

Abstract
Oxidized low-density lipoprotein (ox-LDL) is a major and critical mediator of atherosclerosis, and the underlying mechanism is thought to involve the ox-LDL-induced dysfunction of endothelial cells (ECs). MicroRNAs (miRNAs), which are a group of small non-coding RNA molecules that post-transcriptionally regulate the expression of target genes, have been associated with diverse cellular functions and the pathogenesis of various diseases, including atherosclerosis. miRNA-98 (miR-98) has been demonstrated to be involved in the regulation of cellular apoptosis; however, the role of miR-98 in ox-LDL-induced dysfunction of ECs and atherosclerosis has yet to be elucidated. Therefore, the present study aimed to investigate the role of miR-98 in ox-LDL-induced dysfunction of ECs and the underlying mechanism. It was demonstrated that miR-98 expression was markedly downregulated in ox-LDL-treated human umbilical vein ECs (HUVECs) and that miR-98 promoted the proliferation and alleviated apoptosis of HUVECs exposed to ox-LDL. In addition, the results demonstrated that lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) was a direct target of miR-98 in HUVECs, as indicated by a luciferase assay. The results of the present study suggested that miR-98 may inhibit the uptake of toxic ox-LDL, maintain HUVEC proliferation and protect HUVECs against apoptosis via the suppression of LOX-1.
Keywords: microRNA-98, lectin-like oxidized low-density lipoprotein receptor 1, human umbilical vein endothelial cells, proliferation, apoptosis
Introduction
The health and homeostasis of blood vessels is dependent on the integrity of the endothelial lining (1). Endothelial cells (ECs) have an important role in vascular homeostasis and have been implicated in various diseases; their dysfunction marks an early stage of atherosclerosis (2). Cardiovascular risk factors, including hyperlipidemia, insulin resistance and vascular inflammation, may induce endothelial injury and apoptosis during atherosclerosis (3). Endothelial injury and dysfunction may be repaired by adjacent viable ECs in a process involving cellular proliferation, migration and apoptosis (4,5). An effective strategy for the prevention or treatment of atherosclerosis is the maintenance of endothelial barrier integrity, which is achieved by preserving the proliferative capacity and inhibiting the apoptosis of ECs (6). Oxidized low-density lipoprotein (ox-LDL) is a critical marker of atherosclerosis (7). Ox-LDL induces vascular EC activation and dysfunction, which induces the pro-adhesive properties of ECs and promotes the recruitment of monocytes, leading to subsequent inflammation and the accumulation of lipid-rich macrophages and proinflammatory lymphocytes within the intima of artery walls (7). This ultimately leads to the formation of atherosclerotic plaques (7). Ox-LDL induces pro-inflammatory responses, pro-oxidative conditions and EC apoptosis (8,9). Since ox-LDL is a major risk factor for the onset and development of atherosclerosis (7), the present study used ox-LDL-treated human umbilical vein ECs (HUVECs) as an experimental model to identify the specific microRNAs (miRNAs) associated with endothelial dysfunction in atherosclerosis.
miRNAs are a family of highly conserved, small non-coding RNAs that post-transcriptionally repress the expression of their target genes by inducing the degradation of their target mRNA or inhibiting their translation (10). miRNAs are involved in diverse cellular functions, including differentiation, growth, proliferation and apoptosis (10). Some specific miRNAs have previously been implicated in the pathogenesis of atherosclerosis by regulating various cellular functions and the properties of vascular cells (11). In addition, miRNAs have been demonstrated to be differentially expressed at the various stages of atherosclerosis (12), and abnormally expressed miRNAs have been closely associated with the pathogenesis of atherosclerosis (12). For example, a previous study demonstrated that miR-21 was involved in atherosclerosis by regulating the function of arterial smooth muscle cells (13). Furthermore miR-133a was shown to regulate the functions of arterial smooth muscle cells by targeting RhoA, and was involved in the pathogenesis of arterial sclerosis occlusion, which is a type of atherosclerosis affecting the lower extremities (14). In our previous study, a miRNA microarray analysis demonstrated that miR-98 was downregulated in atherosclerotic tissue (13), thus suggesting that miR-98 may be involved in the pathogenesis of atherosclerosis. Furthermore, miR-98 was identified as a modulator of proliferation and apoptosis in numerous types of cancer cells (15). miR-98 was demonstrated to suppress the P53 tumor suppressor gene and has been suggested to be involved in the potential therapeutic effect of (−)-epigallocatechin-3-gallate for the treatment of various types of non-small-cell lung cancers (16). Based on these results, the authors of the present study hypothesized that miR-98 may be a regulator of endothelial function.
Lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), which has a C-type lectin-like extracellular domain and a short cytoplasmic tail, is a novel type II membrane protein receptor for ox-LDL (17). In addition, LOX-1 is a key molecule involved in the pathogenesis of atherosclerosis (17), and is a critical receptor that induces the uptake of toxic ox-LDL by ECs, which subsequently results in a series of vascular pathological processes (18). Under certain pathological conditions, high LOX-1 expression may be harmful to vascular cells, in particular ECs, inducing vascular cell injury by decreasing cellular viability, reducing cell growth and proliferation and promoting cell death (19,20). Therefore, LOX-1 may be considered a potential target for the regulation of EC turnover. The present study aimed to investigate the role of miR-98 in ox-LDL-induced dysfunction of ECs and the underlying mechanism.
Materials and methods
Reagents
miR-98 mimics and inhibitor, and their control oligonucleotides (oligos), were purchased from Guangzhou RiboBio, Co., Ltd. (Guangzhou, China). Lipofectamine® RNAiMAX transfection reagent was purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Ox-LDL and 1,1′-dioctadecyl-3,3,3′3′-tetra-methylindocyanide perchlorate (Dil)-labeled ox-LDL (Dil-ox-LDL) were obtained from Yiyuan Biological Technology, Co., Ltd. (Guangzhou, China). The Cell Counting kit-8 (CCK-8) Assay kit and the Annexin V/Propidium Iodide (PI) Flow Cytometry Detection kit were purchased from Dojindo Molecular Technologies, Inc. (Shanghai, China). The EdU Assay and Hoechst 33342 Assay kits were purchased from Guangzhou RiboBio, Co., Ltd. Rabbit anti-human LOX-1 monoclonal antibody (cat. no. ab175922) was purchased from Abcam (Cambridge, UK). Rabbit anti-human B-cell lymphoma-2 (Bcl-2) monoclonal antibody (cat. no. 2870S), rabbit anti-Bcl-2-associated X protein (Bax) polyclonal antibody (cat. no. 2772S), rabbit anti-caspase-3 polyclonal antibody (cat. no. 9662S) and rabbit anti-β-actin monoclonal antibody (cat. no. 8457S) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Primers for LOX-1, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), miR-98 and U6 were obtained from Takara Biotechnology, Co., Ltd. (Dalian, China).
Cell culture
HUVECs were purchased from Fuxiang Biotechnology, Co., Ltd. (Shanghai, China) and cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) and 100 µg/ml penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.).
RNA interference
miR-98 mimics were used to imitate miR-98 and an miR-98 inhibitor to deplete endogenous miR-98. HUVECs were seeded into wells 24 h prior to transfection. The cells were transfected with the miR-98 mimics, miR-98 inhibitor or their control oligos (60 nmol/l) using Lipofectamine® RNAiMAX transfection reagent, according to the manufacturer's protocol. The transfection medium was replaced with fresh DMEM at 8 h following transfection.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
HUVECs (2×105 cells/well) were seeded into 6-well plates 12 h prior to transfection. Ox-LDL was added to the cells 24 h following transfection and the cells were incubated for an additional 48 h. After washing the cells twice with phosphate-buffered saline (PBS), total RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Total RNA was reverse-transcribed into cDNA using the PrimeScript RT Reagent kit (Takara Biotechnology, Co., Ltd.). cDNA was amplified by qPCR using SYBR Premix Ex Taq II (Takara Biotechnology, Co., Ltd.) on a CFX96 thermocycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA), according to the manufacturer's protocol. For miR-98 amplification, the PCR conditions were as follows: One cycle at 95°C for 10 sec; 39 cycles at 95°C for 5 sec and 60°C for 20 sec; one cycle at 95°C for 15 sec and 65°C for 10 sec; and a final increase to 95°C. For LOX-1 mRNA amplification, the PCR conditions were as follows: One cycle at 95°C for 30 sec; 39 cycles at 95°C for 3 sec and 60°C for 30 sec, one cycle at 95°C for 15 sec and 60°C for 10 sec; and a final increase to 95°C. The relative LOX-1 and miR-98 expression levels were calculated using the 2−ΔΔCq method (21), following normalization to the GAPDH and U6 small nuclear RNA internal controls, respectively. The sequences of the primers were as follows: LOX-1 forward, 5′-GTCCATAGGGCACTTCCAGAAA-3′ and reverse, 5′-TGCTCGGAAGCTGAATGAGAA-3′; GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′; miR-98, 5′-CCGAGGTAGTAAGTTGTATTGTT-3′; and U6, 5′-ACGCAAATTCGTGAAGCGTT-3′ (Takara Biotechnology, Co., Ltd.).
