CP009296 | Choi et al., 2015 (81) |
| Novosphingobium sp. PP1Y | Marine water and oil interface | 5,313,905 | One Chr and three plasmids | 63.3 | 5,135 | 106 | 99.07 | 181,419 | FR856862.1, FR856859.1, FR856860.1, FR856861.1 | D’Argenio et al. (6) |
| N. subterraneum DSM12447 | Southeast coastal plain, subsurface core at 180-m depth | 4,885,942 | 54 | 63.2 | 4,838 | 106 | 99.07 | 148,732 | ZRVC00000000.1 | Unpublished data |
| Novosphingobium sp. P6W | Isolated from the plant rhizosphere | 6,537,300 | 65 | 63.7 | 6,279 | 105 | 98.13 | 322,771 | JXZE00000000 | Unpublished data |
| Novosphingobium sp. AP12 | Rhizosphere of Populus deltoides | 5,611,617 | 187 | 65.9 | 5,367 | 105 | 98.13 | 435,323 | AKKE00000000 | Unpublished data |
| N. rosa NBRC 15208 | Isolated from root of plant Rosa sp. 3-ketolactose-forming bacteria | 6,952,763 | 194 | 64.5 | 6,330 | 104 | 97.20 | 636,815 | BCZE01000000 | Unpublished data |
| N. barchamii LL02 | Hexachlorocyclohexane-contaminated soil | 5,307,348 | 26 | 64 | 5,220 | 104 | 97.20 | 264,580 | JACU01000000 | Pearce et al. (4) |
| Novosphingobium sp. KN65.2 | Carbofuran-exposed agricultural soil | 5,024,847 | 243 | 63.1 | 5,036 | 106 | 99.07 | 328,926 | CCBH000000000 | Nguyen et al. (2) |
| N. lindaniclasticum LE124 | Hexachlorocyclohexane-contaminated soil | 4,857,928 | 156 | 64.6 | 4,749 | 105 | 98.13 | 292,630 | ATHL00000000 | Saxena et al. (60) |
| N. naphthalenivorans NBRC102051 | Isolated from polychlorinated-dioxin-contaminated environments | 5,236,092 | 234 | 63.8 | 5,224 | 106 | 99.07 | 342,845 | BCTX00000000.1 | Unpublished data |
| Novosphingobium sp. ST904 | Rhizosphere of Acer pseudoplatanus, growing at a 2,4,6-trinitrotoluene-contaminated forest site | 6,269,463 | 166 | 64.5 | 6,945 | 100 | 93.46 | 303,084 | LGJH00000000 | Unpublished data |
| Novosphingobium sp. B-7 | Steeping fluid of eroded bamboo slips | 4,909,165 | 491 | 65.1 | 4,715 | 104 | 97.20 | 825,534 | APCQ00000000 | Unpublished data |
| Novosphingobium sp. Leaf2 | Derived from an Arabidopsis leaf | 3,715,735 | 22 | 64.1 | 3,675 | 106 | 99.07 | 235,969 | LMJY00000000 | Unpublished data |
| N. nitrogenifigens DSM 13790 | New Zealand pulp mill effluent | 4,148,048 | 77 | 64 | 3,867 | 106 | 99.07 | 255,167 | AEWJ0000000 | Unpublished data |
| N. resinovorum KF1 | Biofilm of a bioreactor fed with polychlorinated phenols | 6,304,486 | 115 | 65.1 | 6,079 | 106 | 99.07 | 279,955 | JFYZ00000000.1 | Unpublished data |
| Novosphingobium sp. Rr2-17 | Grapevine crown gall tumor | 4,539,029 | 166 | 62.7 | 4,513 | 104 | 97.20 | 295,587 | AKFJ00000000 | Gan et al., 2012 (19) |
| N. tardaugens NBRC 16725 | Isolated from activated sludge of sewage treatment plant | 4,291,514 | 54 | 61.3 | 4,223 | 105 | 98.13 | 319,713 | BASZ00000000.1 | Unpublished data |
| N. capsulatum NBRC12533 | Not available | 4,836,455 | 70 | 65.7 | 4,452 | 106 | 99.07 | 612,524 | BCYV00000000.1 | Unpublished data |
| N. lentum NBRC 107847 | Isolated from a cold fluidized-bed process treating chlorophenol-contaminated groundwater | 4,407,848 | 53 | 65.7 | 4,266 | 105 | 98.13 | 528,688 | BCTW00000000.1 | Unpublished data |
The number of contigs/replicons or the number of chromosomes (Chr) and plasmids is shown.
Many Novosphingobium genomes are now available in public repositories (e.g., GenBank), and recently, Gan and colleagues (19) performed comparative genomic analysis where six Novosphingobium genomes were compared to elucidate the mechanism of salt tolerance, cell-cell signaling, and aromatic compound biodegradation. To further enhance our understanding of the metabolic versatility of this genus and to determine how this versatility is distributed by phylogeny and habitat, we selected 27 Novosphingobium genomes from diverse habitats and classified a subset of these strains into four different ecological groups—rhizosphere, contaminated soil, freshwater, and marine water. We then determined whether core metabolic trait distribution was influenced more by habitat or phylogenetic clustering.
RESULTS AND DISCUSSION
General genomic organization of Novosphingobium strains.
The 27 Novosphingobium strains had an average genome size of 4.97 Mbp. The largest genome was 6.95 Mbp, belonging to Novosphingobium rosa NBRC 15208 isolated from rhizospheric soil. The smallest genome was 3.71 Mbp, belonging to N. acidiphillum DSM19966, which was isolated from the acidic lake water. In order to investigate whether certain adaptive traits follow the environment-specific or habitat-specific phenotype, 27 Novosphingobium strains were grouped based on their isolation habitat. Of these 27 strains, 19 strains were grouped in one of the four different habitats, i.e., rhizosphere (strains AP12, P6W, and NBRC15208), contaminated soil (strains LL02, LE124, NBRC102051, KN65.2, and ST904), freshwater (strains AAP1, AAP83, AAP93, FNE08-7, DSM12444, and DSM19966), and marine water (strains MBES04, Musc273, DSM12447, US6-1, and PP1Y). The remaining eight strains (B-7, Leaf2, DSM13790, KF1, Rr2-17, NBRC 16725, NBRC 12533, and NBRC 107847) were excluded, as either there was no information available on their isolation site or less than three representatives were available to represent a habitat (Table 1). Focusing on the habitats, the largest genomes were found in the rhizosphere (6.37 ± 0.56 Mbp; n = 3), followed by contaminated soil (5.34 ± 0.55 Mbp; n = 5), marine water (5.21 ± 0.24 Mbp; n = 5), and freshwater (4.20 ± 0.34 Mbp; n = 6). Average genome size differed significantly between habitats (F3,15 = 16.89 and P < 0.0001 by analysis of variance [ANOVA]); it has previously been correlated with environmental complexity where the largest genomes are found in rhizospheric soil (18).
Previous studies based entirely on 16S rRNA gene sequencing predicted that the GC content in Novosphingobium varied between 62 and 67% (1, 12, 19). However, GC content of 27 Novosphingobium genomes in this study ranged from 59.4% in Novosphingobium sp. strain AAP83 to 65.9% in Novosphingobium sp. strain AP12. Based on essential marker gene analysis, the genomes of strain AP12 and AAP83 were >98% complete (Table 1); thus, the GC content range for the genus Novosphingobium as defined previously by DNA-DNA hybridization (DDH) should be reclassified to 59% to 67%. A previous study suggested that GC content is predicted to significantly influence the functional potential and hence ecological adaptation of an organism (20). However, the variability in percent GC content for Novosphingobium was not significant between the four habitats (F3,15 = 0.308 and P < 0.82 by ANOVA), suggesting that the ecological adaptations of Novosphingobium spp. are not influenced by a shift in percent GC content.
Core genome and pangenome analysis.
Bacterial pangenomes typically consist of distinct core and accessory gene complements (21). Novosphingobium maintained a core gene complement of 220 genes (query coverage of ≥75% and nucleotide Identity of ≥75%) for the 27 genomes analyzed. As expected, these orthologs include components of regulatory pathways such as DNA replication, basic transcriptional machinery, translation, mismatch repair, nucleotide excision repair, homologous recombination, signal transduction, bacterial secretion system and protein export. In addition, citric acid cycle, fatty acid biosynthesis and elongation, amino acid biosynthesis and purine metabolism were also present. However, only 128 of the 220 orthologous genes could be reliably annotated as “essential” against the DEG database (22), whereas the remaining 92 accessory genes still coded for basic metabolic functions.