Western blot analysis
HUVECs (2×105 cells/well) were seeded into 6-well plates 12 h prior to transfection. Ox-LDL was added to the cells 24 h following transfection and the cells were incubated for an additional 48 h. The cells were washed twice with PBS and lysed in radioimmunoprecipitation assay lysis buffer (Cell Signaling Technology, Inc.), supplemented with protease inhibitors (Roche Diagnostics, Basel, Switzerland). The total protein concentrations were determined using a BCA Protein Assay kit (Thermo Fisher Scientific, Inc.). The protein lysates were separated on an 8–12% gradient gel by SDS-PAGE and transferred to a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). The membrane was blocked with 5% non-fat dry milk in PBS buffer for 1 h at room temperature, then incubated with rabbit anti-human LOX-1, Bcl-2, Bax, β-actin, and caspase-3 primary antibodies (dilutions, 1:1,000) overnight at 4°C. Subsequently, the membrane was incubated with the secondary antibody for 1 h at room temperature and the immunoblotting signal was developed using an Enhanced Chemiluminescence Western Blotting Analysis kit (Invitrogen; Thermo Fisher Scientific, Inc.) and exposed on X-ray films (Kodak, Rochester, NY, USA). The relative protein expression levels were normalized to β-actin and analyzed using ImageJ software (https://imagej.nih.gov/ij/).
Cell proliferation
HUVECs (1×104 cells/well) were seeded into 96-well plates in triplicate 12 h prior to transfection. Ox-LDL (80 µg/ml) was added to the cells 24 h following transfection and the cells were incubated for an additional 48 h. For the CCK-8 assay, 10 µl CCK-8 solution was added to each well and incubated for 1 h, after which the absorbance values were measured at 450 nm using a spectrophotometer. For the EdU assay, the cells were incubated with 50 µM EdU for 2 h and then fixed using 4% paraformaldehyde for 15 min. The cells were subsequently treated with 100 µl Apollo reaction cocktail for 30 min, followed by 50 µl 1% Hoechst 33342 for 30 min for nuclei staining. Finally, an inverted fluorescence microscope was used for visualization. A total of 10 random microscope fields in each well were imaged, and the number of Apollo-positive nuclei was divided by the total number of nuclei stained with Hoechst 33342. The fluorescence images were analyzed using Image-Pro® Plus software 6.0 (Media Cybernetics, Rockville, MD, USA).
Fluorescence microscopy analysis of Dil-ox-LDL uptake and cellular apoptosis
HUVECs (1×104 cells/well) were seeded into 96-well plates in triplicate 12 h prior to transfection. Ox-LDL (80 µg/ml) was added to the cells 24 h following transfection and the cells were incubated for an additional 48 h. To visualize and assess ox-LDL uptake, the HUVECs were incubated with 20 µg/ml Dil-ox-LDL for 3 h at 37°C. Following incubation, the cells were gently washed three times with PBS and immediately imaged using an inverted fluorescence microscope. To visualize the morphological changes in the nuclear chromatin of apoptotic cells, the cells were washed three times with PBS and fixed using 4% paraformaldehyde for 15 min, followed by another three washes with PBS. The fixed cells were incubated with 100 µl Hoechst 33342 solution (5 µg/ml dissolved in PBS) for 20 min in the dark. HUVEC nuclear staining was examined using an inverted fluorescence microscope. Viable HUVEC nuclei exhibited a pale blue color with organized structures, whereas the nuclei of apoptotic HUVECs exhibited a bright blue color and were reduced in size.
Flow cytometric analysis of cellular apoptosis
HUVECs (2×105 cells/well) were seeded into 6-well plates in triplicate 12 h prior to transfection. Ox-LDL (80 µg/ml) was added to the cells 24 h following transfection and the cells were incubated for an additional 48 h. The cells were washed twice with PBS, digested with EDTA-free trypsin and washed three times with PBS. Subsequently, the cells were double-stained using the Annexin V/PI Flow Cytometry Detection kit, according to the manufacturer's protocol. The percentages of viable cells (Annexin V- and PI-negative), apoptotic cells (Annexin V-positive and PI-negative) and necrotic cells (Annexin V- and PI-positive) were detected using an EPICS XL-MCL™ Flow Cytometer (Beckman Coulter, Inc., Fullerton, CA, USA). The results were analyzed using Kaluza 1.2 software (Beckman Coulter, Inc.).
Luciferase assay
Using TargetScan 6.2, miR-98 was predicted a potential target region in position 447–453 of LOX-1 mRNA 3′-untranslate dregion (UTR). The 3′-UTR miR-98 binding sites of LOX-1 mRNA were amplified by PCR and cloned into the pLUC luciferase reporter vector (Guangzhou Liang Zi Kang Biotechnology, Co., Ltd., Guangzhou, China) to construct the fluorescent reporter system. A pLUC vector containing the LOX-1 mRNA 3′-UTR with mutated binding sites for miR-98 was also generated. HEK293T cells (Guangzhou Liang Zi Kang Biotechnology, Guangzhou, China) were cultured in 96-well plates in triplicate at 37°C until 70% confluence was reached, after which the cells were co-transfected with the wild-type or mutant LOX-1 3′-UTR vectors and the miR-98 mimics or control oligos (100 nmol/l) using FuGENE® HD (Roche Diagnostics), according to the manufacturer's protocol. After 48 h, the cells were lysed and the luciferase activity was measured using the Dual-Glo™ Luciferase Assay kit (Promega Corporation, Madison, WI, USA), according to the manufacturer's protocol.
Statistical analysis
Data are presented as the mean ± standard deviation of at least three repeated experiments and were analyzed using Student's t-test to compare two conditions. One-way analysis of variance was used for comparing three or more conditions. Statistical analyses were performed using GraphPad Prism software, version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Downregulation of miR-98 expression upregulates the expression of LOX-1 in ox-LDL-treated HUVECs
HUVECs were exposed to ox-LDL at various concentrations (20–80 µg/ml) for 48 h or 40 µg/ml ox-LDL for various durations (0–48 h), and the expression levels of miR-98 and LOX-1 mRNA were determined by RT-qPCR. Incubation of HUVECs with 0–80 µg/ml ox-LDL for 48 h resulted in a dose-dependent downregulation of miR-98 expression (Fig. 1A). In addition, miR-98 expression was gradually downregulated during 40 µg/ml ox-LDL treatment, decreasing by >75% at the 48 h time-point (Fig. 1B). In HUVECs treated with 0–80 µg/ml ox-LDL, a dose-dependent increase in LOX-1 mRNA and protein expression levels was observed (Fig. 1C and D). Incubation of HUVECs with 40 µg/ml ox-LDL for 0–48 h resulted in a time-dependent increase in LOX-1 mRNA and protein expression levels (Figs. 1E and F).
Figure 1.
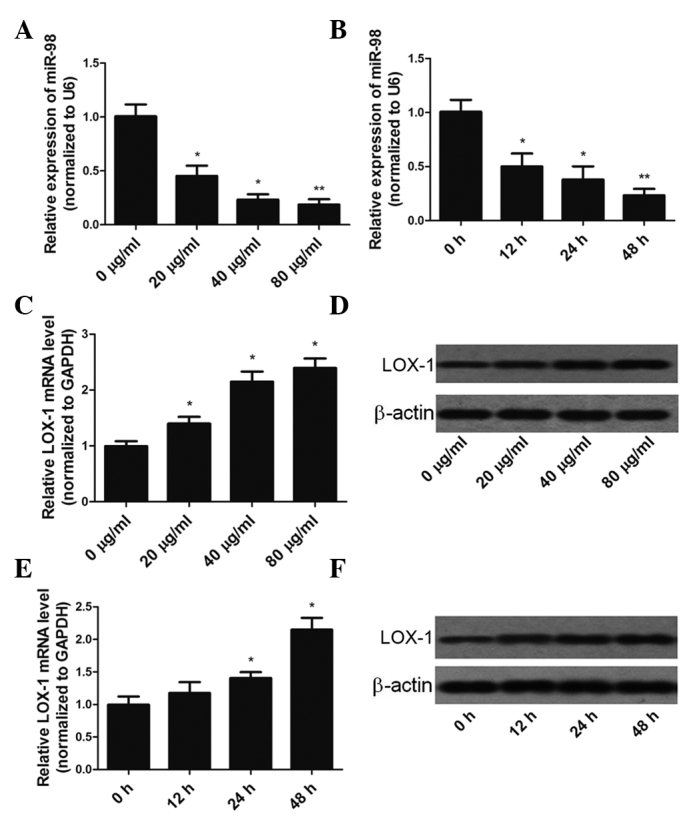
ox-LDL treatment altered the expression of miR-98 and LOX-1 in HUVECs. (A) HUVECs treated with 0–80 µg/ml ox-LDL for 48 h exhibited a dose-dependent downregulation of miR-98 expression levels. (B) miR-98 was downregulated in a time-dependent manner following treatment of HUVECs with 40 µg/ml ox-LDL. (C and D) Ox-LDL treatment of HUVECs increased the mRNA and protein expression levels of LOX-1 in a dose-dependent manner. (E and F) Treatment of HUVECs with 40 µg/ml ox-LDL increased the mRNA and protein expression levels of LOX-1 in a time-dependent manner. The relative levels of miR-98 and LOX-1 mRNA in HUVECs were analyzed by reverse transcription-quantitative polymerase chain reaction. Relative LOX-1 protein levels were analyzed by western blotting. Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. the 0 µg/ml or 0 h groups. ox-LDL, oxidized low-density lipoprotein; miR-98, microRNA-98; LOX-1, lectin-like oxidized low-density lipoprotein receptor 1; HUVECs, human umbilical vein endothelial cells; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
miR-98 regulates ox-LDL uptake by HUVECs
To investigate the effects of miR-98 on the uptake of ox-LDL by HUVECs, HUVECs were pretreated with ox-LDL (80 µg/ml) for 48 h, followed by treatment with Dil-ox-LDL (20 µg/ml) for 3 h at 37°C and visualization. As is shown in Fig. 2, ox-LDL enhanced the uptake of Dil-ox-LDL. Furthermore, the miR-98 mimics blocked ox-LDL uptake, whereas the miR-98 inhibitor increased ox-LDL uptake.