Pangenome analysis of the 27 Novosphingobium strains (Fig. 1) identified 21,915 nonredundant (nonrepetitive) genes in the pangenome, out of 128,647 total genes. The genome curve displayed an asymptotic trend, indicating that 27 genomes were insufficient to describe the complete gene repertoire of the genus Novosphingobium. Analysis of the core genome was also asymptotic, with 714 core genes after the addition of the 27th genome; however, this trend suggests that further Novosphingobium genomes will result in only minor changes in the core genome of this genus (Fig. 1).
FIG 1 .
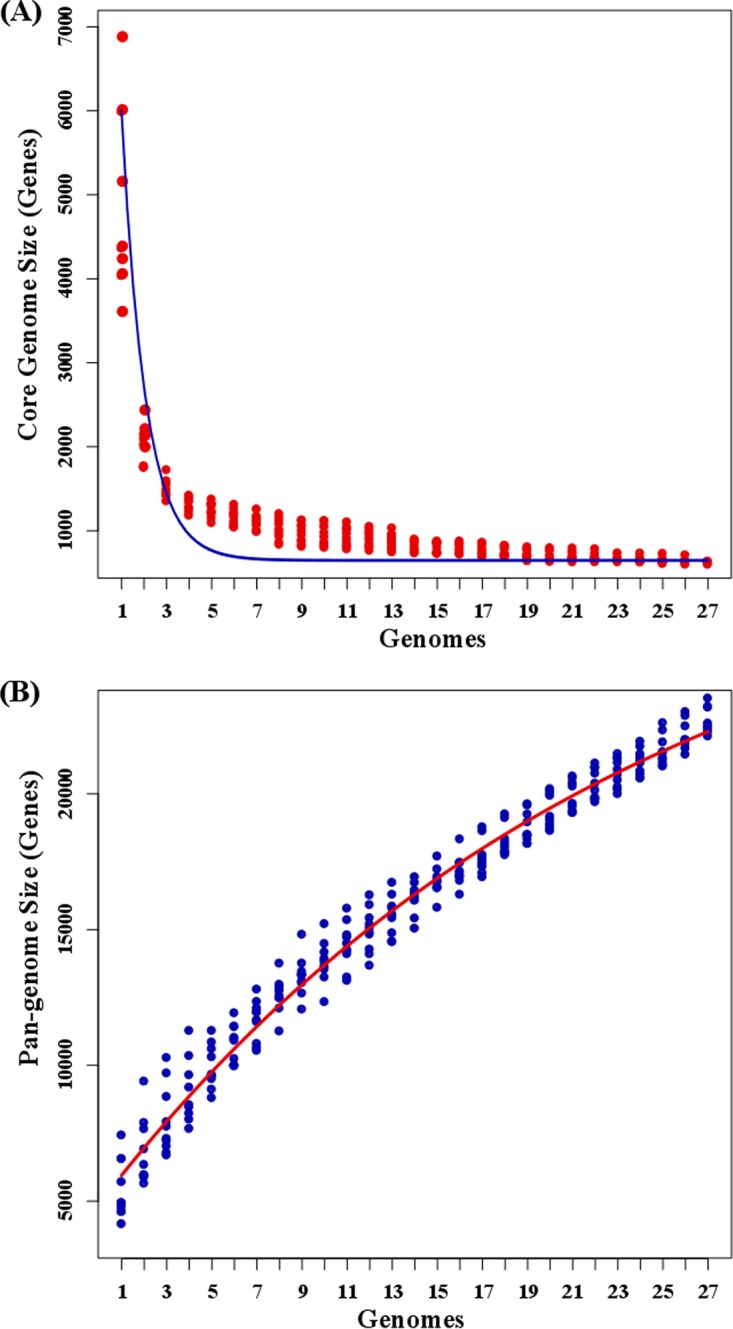
Core and pangenome of 27 Novosphingobium strains plotted against the number of genomes. (A) Core genome. The x axis shows the number of genomes, and the y axis shows the core genome size (number of genes) of Novosphingobium spp. (B) Pangenome. The x axis shows the number of genomes added, and the y axis shows the increase in pangenomic content of Novosphingobium spp. with the addition of genomes. The sizes of the core and pangenome clusters were computed using the BDBH algorithm. For the robustness of the calculation, the built-in program runs the sampling experiments (n = 10), where genomes are randomly added to estimate the stability of the core and pangenome. The best-fit Tettelin curve represents the regression line for the core and pangenome.
Habitat-specific traits.
The orthologous gene contents for Novosphingobium strains in four habitats were identified, and a pairwise comparison was performed to obtain habitat-specific genes. Out of 17,976 redundant orthologous genes, 1,943 gene sets were core genome for rhizosphere, 1,530 for contaminated soil, 1,485 for freshwater, and 1,546 for marine water. Further, comparison of the core genome of each habitat with respect to another revealed the presence of 438 specific genes for rhizosphere, 346 for contaminated soil, 143 for marine water, and 297 for freshwater. These habitat-specific genes were annotated against the KAAS server (23), but only 211 rhizospheric, 125 contaminated soil, 54 marine, and 150 freshwater genes could be annotated with a KEGG Orthology (KO) identifier. These KO identifiers were mapped against metabolic pathways using iPath (24), and the differences were mostly observed in amino acid metabolism, suggesting different amino acid availabilities in these environments (see Fig. S1 in the supplemental material). Rhizosphere-specific gene content consists of genes encoding components involved in glycine, serine, and threonine metabolism. Contaminated-soil-specific gene content consists of genes encoding components involved in tyrosine and phenylalanine metabolism. Freshwater-specific gene content contain genes encoding components involved in alanine, aspartate, and glutamate metabolism, and marine water-specific gene content contain genes encoding components involved in the bacterial chemotactic regulatory pathway, which could be involved in nutrient acquisition in this normally oligotrophic environment. Genes related to terpenoid backbone biosynthesis were present only in the core genomes of rhizospheric strains, which has been shown to play a role in the stability of bacterial cell membranes and root interaction in rhizospheric strains (25). Therefore, the analysis has put forward the differences between Novosphingobium strains based on differences in the metabolic preferences for amino acids in their respective habitats, representing the resultant adaptive changes in response to the environment.
Mapping of habitat-specific metabolic pathways. The habitat-specific metabolic pathways were mapped using iPath v2. In the map, different habitats were represented by different colors: green for rhizosphere, yellow for contaminated soil, blue for marine water, and red for freshwater. Download FIG S1, TIF file, 8.3 MB (8.5MB, tif) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Distribution of Novosphingobium strains along their phylogenetic clade.
The consensus phylogeny of Novosphingobium spp. has shown the mixed trend of phylogenetic clustering of strains isolated from a similar environment. For instance, N. barchamii strain LL02 (contaminated soil), Novosphingobium sp. strain P6W (rhizosphere), and Novosphingobium sp. strain AP12 (rhizosphere), despite belonging to different environments, clustered together. While Novosphingobium sp. strain ST904 and N. lindaniclasticum LE124, which were both isolated from contaminated soil, form a monophyletic clade (Fig. 2 and Table 1). Notably, strains LL02 (13) and LE124 (3) were isolated from hexachlorocyclohexane (HCH) dumpsites, but in all three methods (conserved marker genes and average nucleotide identity [ANI] on the whole genome and core genome), these strains clustered separately. Similarly, Novosphingobium sp. strain KN65.2 was isolated from carbofuran-contaminated soil but clustered with marine isolates, Novosphingobium sp. strain PP1Y (6) and N. pentaromaticivorans US6-1 (10). This clustering is likely a result of shared metabolic tendency, as strain KN65.2 can degrade carbofuran (2) and strains PP1Y and US6-1 can degrade polyaromatic hydrocarbon (PAH) compounds (6, 10). Further ambiguity in habitat specificity was observed from the clustering of strains of marine, contaminated soil, and freshwater habitats (N. malaysiense Musc273 [marine], N. naphthalenivorans NBRC102051 [contaminated soil], N. fuchskuhlense FNE08-7 [freshwater], Novosphingobium sp. AAP93 [freshwater], N. subterraneum DSM12447 [marine], N. aromaticivorans DSM12444 [freshwater], and Novosphingobium sp. AAP83 [freshwater]). The results indicated that the phylogenetic clustering of genomes was apparently different from the habitat-specific grouping of these strains. This may be because Novosphingobium spp. have varied metabolic preferences, suggesting that habitat-specific factors are probably masked by the microenvironment in shaping the Novosphingobium genomes. Also, the differences in tree topology using these two methods, i.e., ANI (whole genome based) and 400 conserved bacterial marker genes, could be due to the inclusion of pangenomic content in the case of the whole genome (ANI) rather than the conserved marker genes. Further, to check the impact of the missing gene content from draft genomes, the phylogeny was constructed on the core genome using ANI. The result suggested that the least complete genome (≅93.46% [Table 1]), i.e., Novosphingobium sp. strain ST904 grouped with N. lindaniclasticum LE124 by all three methods. Thus, it can be inferred that the missing gene content will have the least impact on the change in phylogeny among the Novosphingobium strains.