Figure 2.

miR-98 regulates Dil-ox-LDL uptake in HUVECs, as determined by inverted fluorescence microscopy. Ox-LDL enhanced the uptake of Dil-ox-LDL. miR-98 mimics blocked ox-LDL uptake, whereas the miR-98 inhibitor increased ox-LDL uptake. miR-98, microRNA-98; ox-LDL, oxidized low density lipoprotein; Dil-ox-LDL, 1,1′-dioctadecyl-3,3,3′3′-tetra-methylindocyanide perchlorate-labeled ox-LDL; HUVECs, human umbilical vein endothelial cells.
miR-98 regulates proliferation in ox-LDL-treated HUVECs
To elucidate the role of miR-98 in HUVEC proliferation, CCK-8 and EdU assays were performed. HUVECs were transfected with the miR-98 mimics, miR-98 inhibitor or control oligos and were then exposed to 80 µg/ml ox-LDL for 48 h. Exposure of HUVECs to ox-LDL inhibited cell proliferation, which was reversed by transfection with the miR-98 mimics. Conversely, the miR-98 inhibitor suppressed the proliferation of HUVECs (Fig. 3A-C).
Figure 3.

Effects of miR-98 mimics and inhibitor on the proliferation of ox-LDL-treated HUVECs. (A) The miR-98 mimics significantly promoted the proliferation of ox-LDL-treated HUVECs, whereas the miR-98 inhibitor exerted the opposite effect, as determined by cell counting kit-8 assays. (B and C) The miR-98 mimics significantly promoted the proliferation of ox-LDL-treated HUVECs, whereas the miR-98 inhibitor exerted the opposite effect, as determined by EdU assays. The second group of “vehicle” were treated with ox-LDL while the first group of “vehicle” not. *P<0.05 vs. the vehicle and ox-LDL treatment group. miR-98, microRNA-98; ox-LDL, oxidized low-density lipoprotein; HUVECs, human umbilical vein endothelial cells; oligo, oligonucleotide.
miR-98 inhibits apoptosis in ox-LDL-treated HUVECs
To assess the effects of miR-98 on ox-LDL-induced apoptosis of ECs, HUVECs were transfected with miR-98 mimics, miR-98 inhibitor or control oligos and were then incubated with 80 µg/ml ox-LDL for 48 h. Subsequently, the levels of apoptosis were assessed by determining the protein expression levels of LOX-1, Bax, Bcl-2, caspase-3 and activated caspase-3 (p17). As is shown in Fig. 4A, the expression levels of LOX-1, Bax and activated caspase-3 (p17) were significantly increased, and those of Bcl-2 were significantly decreased, in HUVECs treated with 80 µg/ml ox-LDL for 48 h, as compared with the vehicle group. In addition, the effects of ox-LDL on HUVECs were attenuated by miR-98 mimics and aggravated by the inhibitor. Subsequently, the levels of apoptosis were examined using FITC-conjugated Annexin V/PI double-staining and flow cytometry. As is shown in Fig. 4B and C, treatment with ox-LDL induced apoptosis in HUVECs, and this was alleviated by the miR-98 mimics and exacerbated by the miR-98 inhibitor. To examine the morphological changes in nuclear chromatin, an inverted fluorescence microscope was used to visualize Hoechst 33342 staining. The majority of nuclei in the vehicle group exhibited a uniform pale blue color and had an organized structure, whereas ox-LDL treatment resulted in nuclei with bright blue staining, indicating pyknosis. The miR-98 mimics alleviated cell death, whereas the miR-98 inhibitor enhanced apoptosis (Fig. 4D).
Figure 4.

Effects of miR-98 mimics and a miR-98 inhibitor on the apoptosis of ox-LDL-treated HUVECs. (A) The protein expression levels of Bcl-2 were upregulated, and those of LOX-1, Bax and cleaved caspase-3 were downregulated, in HUVECs transfected with miR-98 mimics, as determined by western blotting. (B and C) Ox-LDL-induced apoptosis levels were decreased in HUVECs transfected with miR-98 mimics, whereas they were increased in HUVECs transfected with a miR-98 inhibitor, as shown by an Annexin V/PI dual-staining assay. (D) miR-98 mimics alleviated, whereas a miR-98 inhibitor enhanced, cellular apoptosis and pyknosis in ox-LDL-treated HUVECs, as demonstrated by the Hoechst 33342 staining assay. *P<0.05, vs. the vehicle and ox-LDL treatment group. Oligo, oligonucleotide; miR-98, microRNA-98; ox-LDL, oxidized low-density lipoprotein; HUVECs, human umbilical vein endothelial cells; Bcl-2, B-cell lymphoma-2; LOX-1, lectin-like oxidized low-density lipoprotein receptor 1; Bax, Bcl-2-associated X protein; PI, propidium iodide.
LOX-1 is a direct target of miR-98
LOX-1 was selected as a potential target of miR-98, since its mRNA 3′-UTR was complementary to miR-98 (predicted by TargetScan 6.2) (Fig. 5A). The protein expression levels of LOX-1 were decreased in HUVECs transfected with miR-98 mimics, as compared with cells transfected with control oligos (Fig. 5B). Conversely, the protein expression levels of LOX-1 were increased in HUVECs transfected with miR-98 inhibitor (Fig. 5C). Notably, the effect of the miR-98 mimics on LOX-1 mRNA expression levels was similar to that on LOX-1 protein expression levels (Fig. 5D). These results suggest that miR-98 may bind to the 3′-UTR of LOX-1 mRNA and induce mRNA degradation, resulting in the downregulation of LOX-1 expression at the mRNA and protein levels. In order to determine whether miR-98 directly binds to the 3′-UTR of LOX-1 mRNA a luciferase reporter assay was performed. The miR-98 mimics significantly reduced luciferase activity in the presence of the LOX-1 3′-UTR (wild-type 3′-UTR), as compared with the negative control (Fig. 5E). However, when the 3′-UTR of the LOX-1 mRNA was mutated to disrupt the miR-98 binding site (Fig. 5A), the inhibitory effect of miR-98 on the relative luciferase activity was partially reversed (Fig. 5E). These results suggest that LOX-1 is a direct target of miR-98.
Figure 5.

LOX-1 is a direct target of miR-98. (A) Depiction of the miR-98 seed sequence in the wild-type 3′-UTR of LOX-1 and the mutated site. (B and C) The protein expression levels of LOX-1 were downregulated by the miR-98 mimics (60 nmol/l) and upregulated by the miR-98 inhibitor (60 nmol/l), as assessed by western blotting. (D) The mRNA expression levels of LOX-1 were downregulated by miR-98 mimics and upregulated by the miR-98 inhibitor (60 nmol/l), as assessed by reverse transcription-quantitative polymerase chain reaction. *P<0.05, **P<0.01, vs. the vehicle group. (E) Co-transfection of miR-98 with the wild-type LOX-1 3′-UTR in HEK293T cells led to a marked decrease in luciferase activity, whereas co-transfection of miR-98 with the mutant LOX-1 3′-UTR had no effect on luciferase activity. The luciferase activities were measured using the dual-luciferase reporter assay. **P<0.01, vs. the mimics control group. LOX-1, lectin-like oxidized low-density lipoprotein receptor 1; miR-98, microRNA-98; 3′-UTR, 3′-untranslated region; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; oligo, oligonucleotide.
Discussion
The present study demonstrated that miR-98 expression was essential for the replicative response of HUVECs to ox-LDL injury. In addition, miR-98 was shown to be essential for the protective effect against ox-LDL-induced apoptosis by targeting the ox-LDL membrane receptor, LOX-1. The ability of miR-98 to preserve EC proliferation and inhibit apoptosis may be responsible for the reduction in lesion formation at regions where endothelial permeability is increased due to hyperlipidemic stress.
Endothelial dysfunction, which includes cell proliferation defects and aberrant apoptosis, is a key characteristic of atherosclerosis (22). Hyperlipidemia in atherosclerosis typically leads to EC injury and death, as well as detachment of ECs from the vascular wall, loss of anti-adhesive properties toward leukocytes and an increased permeability of the endothelial wall to circulating lipids, all of which lead to atherosclerotic lesion formation (23). Therefore, the proliferative capacity of ECs is essential for maintaining endothelial wall integrity by repairing the endothelial lining of the vessel in response to injury. Elevated levels of ox-LDL as a result of hyperlipidemia have been demonstrated to inhibit the proliferation of ECs by suppressing basic fibroblast growth factor expression, which is essential for endothelial proliferation (24). The mitogen-activated protein kinase/extracellular-signal-regulated kinase 1/2 signaling pathway is involved in the cytotoxic effects of ox-LDL that inhibit the proliferation of HUVECs (25). The cytotoxicity of ox-LDL in HUVECs is dependent on the LOX-1 receptor (26). Therefore rescuing EC proliferation, which represents a potential therapeutic approach for limiting the development of atherosclerosis (27), may be achieved by inhibiting the expression of LOX-1.