FIG 2 .

Phylogenetic clustering of 27 Novosphingobium strains. (A) Phylogeny based on 400 conserved marker genes with 1,000 bootstraps by using S. indicum B90A as an outgroup. (B and C) Average nucleotide identity (ANI)-based phylogeny was constructed with 220 orthologous genes and the whole genome, respectively. The bars represent 1 nucleotide substitution per position.
Habitat-specific protein identification and their protein-protein interaction analysis.
The phylogenomics of the different strains did not reflect their habitat specificity, which suggests that the functional repertoire of these strains may supersede evolutionary relatedness. Protein-protein interaction (PPI) networks enable biological characteristics and protein function to be taken into consideration for each strain (26) and can be used to identify habitat-specific adaptations (27). To confirm that the proteome interaction with the environment, particularly for the uptake and secretion of molecules, is highly habitat specific, we aimed for the identification of putative outer membrane proteins involved in the transport of metabolites and toxins, as well as membrane biogenesis (28). We focused on proteins characterized as trans-membrane beta-barrel proteins (TMBbps) in Novosphingobium proteomes. The analysis showed the presence of different numbers of TMBbps in each strain of Novosphingobium across the four habitats. The identified TMBbp sequences of different strains clustered together based on habitat, when subjected to protein sequence similarity analysis. The proteins with the highest percentage of similarity were further referred to as habitat-specific proteins (HSPs). To validate their specificity toward the habitat, amino acid sequences of these TMBbps were subjected to phylogenetic analysis, which demonstrated habitat-specific clustering (Fig. S2). To confirm the stability of these proteins as key regulatory molecules, PPI interaction networks were established based on the core genome. To identify the key molecules, networks for each habitat were constructed and analyzed (Fig. 3A to D). The hub proteins for each strain in all four habitats were identified (Table S1). To understand the topological properties of these networks, the probability of degree distribution P(k) showed that each network followed a power law scaling behavior
FIG 3 .

The protein-protein interaction (PPI) network of four habitats, i.e., freshwater, marine water, rhizosphere, and soil. Expanded view of the network imported from Cytoscape, where nodes represent proteins and edges represent physical interactions. The nodes in all four habitats (freshwater, marine water, rhizosphere, and contaminated soil) were represented as filled circles that were light red, green, dark blue, and light blue, respectively. The edges in all habitats were represented in the form of grey lines. The significant existence of sparsely distributed hubs in four habitat networks were represented by colored circles as purple (freshwater), dark blue (marine), orange (rhizosphere), and pink (contaminated soil).
Phylogenetic analysis based on habitat-specific beta-barrel protein using the neighbor-joining method and 1,000 bootstrap replicates. The evolutionary distances were computed by the Poisson correction method. Download FIG S2, EPS file, 1.5 MB (1.6MB, eps) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of regulatory hub proteins in four different habitats of genus Novosphingobium. Download TABLE S1, DOCX file, 0.02 MB (16.8KB, docx) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
| (1) |
with the values of the degree exponent γ were ∼0.52, 1.0, 0.43, and 0.59 in freshwater, marine water, rhizosphere, and contaminated soil habitats, respectively (Fig. 4A). The small value of γ (γ < 2) indicated that the network was hierarchical (29), signifying the emergence of hierarchical modules and/or communities (30), with a sparse distribution of highly connected hubs (31). The fact that these few highly connected hubs were connected to many low-degree nodes was indicative of a regulatory power of the hubs over these nodes. For further analysis of this topological feature of the network (30), the average clustering coefficient C(kn) was calculated as a function of the number of neighbors kn:
FIG 4 .

Topological properties of the PPI networks in the four habitats (freshwater, marine water, rhizosphere, and soil). The Pearson correlation coefficient values (r2) and probability of degree distributions P(k) (A), average clustering coefficient (B), and average neighborhood connectivity of the PPI network (C) are shown. All these properties follow the power law distribution and show the nature of scale-free network, suggesting a hierarchical organization in the network.
| (2) |
Again, this followed the power scaling law with β values of ∼0.31, 0.40, 0.73, and 0.36 in freshwater, marine water, rhizosphere, and contaminated soil habitats, respectively, which supported that the network falls in a hierarchical network (Fig. 4B).
The average neighborhood connectivity Cn(kn) was constructed as a function of kn as follows:
| (3) |
with values of ~0.42, 0.24, 0.36, and 0.33 in freshwater, marine water, rhizosphere, and soil habitats, respectively (Fig. 4C), also indicating that the network falls in a hierarchical network (30, 31), the hub proteins in each habitat network are likely indicative of key molecules for habitat adaptation in each genome (32), and these proteins had the highest degree of interactions in these hierarchical networks. Hub proteins of each habitat were identified, and these proteins include the Saro_1868 protein (TonB-dependent receptor) for the freshwater habitat (Fig. 3A), PP1Y_AT17644 protein (hypothetical protein with porin domain) for the marine habitat (Fig. 3B), PMI02_00367 protein (TonB-dependent receptor) for the rhizosphere habitat (Fig. 3C), and V474_17210 protein (TonB-dependent receptor) for the soil habitat (Fig. 3D). As these β-barrel outer membrane proteins are present on the surfaces of Gram-negative bacteria and perform a variety of functions such as active ion transport, passive nutrient uptake, membrane anchors, membrane-bound enzymes, and in defense (33), they are likely crucial for the adaptation of the Novosphingobium strains in their respective environments.
Sulfur uptake and metabolism are different between habitats.
The sulfur metabolism pathway in prokaryotes involves the uptake and utilization of environmental sulfur derivatives for the synthesis of proteins, sulfate esters of polysaccharides, phenols, steroids, and coenzymes. In general, there are three different routes for the assimilation of environmental sulfur (Fig. 5). The first and predominant mode includes the uptake and metabolism of sulfates in the form of inorganic sulfur (sulfates and thiosulfates) which is carried out by proteins encoded by cysPAUW (transport system) (34) and cysD and cysNC (activation and utilization) (35) followed by cysteine biosynthesis genes cysE, cysK, and cysQ. The second route involves the uptake and utilization of environmental sulfonates, characterized by the presence of the ssuABC (transport system) and ssuD (FMNH2-dependent alkane sulfonate monooxygenase) genes. The alkane sulfonates comprise the major portion of carbon-bonded environmental sulfur (68%) (36) and 20 to 40% of organic sulfur present near marine sediments (37). The third route of sulfur assimilation involves taurine transport and metabolism encoded by the tauABC (transport system) and tauD (taurine dioxygenase) genes, respectively.
FIG 5 .

Schematic representation of different modes of environmental sulfur uptake and utilization within the Novosphingobium genus. The three different routes for sulfur assimilation are shown. Sulfur assimilation as inorganic sulfur (sulfates and thiosulfates) (A), via ssuABC (transport system) and ssuD (FMNH2-dependent alkane sulfonate monooxygenase) (B), and via taurine transport and metabolism by tauABC (transport system) and tauD (taurine dioxygenase) (C). APS, adenosine phosphosulfate; PAPS, phosphoadenosine phosphosulfate; SUOX, sulfite oxidase.
Studies related to sulfur assimilation in bacteria isolated from different habitats have revealed the coexistence of these routes in the same species (38), but to date, no study has determined the distribution of these three pathways across different habitats. To determine this for Novosphingobium in rhizosphere, contaminated soil, freshwater, and marine water, the genes involved in sulfur metabolism were identified and strains were clustered according to their sulfur assimilation repertoire. Four resultant clades were designated: clade I, clade II, clade III, and clade IV (Fig. 6). Although clustering of the strains based on habitat was not observed, the pattern of differentiation of pathways was clearly demarcated. For instance, sulfate metabolism, the most predominant mode of environmental sulfur assimilation, was found only in clade I (strains MBES04, LE124, FNE08-7, and AAP93) and clade II (strains ST904, AP12, P6W, LL02, and NBRC15208) (Fig. 6). Further, the complete pathway of alkane sulfonate assimilation was found exclusively in strains clustered in clade II, which comprised only soil isolates (rhizosphere and contaminated soil). Earlier, the alkane sulfonate assimilation system had been reported in freshwater isolates (38), but none of the freshwater isolates we studied maintained the system. In addition to this, tauD coding for taurine dioxygenase was identified in all of the Novosphingobium strains, while the taurine transport system was absent. The two other clades, clades III (comprised of mainly aquatic isolates) and IV, lacked a complete sulfur transport system, instead maintaining a mosaic of genes encoding components involved in sulfate oxidation, taurine oxidation, and sulfonate oxidation, which suggests the use of multiple sulfur derivatives. Interestingly, the strains isolated from contaminated soil were found in all four clades and therefore maintained a diverse array of sulfate metabolism. This suggests that the modes of sulfur assimilation in Novosphingobium spp. were not confined to a certain habitat but might relate to the availability of different types of environmental sulfur compounds in their respective habitats.