LOX-1 is located at the EC membrane and is highly expressed under specific pathological conditions, including during the early stage of atherosclerosis (19). Under acute inflammatory conditions, the lectin-like extracellular domain of LOX-1 may be released from the cell membrane into circulation as the soluble LOX-1 (sLOX-1) protein (28,29). Therefore, sLOX-1 may be considered a potential diagnostic and prognostic marker for atherosclerosis-associated events, including acute coronary syndrome (28,29). LOX-1 is the major receptor for ox-LDL uptake by ECs and induces a series of pathological processes in ECs (30). A previous study demonstrated that endothelial-specific LOX-1 overexpression enhanced aortic ox-LDL levels, resulting in endothelial dysfunction, vascular inflammation and plaque formation (31).
Stimulation of ECs with ox-LDL has previously been demonstrated to promote the expression of LOX-1 (32), and this was consistent with the present study; pretreatment of HUVECs with ox-LDL upregulated the LOX-1 receptor at the transcriptional level in a time- and concentration-dependent manner. In turn, elevated levels of LOX-1 facilitated ox-LDL uptake, resulting in pathological processes, including uptake of ox-DL and HUVEC apoptosis. LOX-1 has a critical role in the ox-LDL/LOX-1 positive feedback loop (33). In a previous study, ox-LDL was demonstrated to be taken up by LOX-1 and subsequently induced pathological changes in HUVECs, including senescence and apoptosis (34). These results suggested that LOX-1 may be a promising target for the treatment of vascular endothelial dysfunction.
LOX-1 mediates ox-LDL-induced apoptosis in ECs (35). Cellular apoptosis is induced via two major signaling pathways involving death receptors or the mitochondrial signaling pathway. In a previous study, LOX-1 was shown to mediate the ox-LDL-induced increase in Fas-mediated apoptosis in HUVECs via the death receptors pathway (36). Conversely, LOX-1 triggered the apoptotic cascade in human coronary artery ECs via the mitochondrial pathway (37). In the mitochondrial apoptotic signaling pathway, the cell membrane potential is altered, leading to the release of cytochrome c and activation of the caspase signaling pathway (37). It has also been suggested that LOX-1 may mediate ox-LDL-induced apoptosis in ECs via the endoplasmic reticulum stress signaling pathway (38). All apoptosis pathways eventually converge on caspase-3 activation (39), and thus caspase-3 is considered a critical factor or regulator in the apoptotic process. Caspase-3 can be cleaved to form two mature subunits, including p17 (17 kDa) and p12 (12 kDa) (40). The level of cleaved caspase-3 is an indicator of the level of activated caspase-3 and, in turn, the level of cellular apoptosis (40). The present study measured the levels of p17 (17 kDa) by western blotting in order to assess the apoptosis of HUVECs. Apoptosis is regulated by several apoptosis-associated proteins, including Bcl-2, which serves as an anti-death factor (41). Bcl-2 is an integral mitochondrial membrane protein that forms heterodimers with Bax to prevent mitochondrial alterations and the subsequent release of cytochrome c and other pro-apoptotic factors from the mitochondria (41). Conversely, polymerized Bax reduces the mitochondrial membrane potential, thereby inducing cytochrome c release and caspase activation, and leading to apoptosis (42). Therefore, the expression ratio of Bax/Bcl-2 regulates the induction of caspase activation pathways by ox-LDL (43). LOX-1 mediates ox-LDL-induced apoptosis in ECs and thus may be a potential target for blocking the atherosclerosis process at an early stage (44).
Considering the role of LOX-1 in suppressing the proliferation and inducing the apoptosis of ECs in response to ox-LDL, the present study assessed whether miRNAs were able to regulate LOX-1 expression and activity in ox-LDL-treated HUVECs. A bioinformatics analysis suggested that LOX-1 was a potential target of miR-98. Therefore, in order to investigate the association between miR-98 and LOX-1, the present study used miR-98 mimics and a miR-98 inhibitor to regulate LOX-1 expression in HUVECs. The miR-98 mimics decreased the expression of LOX-1 at the mRNA and protein levels, thus suggesting that miR-98 may bind to a seed site in the 3′-UTR of LOX-1 and promote LOX-1 mRNA degradation. In order to determine whether LOX-1 is a direct target of miR-98, a dual luciferase reporter assay was performed, which confirmed that miR-98 was able to directly bind to the 3-UTR seed sequence of LOX-1 mRNA. These results suggested that, by suppressing LOX-1 expression, miR-98 blocked ox-LDL uptake in HUVECs and alleviated LOX-1-mediated HUVEC apoptosis.