FIG 6 .

Matrix and dual dendrogram based on the presence/absence of sulfur metabolism genes was constructed in 19 Novosphingobium genomes belonging to four different habitats, viz., contaminated soil (C), rhizosphere (R), freshwater (F), and marine water (M). The colored and white boxes represent the presence and absence of a gene, respectively. A dendrogram based on the matrix of sulfur metabolism genes was constructed using Pearson correlation and hierarchical clustering.
Mechanisms for survival in marine environments are also observed in contaminated soils.
In general, there are two different strategies that are known to confer bacterial survival in a saline environment. These strategies include accumulation of inorganic components in the cytoplasm, which counterbalances the salinity (39), and synthesis of the organic osmolytes that do not increase the ionic concentration but maintain the osmotic pressure (40). Two such osmotic solutes are ectoine (1,4,5,6-tetrahydro-2-methyl-4-pyrimidine carboxylic acid) and hydroxyectoine, which are common osmolytes in marine and halotrophic bacteria (41–43). The ectoine biosynthesis pathway involves components encoded by the ectA (L-2,4-diaminobutyric acid acetyltransferase), ectB (L-2,4-diaminobutyric acid transaminase), and ectC (l-ectoine synthase) genes (44). In addition to this, the protein encoded by ectD (ectoine hydroxylase) catalyzes the conversion of ectoine into hydroxyectoine (45).
Ectoine biosynthesis is considered to be an adaptation of marine Novosphingobium strains, such as Novosphingobium sp. strain PP1Y and N. pentaromaticivorans US6-1, which were previously reported to possess the ectoine biosynthesis pathway (19). However, we found that among the marine isolates, only N. malaysiense Musc273 along with PP1Y and US6-1 maintained a complete ectoine biosynthesis pathway, while two other marine isolates, N. subterraneum DSM12447 and Novosphingobium sp. strain MBES04, did not possess any of the ectoine pathway genes. The complete absence of the ectoine pathway in marine strains MBES04 and DSM12447 suggested that these strains might use different routes to compensate for high-salt conditions of marine water. Another possible reason might be that both strains are not truly marine, as the former was isolated from sunken wood (5) while the latter was isolated from coastal plains at a depth of 180 m (unpublished). Interestingly, strains isolated from other habitats were found to possess genes for ectoine biosynthesis, such as Novosphingobium sp. KN65.2, a carbofuran-contaminated soil isolate, which possessed the complete ectoine biosynthesis pathway. In addition to this, ectA and ectB were identified in N. barchamii LL02 and Novosphingobium sp. ST904, isolated from hexachlorocyclohexane- and 2,4,6-trinitrotoluene-contaminated soil, respectively. Also, rhizospheric strains, Novosphingobium sp. P6W and N. rosa NBRC 15208 were found to possess ectA and ectB, respectively, while freshwater strains were completely devoid of genes for ectoine biosynthesis. The occurrence of ectoine pathway genes in strains from contaminated soil and rhizosphere habitats implies that ectoine synthesis may not be a habitat-specific trait but it may instead be acquired and maintained by strains from different ecological niches, likely driven by environmental stress, or that the pathway is not useful but simply maintained in the contaminated soil environment.
Degradation potential of Novosphingobium strains across four different habitats.
Sphingomonads have been widely reported as efficient degraders of xenobiotic compounds such as hexachlorocyclohexane, chlorophenol, phenol, homogentisate, anthranilate, and other polyaromatic hydrocarbons (46, 47). Of the sphingomonads, Sphingobium and Sphingomonas strains have been extensively studied with respect to their xenobiotic degradation potential (12, 48), while less is known about Novosphingobium spp. A comparative genomic study on six Novosphingobium strains was carried out earlier (19), but the focus was on overall genomic repertoire. Here we analyzed Novosphingobium genomes for the presence of aromatic compound degradation pathway genes. The analysis revealed that the genes encoding PAH and components involved in xenobiotic degradation were enriched in Novosphingobium strains (Fig. 7) among which freshwater strains showed similarity in genes encoding mono- and dioxygenases, with very similar metabolic profiles, while strains from the other three habitats clustered separately (Fig. 7A and B). Of note, N. rosa NBRC15208, a rhizospheric isolate, was found to harbor the highest number of genes (n = 157) for aromatic compound degradation, especially for gentisate, protocatechuate, and catechol. The two other rhizospheric strains, Novosphingobium sp. P6W (n = 45) and Novosphingobium sp. AP12 (n = 59), contained only 33% of the N. rosa NBRC15208 gene complement. Following this, Novosphingobium sp. KN65.2 (contaminated soil) and Novosphingobium sp. PP1Y (marine) with 124 genes each, had the second highest metabolic repertoire. Novosphingobium sp. KN65.2 possessed genes mainly for gentisate, biphenyl, homogentisate, and protocatechuate degradation, while Novosphingobium sp. PP1Y possessed a high number of gentisate and biphenyl degradation genes. Interestingly, strains from sites contaminated with HCH, polychlorinated dioxin, pulp mill effluent, and carbofuran contained comparably fewer genes for aromatic compound degradation, which suggested that particular contaminants might lead to genome streamlining under environmental stress. Also, genes for gentisate, catechol, and protocatechuate catabolism were found in abundance, projecting their ability to degrade a variety of aromatic compounds (49).
FIG 7 .

Correlation between the ability of Novosphingobium strains from four different habitats to degrade aromatic and xenobiotic compounds. (A) The heat map represents clustering of genomes based on the presence of different aromatic degradation pathways. (B) Principal-component analysis (PCA) plot using strain-specific degradation pathways. (C) Distribution of mono- and dioxygenase genes within Novosphingobium genomes.
The presence of mono- and dioxygenase family proteins in Novosphingobium spp. (50), i.e., enzymes known for aromatic ring cleavage, was also determined. Novosphingobium sp. PP1Y showed the greatest number of genes coding for mono- and dioxygenases (114 genes) (Fig. 7C). The most predominant types of monooxygenases in Novosphingobium strains include cyclohexanone monooxygenase, nitrotriacetate monooxygenase, vanillate monooxygenase, alkanal monooxygenase, toluene-4-monooxygenase, alkane sulfonate monooxygenase, and choline monooxygenases. Of these monooxygenases, the most abundant was alkane sulfonate monooxygenase (17 copies) in strain P6W. Major dioxygenases include alpha-ketoglutarate-dependent taurine dioxygenase, benzoate 1,2-dioxygenase, catechol 1,2-dioxygenase, catechol 2,3-dioxygenase, phenylpropionate dioxygenase, and protocatechuate 4,5-dioxygenase, while alpha-ketoglutarate-dependent taurine dioxygenases (15 copies) were the most abundant and observed in strain AP12. This high diversity of mono- and dioxygenases in Novosphingobium strains suggests their hidden potential to metabolize a wide variety of aromatic hydrocarbons. Also, the abundance of alkane sulfonate monooxygenases and taurine dioxygenases indicates an ability to utilize environmental sulfur via environmental alkane sulfonate and taurine, respectively.
Phage integration and genomic adaptation.
Phage/virus integration in bacterial genomes is often considered a genomic adaptation mechanism of bacterial strains which enables novel gene acquisition and might be critical for survival. It has been reported that integrated prophages can constitute up to 20% of a bacterial genome (51), which eventually leads to strain emergence and diversification. Such genomic reservoirs have been shown to be highly diverse across aquatic and terrestrial ecosystems (52). In this study, 29 intact phages in Novosphingobium genomes (Table 2) were identified, with the greatest diversity in strains from contaminated soil (12 phages). Although most of the proteins encoded by the integrated phage were either phage related or hypothetical, a few of the annotated proteins, such as arsenic resistance, NADH-dependent flavin mononucleotide (FMN) reductases, dioxygenase, and permease, could provide improved resistance and degradation of polyaromatic hydrocarbons (PAHs) (Table 2).
TABLE 2 .
Characteristic features of predicted phages within the genus Novosphingobium
| Strain | Putative attL or attR sequence | Region length (kbp) | % GC | Total no. of CDSa | No. of phage proteins | No. of hypothetical proteins | Presence (no.)/absence of genes encodingb: |
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Integrase/transposes | Short-chain dehydrogenase/reductase SDR | Translation initiation factor IF-2 | Transcription elongation factor NusA | CopG/Arc/MetJ/Ars family transcriptional regulator | Phage shock protein PspC | Methylase | Hsp33 protein | NADPH-dependent FMN reductase | LexA repressor | Putative lipoprotein | 5-Oxoprolinase | OmpA/MotB | Nuclease | Plasmid pRiA4b ORF-3 family protein | Serine o-acetyltransferase | ATPase subunit C | Dioxygenase | Protein-tyrosine-phosphatase | Arsenical resistance | Membrane dipeptidase | Amino acid permease | |||||||
| Novosphingobium sp. KN65.2 | GCGCCTGATGCGC | 57.2 | 61.40 | 63 | 31 | 32 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CTCCCGCTCCGCCA | 39.2 | 62.16 | 55 | 33 | 21 | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| Unresolved | 23.8 | 65.32 | 30 | 20 | 8 | − | − | − | − | − | − | − | 1 | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| Unresolved | 12 | 63.81 | 17 | 13 | 4 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| N. barchamii LL02 | CAAGGCAGGGAA | 34.9 | 63.00 | 49 | 22 | 25 | − | − | − | − | − | − | − | − | − | 1 | 1 | − | − | − | − | − | − | − | − | − | − | − |
| Unresolved | 16.8 | 69.11 | 20 | 11 | 8 | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| N. lindaniclasticum LE124 | TGCGCGGCGCCTT | 35.2 | 63.73 | 48 | 30 | 18 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Novosphingobium sp. ST904 | Unresolved | 16.7 | 68.88 | 24 | 13 | 10 | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − |
| GGGCGGTTAGCTCAGTTGGTAGAGCATCTCGTTTACAC | 40.3 | 63.01 | 55 | 33 | 22 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| GACGGCGCCGAGCAT | 40.5 | 65.26 | 37 | 27 | 10 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| N. napthalenivorans NBRC102051 | Unresolved | 35.7 | 64.76 | 54 | 28 | 21 | − | − | − | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | 1 | 1 | 1 | − | − |
| TTCGGATCAGGCTCT | 25.9 | 61.12 | 26 | 14 | 7 | 3 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | 1 | 1 | |
| Novosphingobium sp. MBES04 | GAGGGTGAGATG | 36.1 | 61.61 | 27 | 13 | 9 | 2 | − | − | − | − | − | − | − | − | − | − | 2 | − | − | − | − | − | − | − | − | − | − |
| Unresolved | 19.3 | 68.18 | 27 | 15 | 7 | − | − | − | − | − | − | − | − | 1 | − | − | − | 2 | − | − | − | − | − | − | − | − | − | |
| Novosphingobium sp. PP1Y | CGCCGCCGCTGGTCG | 49.9 | 61.54 | 46 | 29 | 15 | 1 | − | − | − | − | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | − |
| Unresolved | 18.5 | 63.16 | 24 | 14 | 5 | 3 | − | − | − | 1 | − | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | |
| N. subterraneum DSM12447 | CCGACCAAAGCACGAACCCGCTCCGCGGGAGAGTCGCTTGGGGTGCCGTAGCGTAGTATTGTTCAGGCTTTGCGTGCGGC | 24.6 | 62.74 | 31 | 14 | 12 | − | − | 1 | 1 | 1 | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| N. pentaromaticivorans US6-1 | Unresolved | 23.3 | 63.07 | 29 | 24 | 5 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Novosphingobium sp. P6W | AGGAGCCCACGC | 35.3 | 62.47 | 43 | 29 | 14 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| N. rosa NBRC15208 | Unresolved | 23.8 | 64.75 | 31 | 23 | 8 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Novosphingobium sp. AAP93 | Unresolved | 13.5 | 64.21 | 15 | 12 | − | 2 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − |
| N. fuschkulense FNE08-7 | Unresolved | 30.9 | 62.27 | 43 | 28 | 15 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| N. nitrogenifigens DSM 13790 | Unresolved | 27.9 | 64.58 | 34 | 26 | 7 | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| N. resinovorum KF1 | Unresolved | 19.6 | 68.73 | 25 | 15 | 9 | − | − | − | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| N. tardaugens NBRC 16725 | GATCAGCTTGCTATGGACAAGACAACCACACGGCC | 23.5 | 59.62 | 23 | 13 | 9 | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Novosphingobium sp. Leaf2 | CGGATTTTAAGTCCGCAGCGTCTACCATTCCGCCACGCCCGCAC | 37.4 | 63.76 | 51 | 34 | 15 | − | − | − | − | − | − | − | − | − | 1 | − | − | − | 1 | − | − | − | − | − | − | − | − |
| Unresolved | 25.1 | 66.40 | 29 | 19 | 8 | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | |
| Novosphingobium sp. Rr2-17 | Unresolved | 16.3 | 67.62 | 21 | 11 | 9 | − | − | − | − | − | − | − | − | 1 | − | − | − | − | − | − | − | − | − | − | − | − | − |
| TTGATGGCGACGC | 52.3 | 60.80 | 37 | 27 | 10 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
CDS, coding sequences.
The presence or absence (−) of genes encoding the indicated protein or characteristic is shown. If the gene is present, the number of genes is shown.
Novosphingobium strains from marine habitats had the second greatest abundance of phage content. This may be due to the fact that viruses are very common in oligotrophic marine environments (53, 54). Interestingly, Novosphingobium sp. MBES04 acquired the gene encoding 5-oxoprolinase via phage-mediated horizontal gene transfer, which catalyzes 5-oxoproline conversion into glutamate. Pyroglutamic acid or 5-oxoproline is an osmolyte that helps in the maintenance of osmotic balance along with sucrose and ectoine, predominantly characterized in bacteria inhabiting environments with high salt concentrations (55). Further, studies have also shown the role of glutamate in osmoregulation (56). Although the complete pathway for pyroglutamic acid synthesis was absent, the strain MBES04 might be using an alternative pathway and thus acquiring these features for streamlining the genome with respect to the habitat. Apart from this, marine strains have shown the acquisition of ompA and motB genes (MBES04), generally found in the outer membranes of Gram-negative bacteria (57) and known to influence bacterial attachment (58). Hence, this is predicted to further boost the chemotactic behavior of marine bacteria. Further, the acquisition of phage-mediated transcription initiation factor, elongation factor, and regulators may help in activation of adaptive genes (59). Thus, Novosphingobium strains have shown the well-developed phage acquisition-adaptation machinery that might play an important role in combating stress from the environment they inhabit.
Conclusions.
The phylogenetic relationship among Novosphingobium strains was not completely concordant with habitat, as only some strains clustered with strains from similar habitats. The overall genetic repertoire played a significant role in structuring the similarity between strains, suggesting that habitat has little influence on the phylogenetic relationship. However, a systems biology approach revealed habitat-specific protein hubs that were able to delineate Novosphingobium strains based on their habitats. Further, metabolic genes with significant habitat-specific delineation were determined. For instance, sulfur acquisition was differentially encoded among strains and habitats, while the alkane sulfonate assimilation pathway was common among all rhizospheric strains. The ectoine biosynthesis pathway, predominantly identified for osmoregulation in marine bacteria, was also identified in strains isolated from other habitats, suggesting its significance beyond the marine habitat. Aromatic compound degradation and abundance of mono- and dioxygenase genes across all strains in all habitats suggest that Novosphingobium represents an untapped resource for the field of biotechnology. Abundance of integrated phage and resultant acquisition of genes that confer stability in their habitat are signs of well-developed phage gene acquisition machinery in Novosphingobium.
MATERIALS AND METHODS
Gene prediction and annotation.
Novosphingobium genomes, including both draft and complete genomes, were retrieved from the NCBI database (Table 1). One strain, Novosphingobium lindaniclasticum LE124, was sequenced by our laboratory using an Illumina genome analyzer and 454 GS FLX titanium platform, and reads were assembled into 156 contigs (60). For all of the Novosphingobium strains, genome annotations were carried out using RAST version 2.0 (61) and gene caller Glimmer-3 (62). Orthologs were predicted using the sequence clustering algorithm, COGtriangles (63) available in GET_HOMOLOGUES software package (64) with both identity and query coverage of ≥75%, using amino acid sequences. Further, the presence of essential genes in the orthologs was identified using the Database of Essential Genes (DEG) version 13.3 (22). The genome completeness was estimated by analyzing the presence of 107 essential copy genes using the Comprehensive Microbial Resource as a database, where 107 hidden Markov models (HMMs) of essential copy genes were analyzed in all of the Novosphingobium strains (65).
Pangenomic and core genomic trend analysis.
For each genome, amino acid sequences were retrieved from RAST version 2.0 (61) and were used for pangenome and core genome trend analysis using the bidirectional best-hits (BDBH) clustering algorithm (64) at default parameters. Thereafter, the number of genes was plotted against the number of genomes added in the analysis, with Tettelin fitted curve (82).
Phylogenetic analysis.
In order to obtain congruency in the phylogeny of Novosphingobium strains, three different methods were used. In the first method, phylogenetic clustering was performed based on protein sequences of 400 marker genes of Novosphingobium strains (66). The maximum likelihood methodology was used for the construction of the phylogenetic tree, using S. indicum B90A as an outgroup. In order to further demarcate the phylogeny of Novosphingobium strains, two other methods based on pairwise average nucleotide identity (ANI) (67) were used; the first method involves pairwise ANI comparison between 220 orthologous genes, and the second method employed whole-genome sequences to account for both core and accessory genome content. Two-way matrices were prepared, and dendrograms were constructed by the Pearson correlation method and hierarchical clustering using MeV (68).
Habitat-specific genes and their metabolic pathways.
In order to identify the habitat-specific traits of the genus Novosphingobium, we divided the genomes into four different habitats, rhizosphere, contaminated soil, marine water, and freshwater (Table 1). Strains belonging to these habitats were included for further analysis. The strains isolated from the rhizosphere were Novosphingobium sp. AP12, Novosphingobium sp. P6W, and N. rosa NBRC 15208. The strains isolated from contaminated soil were N. barchamii LL02, N. lindaniclasticum LE124, N. naphthalenivorans NBRC102051, Novosphingobium sp. KN65.2, and Novosphingobium sp. ST904. The strains isolated from freshwater were Novosphingobium sp. AAP1, Novosphingobium sp. AAP83, Novosphingobium sp. AAP93, N. fuchskuhlense FNE08-7, N. aromaticivorans DSM12444, and N. acidiphillum DSM19966. The strains isolated from marine water were Novosphingobium sp. MBES04, N. malaysiense Musc273, N. subterraneum DSM12447, N. pentaromaticivorans US6-1, and Novosphingobium sp. PP1Y. Initially, the core genome content of each habitat was predicted by clustering the genomes with the COGtriangles algorithm (as described above). Then, the core genome of each habitat was compared to identify the cloud content (i.e., genes that were present in ≤2 habitats). Further, habitat-specific genes were retrieved manually, mapped for metabolic pathways using KAAS (23), and visualized using iPATH version 2 (24).
Identification of habitat-specific proteins and their protein-protein interactions.
To identify habitat-specific proteins (HSPs), the trans-membrane beta-barrel proteins (TMBbps) (28) were predicted based on the BOMP (Beta-barrel Outer Membrane protein Predictor) program (69). Then, protein sequences of all strains were subjected to TMBbp prediction, and potential proteins were selected for further analysis. All TMBbp sequences of each habitat group were compared using BLASTp, so that the similar proteins could be used for hub identification (70). The TMBbp sequence comparison identified similar sequences present in all of the strains from these four habitats. The topmost sequence is considered a habitat-specific protein (HSP) and subjected to validation using phylogenetic analysis. In order to construct the protein-protein interactions (PPIs), HSP sequences of Novosphingobium strains were searched against the STRING Database (v10) (71). Strains from freshwater and marine water habitats were searched against Novosphingobium aromaticivorans and Novosphingobium sp. strain PPIY, respectively, while the soil and rhizosphere strains sequences were queried against Novosphingobium nitrogenifigens. The STRING v10 database consisted of known and predicted PPIs, which included both direct (physical) and indirect (functional) associations. The associations were integrated with different sources such as genomic context, high-throughput experimental data, database and literature mining, and analysis of coexpressed genes. This allowed an agile exploration of the interactome network and included certain calculated parameters that weighed the reliability of a given interaction (i.e., the “edges” of the interactome network) between two proteins and also qualified the functional environment around any given protein and their interacting partners (i.e., the “nodes” of the interactome network) (72). The PPI networks were visualized using Cytoscape version 3.0.1 (73). The hubs are proteins having a high degree of interactions, randomly placed in the network, and have important functional roles. In the current study, the hubs were identified using network analyzer and Perl programming version 5.18.2.2.
Statistical analysis of the network.
The statistical and functional significance of the network was measured using various statistical parameters, namely, probability of degree distribution, average clustering coefficient, and average neighborhood connectivity (74). The degree of probability distribution, P(k), of a network defined by P(k) = nk/N, which is the ratio of the number of nodes having a k degree in the network (nk) to the size of the network (N), was used to capture the network structure, identification of hubs, and modular organization of the network. The network we constructed obeyed the power law, P(k) ∼ k−γ, indicating the scale-free nature of the network, where γ is an order parameter that identified the different topological structure of a scale-free network. The clustering coefficient C(k), which is defined by
| (4) |
is the ratio of the number of edges E of the node having a k degree with neighbors to the total possible number of such edges,
| (5) |
which is a measure of the topological structure of the network (75). The average clustering coefficient C(k) identifies overall organization of formation of clusters in the network. Similar to P(k), C(k) may depend on network size and characterizes various properties of the network: (i) for scale-free and random networks where C(k) is independent of k, C(k) ∼ constant, and (ii) for hierarchical networks where C(k) follows power law scaling behavior, C(k) ∼ kβ with β ∼ 1. The neighborhood connectivity of a node is the number of neighbors connected to it and characterizes the correlation pattern of connectivity of interacting nodes in the network. This connectivity correlation would be measured by defining a conditional probability
| (6) |
which is the probability of making a link from a node having degree kn to another node of degree k′n (76). Then, the average neighborhood connectivity of nodes with connectivity kn is given by
| (7) |
(76) following a power law scaling behavior with α < 1 for most of the real networks (31, 77). If Cn(kn) is an increasing function of kn (for negative values of α), then the topology of the network shows assortive mixing (78) where nodes with a high number of edges per node (high-degree nodes) have affinity to connect to other high-degree nodes in the network. However, from equation 3 with positive values for α is the signature of the network having hierarchical structure, where low-degree nodes tend to connect high-degree hubs (78) and few high-degree hubs present in the network try to control the low-degree nodes.
Phage and genomic island prediction.
Genomes were searched for phage content using the online server PHAST (79). The phage content was then analyzed for the presence of phage-related, hypothetical, and bacterial genes (Table 2). Further, genomic islands (GIs) were predicted using IslandViewer (80).
ACKNOWLEDGMENTS
This work was supported by grants from the Department of Biotechnology (DBT), R.K., S.H., K.P., A.B., and U.S. gratefully acknowledge the National Bureau of Agriculturally Important Microorganisms (NBAIM), Science and Engineering Research Board (SERB), N-PDF (PDF/2015/000062), (PDF/2015, 000319), University Grant Commission (UGC) for the Dr. D. S. Kothari Postdoctoral Fellowship and UGC for providing fellowships, respectively.
We acknowledge Phoebe Oldach for helpful suggestions and editing the article.
REFERENCES
- 1.Takeuchi M, Hamana K, Hiraishi A. 2001. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int J Syst Evol Microbiol 51:1405–1417. doi: 10.1099/00207713-51-4-1405. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen TPO, Mot RD, Speingael D. 2015. Draft genome sequence of the carbofuran-mineralizing Novosphingobium sp. strain KN65.2. Genome Announc 3:e00764-15. doi: 10.1128/genomeA.00764-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saxena A, Anand S, Dua A, Sangwan N, Khan F, Lal R. 2013. Novosphingobium lindaniclasticum sp. nov., a hexachlorocyclohexane (HCH)-degrading bacterium isolated from an HCH dumpsite. Int J Syst Evol Microbiol 63:2160–2167. doi: 10.1099/ijs.0.045443-0. [DOI] [PubMed] [Google Scholar]
- 4.Pearce SL, Oakeshott JG, Pandey G. 2015. Insights into ongoing evolution of the hexachlorocyclohexane catabolic pathway from comparative genomics of ten Sphingomonadaceae strains. G3 5:1081–1094. doi: 10.1534/g3.114.015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohta Y, Nishi S, Hasegawa R, Hatada Y. 2015. Combination of six enzymes of a marine Novosphingobium converts the stereoisomers of β-O-4 lignin model dimers into the respective monomers. Sci Rep 5:15105. doi: 10.1038/srep15105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Argenio V, Petrillo M, Cantiello P, Naso B, Cozzuto L, Notomista E, Paolella G, Di Donato A, Salvatore F. 2011. De novo sequencing and assembly of the whole genome of Novosphingobium sp. strain PP1Y. J Bacteriol 193:4296. doi: 10.1128/JB.05349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan QX, Hong Q, Han P, Dong XJ, Shen YJ, Li SP. 2007. Isolation and characterization of a carbofuran-degrading strain Novosphingobium sp. FND-3. FEMS Microbiol Lett 271:207–213. doi: 10.1111/j.1574-6968.2007.00718.x. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki S, Hiraishi A. 2007. Novosphingobium naphthalenivorans sp. nov., a naphthalene-degrading bacterium isolated from polychlorinated-dioxin-contaminated environments. J Gen Appl Microbiol 53:221–228. doi: 10.2323/jgam.53.221. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto T, Onda K, Morita T, Luxmy BS, Tada K, Miya A, Murakami T. 2010. Contribution of the estrogen-degrading bacterium Novosphingobium sp. strain JEM-1 to estrogen removal in wastewater treatment. J Environ Eng 136:890–896. doi: 10.1061/(ASCE)EE.1943-7870.0000218. [DOI] [Google Scholar]
- 10.Luo YR, Kang SG, Kim SJ, Kim MR, Li N, Lee JH, Kwon KK. 2012. Genome sequence of benzo(a)pyrene-degrading bacterium Novosphingobium pentaromativorans US6-1. J Bacteriol 194:907. doi: 10.1128/JB.06476-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eguchi M, Ostrowski M, Fegatella F, Bowman J, Nichols D, Nishino T, Cavicchioli R. 2001. Sphingomonas alaskensis strain AFO1, an abundant oligotrophic ultramicrobacterium from the North Pacific. Appl Environ Microbiol 67:4945–4954. doi: 10.1128/AEM.67.11.4945-4954.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aylward FO, McDonald BR, Adams SM, Valenzuela A, Schmidt RA, Goodwin LA, Woyke TA, Currie CR, Suen G, Poulsen M. 2013. Comparison of 26 sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl Environ Microbiol 79:3724–3733. doi: 10.1128/AEM.00518-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niharika N, Moskalikova H, Kaur J, Sedlackova M, Hampl A, Damborsky J, Prokop Z, Lal R. 2013. Novosphingobium barchaimii sp. nov., isolated from a hexachlorocyclohexane (HCH) contaminated soil. Int J Syst Evol Microbiol 63:667–672. doi: 10.1099/ijs.0.039826-0. [DOI] [PubMed] [Google Scholar]
- 14.Gan HM, Buckley L, Szegedi E, Hudson AO, Savka MA. 2009. Identification of an rsh gene from a Novosphingobium sp. necessary for quorum-sensing signal accumulation. J Bacteriol 191:2551–2560. doi: 10.1128/JB.01692-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplan MM. 2004. Novosphingobium aromaticivorans: a potential initiator of primary biliary cirrhosis. Am J Gastroenterol 99:2147–2149. doi: 10.1111/j.1572-0241.2004.41121.x. [DOI] [PubMed] [Google Scholar]
- 16.Rutebemberwa A, Stevens M, Perez M, Smith L, Sanders L, Tuder R, Harris JK. 2014. Novosphingobium spp. in chronic obstructive pulmonary disease in humans and subacute lung inflammation in mice. Ann Am Thorac Soc 11:S76–S77. doi: 10.1513/AnnalsATS.201306-207MG. [DOI] [Google Scholar]
- 17.Du J, Singh H, Yi TH. 2017. Biosynthesis of silver nanoparticles by Novosphingobium sp. THG-C3 and their antimicrobial potential. Artif Cells Nanomed Biotechnol 45:211–217. doi: 10.1080/21691401.2016.1178135. [DOI] [PubMed] [Google Scholar]
- 18.Lagos ML, Maruyama F, Nannipieri P, Mora ML, Ogram A, Jorquera MA. 2015. Current overview on the study of bacteria in the rhizosphere by modern molecular techniques: a mini–review. J Soil Sci Plant Nutr 15:504–523. [Google Scholar]
- 19.Gan HM, Hudson AO, Rahman AYB, Chan KG, Savka MA. 2013. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics 14:431. doi: 10.1186/1471-2164-14-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O. 2014. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci U S A. 111:E4096–E4102. doi: 10.1073/pnas.1321152111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sangwan N, Verma H, Kumar R, Negi V, Lax S, Khurana P, Khurana JP, Gilbert JA, Lal R. 2014. Reconstructing an ancestral genotype of two hexachlorocyclohexane degrading Sphingobium species using metagenomic sequence data. ISME J 8:398–408. doi: 10.1038/ismej.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo H, Lin Y, Gao F, Zhang CT, Zhang R. 2014. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res 42:D574–D580. doi: 10.1093/nar/gkt1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada T, Letunic I, Okuda S, Kanehisa M, Bork P. 2011. iPath2.0: interactive pathway explorer. Nucleic Acids Res 39:W412–W415. doi: 10.1093/nar/gkr313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tholl D, Boland W, Hansel A, Loreto F, Röse USR, Schnitzler JP. 2006. Practical approaches to plant volatile analysis. Plant J 45:540–560. doi: 10.1111/j.1365-313X.2005.02612.x. [DOI] [PubMed] [Google Scholar]
- 26.Kar G, Gursoy A, Keskin O. 2009. Human cancer protein-protein interaction network: a structural perspective. PLoS Comput Biol 5:e1000601. doi: 10.1371/journal.pcbi.1000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallik S, Kundu S. 2013. A comparison of structural and evolutionary attributes of Escherichia coli and Thermus thermophilus small ribosomal subunits: signatures of thermal adaptation. PLoS One 8:e69898. doi: 10.1371/journal.pone.0069898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wimley WC. 2003. The versatile beta-barrel membrane protein. Curr Opin Struct Biol 13:404–411. doi: 10.1016/S0959-440X(03)00099-X. [DOI] [PubMed] [Google Scholar]
- 29.Barabási AL, Oltvai ZN. 2004. Network biology: understanding the cell’s functional organization. Nat Rev Genet 5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 30.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL. 2002. Hierarchical organization of modularity in metabolic networks. Science 297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 31.Pastor-Satorras R, Vespignani A. 2001. Epidemic spreading in scale-free networks. Phys Rev Lett 86:3200–3203. doi: 10.1103/PhysRevLett.86.3200. [DOI] [PubMed] [Google Scholar]
- 32.Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz GE. 2000. Beta-barrel membrane proteins. Curr Opin Struct Biol 10:443–447. [DOI] [PubMed] [Google Scholar]
- 34.Higgins CF. 1992. ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 35.Leyh TS, Taylor JC, Markham GD. 1988. The sulfate activation locus of Escherichia coli K12: cloning, genetic and enzymatic characterization. J Biol Chem 263:2409–2416. [PubMed] [Google Scholar]
- 36.Stanko-Golden KM, Fitzgerald JW. 1991. Sulfur transformations and pool sizes in tropical forest soils. Soil Biol Biochem 23:1053–1058. doi: 10.1016/0038-0717(91)90043-J. [DOI] [Google Scholar]
- 37.Vairavamurthy A, Zhou W, Eglinton T, Manowitz B. 1994. Sulfonates: a new class of organic sulfur compounds in marine sediments. Geochim Cosmochim Acta 58:4681–4687. doi: 10.1016/0016-7037(94)90200-3. [DOI] [Google Scholar]
- 38.King JE, Quinn JP. 1997. The utilization of organosulphonates by soil and freshwater bacteria. Lett Appl Microbiol 24:474–478. doi: 10.1046/j.1472-765X.1997.00062.x. [DOI] [Google Scholar]
- 39.Oren A, Heldal M, Norland S, Galinski EA. 2002. Intracellular ion and organic solute concentrations of the extremely halophilic bacterium Salinibacter ruber. Extremophiles 6:491–498. doi: 10.1007/s00792-002-0286-3. [DOI] [PubMed] [Google Scholar]
- 40.Roesser M, Müller V. 2001. Osmoadaptation in bacteria and archaea: common principles and differences. Environ Microbiol 3:743–754. doi: 10.1046/j.1462-2920.2001.00252.x. [DOI] [PubMed] [Google Scholar]
- 41.Galinski EA, Pfeiffer HP, Trüper HG. 1985. 1,4,5,6-Tetrahydro-2-methyl-4-pyrimidinecarboxylic acid. A novel cyclic amino acid from halophilic phototrophic bacteria of the genus Ectothiorhodospira. Eur J Biochem 149:135–139. doi: 10.1111/j.1432-1033.1985.tb08903.x. [DOI] [PubMed] [Google Scholar]
- 42.Held C, Neuhaus T, Sadowski G. 2010. Compatible solutes: thermodynamic properties and biological impact of ectoines and prolines. Biophys Chem 152:28–39. doi: 10.1016/j.bpc.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 43.Reshetnikov AS, Khmelenina VN, Mustakhimov II, Kalyuzhnaya M, Lidstrom M, Trotsenko YA. 2011. Diversity and phylogeny of the ectoine biosynthesis genes in aerobic, moderately halophilic methylotrophic bacteria. Extremophiles 15:653–663. doi: 10.1007/s00792-011-0396-x. [DOI] [PubMed] [Google Scholar]
- 44.Peters P, Galinski EA, Trüper HG. 1990. The biosynthesis of ectoine. FEMS Microbiol Lett 71:157–162. doi: 10.1111/j.1574-6968.1990.tb03815.x. [DOI] [Google Scholar]
- 45.Inbar L, Lapidot A. 1988. The structure and biosynthesis of new tetrahydropyrimidine derivatives in actinomycin D producer Streptomyces parvulus. Use of 13C- and 15N-labeled l-glutamate and 13C and 15N NMR spectroscopy. J Biol Chem 263:16014–16022. [PubMed] [Google Scholar]
- 46.Cunliffe M, Kertesz MA. 2006. Autecological properties of soil sphingomonads involved in the degradation of polycyclic aromatic hydrocarbons. Appl Microbiol Biotechnol 72:1083–1089. doi: 10.1007/s00253-006-0374-x. [DOI] [PubMed] [Google Scholar]
- 47.Verma H, Kumar R, Oldach P, Sangwan N, Khurana JP, Gilbert JA, Lal R. 2014. Comparative genomic analysis of nine Sphingobium strains: insights into their evolution and hexachlorocyclohexane (HCH) degradation pathways. BMC Genomics 15:1014. doi: 10.1186/1471-2164-15-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tabata M, Ohtsubo Y, Ohhata S, Tsuda M, Nagata Y. 2013. Complete genome sequence of the gamma-hexachlorohexane-degrading bacterium Sphingomonas sp. strain MM-1. Genome Announc 1:e00247-13. doi: 10.1128/genomeA.00247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo JS, Keum YS, Li QX. 2009. Bacterial degradation of aromatic compounds. Int J Environ Res Public Health 6:278–309. doi: 10.3390/ijerph6010278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harayama S, Kok M, Neidle EL. 1992. Functional and evolutionary relationships among diverse oxygenases. Annu Rev Microbiol 46:565–601. doi: 10.1146/annurev.mi.46.100192.003025. [DOI] [PubMed] [Google Scholar]
- 51.Casjens S. 2003. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol 49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- 52.Kimura M, Jia Z, Nakayama N, Asakawa S. 2008. Ecology of viruses in soils: past, present and future perspectives. Soil Sci Plant Nutr 54:1–32. doi: 10.1111/j.1747-0765.2007.00197.x. [DOI] [Google Scholar]
- 53.Jiang SC, Paul JH. 1994. Seasonal and die1 abundance of viruses and occurrence of lysogeny/bactenocinogeny in the marine environment. Mar Ecol Prog Ser 104:163–172. doi: 10.3354/meps104163. [DOI] [Google Scholar]
- 54.Jiang SC, Paul JH. 1998. Significance of lysogeny in the marine environment: studies with isolates and a model of lysogenic phage production. Microb Ecol 35:235–243. doi: 10.1007/s002489900079. [DOI] [PubMed] [Google Scholar]
- 55.Khmelenina VN, Kalyuzhnaya MG, Sakharovsky VG, Suzina NE, Trotsenko YA, Gottschalk G. 1999. Osmoadaptation in halophilic and alkaliphilic methanotrophs. Arch Microbiol 172:321–329. doi: 10.1007/s002030050786. [DOI] [PubMed] [Google Scholar]
- 56.Makemson JC, Hastings JW. 1979. Glutamate functions in osmoregulation in a marine bacterium. Appl Environ Microbiol 38:178–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Way SM, Hosking ER, Braun TF, Manson MD. 2000. Mot protein assembly into the bacterial flagellum: a model based on mutational analysis of the motB gene. J Mol Biol 297:7–24. doi: 10.1006/jmbi.2000.3548. [DOI] [PubMed] [Google Scholar]
- 58.Smith SG, Mahon V, Lambert MA, Fagan RP. 2007. A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol Lett 273:1–11. doi: 10.1111/j.1574-6968.2007.00778.x. [DOI] [PubMed] [Google Scholar]
- 59.Liu T, Li Y, Ye Q, Li H, Liang Y, She Q, Peng N. 2015. Transcriptional regulator-mediated activation of adaptation genes triggers CRISPR de novo spacer acquisition. Nucleic Acids Res 43:1044–1055. doi: 10.1093/nar/gku1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saxena A, Nayyar N, Sangwan N, Kumari R, Khurana JP, Lal R. 2013. Genome sequence of Novosphingobium lindaniclasticum LE124T, isolated from a hexachlorocyclohexane dumpsite. Genome Announc 1:e00715-13. doi: 10.1128/genomeA.00715-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kristensen DM, Kannan L, Coleman MK, Wolf YI, Sorokin A, Koonin EV, Mushegian A. 2010. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26:1481–1487. doi: 10.1093/bioinformatics/btq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Contreras-Moreira B, Vinuesa P. 2013. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL, Lasken RS, Nealson K, Friedman R, Venter JC. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. doi: 10.1038/ismej.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segata N, Börnigen D, Morgan XC, Huttenhower C. 2013. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 4:2304. doi: 10.1038/ncomms3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378. [DOI] [PubMed] [Google Scholar]
- 69.Berven FS, Flikka K, Jensen HB, Eidhammer I. 2004. BOMP: a program to predict integral β-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res 32:W394–W399. doi: 10.1093/nar/gkh351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 71.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hernandez-Toro J, Prieto C, De las Rivas J. 2007. APID2NET: unified interactome graphic analyzer. Bioinformatics 23:2495–2497. [DOI] [PubMed] [Google Scholar]
- 73.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Albert R, Barabási AL. 2002. Statistical mechanics of complex networks. Rev Mod Phys 74:47–97. doi: 10.1103/RevModPhys.74.47. [DOI] [Google Scholar]
- 75.Watts DJ, Strogatz SH. 1998. Collective dynamics of small-world networks. Nature 393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 76.Girvan M, Newman ME. 2002. Community structure in social and biological networks. Proc Natl Acad Sci U S A 99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maslov S, Sneppen K. 2002. Specificity and stability in topology of protein networks. Science 296:910–913. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]
- 78.Almaas E. 2007. Biological impacts and context of network theory. J Exp Biol 210:1548–1558. doi: 10.1242/jeb.003731. [DOI] [PubMed] [Google Scholar]
- 79.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langille MGI, Brinkman FSL. 2009. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25:664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choi DH, Kwon YM, Kwon KK, Kim S-J. 2015. Complete genome sequence of Novosphingobium pentaromativorans US6-1T. Stand Genomic Sci 10:107. doi: 10.1186/s40793-015-0102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc Natl Acad Sci U S A 102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mapping of habitat-specific metabolic pathways. The habitat-specific metabolic pathways were mapped using iPath v2. In the map, different habitats were represented by different colors: green for rhizosphere, yellow for contaminated soil, blue for marine water, and red for freshwater. Download FIG S1, TIF file, 8.3 MB (8.5MB, tif) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic analysis based on habitat-specific beta-barrel protein using the neighbor-joining method and 1,000 bootstrap replicates. The evolutionary distances were computed by the Poisson correction method. Download FIG S2, EPS file, 1.5 MB (1.6MB, eps) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
List of regulatory hub proteins in four different habitats of genus Novosphingobium. Download TABLE S1, DOCX file, 0.02 MB (16.8KB, docx) .
Copyright © 2017 Kumar et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.