In the present study, as well as enhancing the protein expression levels of LOX-1, ox-LDL increased the cleavage and activation of caspase-3 (p17), decreased the expression levels of the anti-apoptotic protein Bcl-2, and increased the expression levels of the pro-apoptotic Bax in HUVECs. Conversely, the miR-98 mimics downregulated LOX-1 and Bax expression levels, elevated the expression levels of Bcl-2 and inhibited the activation of caspase-3. The miR-98 inhibitor produced the opposite effects.
As well as suppressing the uptake of ox-LDL by HUVECs, the miR-98 mimics were able to attenuate ox-LDL-mediated suppression of HUVEC proliferation. These results suggested that miR-98 may be essential for the growth and proliferation of HUVECs, as well as the replicative endothelial regeneration process required to repair the damaged endothelial lining in atherosclerosis.
In conclusion, the present study demonstrated that miR-98 exerted a protective effect on proliferation and anti-apoptotic functions on ECs by suppressing LOX-1 expression in response to ox-LDL. The results of the present study may provide novel insights into the molecular mechanisms underlying endothelial injury and into the pathogenesis of atherosclerosis. The authors of the present study hypothesize that, as the functional roles of other miRNAs are established, miRNAs will emerge as novel targets in therapeutic strategies for the treatment of endothelial dysfunction in atherosclerosis.
Acknowledgements
The present study was supported by the China National Natural Scientific Fund (grant no. 81170293).
References
- 1.Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet. 2007;39:1397–1402. doi: 10.1038/ng.2007.8. [DOI] [PubMed] [Google Scholar]
- 2.Mano T, Masuyama T, Yamamoto K, Naito J, Kondo H, Nagano R, Tanouchi J, Hori M, Inoue M, Kamada T. Endothelial dysfunction in the early stage of atherosclerosis precedes appearance of intimal lesions assessable with intravascular ultrasound. Am Heart J. 1996;131:231–238. doi: 10.1016/S0002-8703(96)90346-4. [DOI] [PubMed] [Google Scholar]
- 3.Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury and death. Annu Rev Pathol. 2009;4:71–95. doi: 10.1146/annurev.pathol.4.110807.092155. [DOI] [PubMed] [Google Scholar]
- 4.Triggle CR, Samuel SM, Ravishankar S, Marei I, Arunachalam G, Ding H. The endothelium: Influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol. 2012;90:713–738. doi: 10.1139/y2012-073. [DOI] [PubMed] [Google Scholar]
- 5.Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat Rev Cardiol. 2012;9:439–453. doi: 10.1038/nrcardio.2012.64. [DOI] [PubMed] [Google Scholar]
- 6.Sun C, Wu MH, Lee ES, Yuan SY. A disintegrin and metalloproteinase 15 contributes to atherosclerosis by mediating endothelial barrier dysfunction via Src family kinase activity. Arterioscler Thromb Vasc Biol. 2012;32:2444–2451. doi: 10.1161/ATVBAHA.112.252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, Megens R, Heyll K, Chun J, Saulnier-Blache JS, et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011;13:592–600. doi: 10.1016/j.cmet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Ishigaki Y, Katagiri H, Gao J, Yamada T, Imai J, Uno K, Hasegawa Y, Kaneko K, Ogihara T, Ishihara H, et al. Impact of plasma oxidized low-density lipoprotein removal on atherosclerosis. Circulation. 2008;118:75–83. doi: 10.1161/CIRCULATIONAHA.107.745174. [DOI] [PubMed] [Google Scholar]
- 9.Galle J, Hansen-Hagge T, Wanner C, Seibold S. Impact of oxidized low density lipoprotein on vascular cells. Atherosclerosis. 2006;185:219–226. doi: 10.1016/j.atherosclerosis.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 11.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 12.Shan Z, Yao C, Li ZL, Teng Y, Li W, Wang JS, Ye CS, Chang GQ, Huang XL, Li XX, et al. Differentially expressed microRNAs at different stages of atherosclerosis in ApoE-deficient mice. Chin Med J (Engl) 2013;126:515–520. [PubMed] [Google Scholar]
- 13.Wang M, Li W, Chang GQ, Ye CS, Ou JS, Li XX, Liu Y, Cheang TY, Huang XL, Wang SM. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol. 2011;31:2044–2053. doi: 10.1161/ATVBAHA.111.229559. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Ouyang M, Shan Z, Ma J, Li J, Yao C, Zhu Z, Zhang L, Chen L, Chang G, et al. Involvement of microRNA-133a in the development of arteriosclerosis obliterans of the lower extremities via RhoA targeting. J Atheroscler Thromb. 2015;22:424–432. doi: 10.5551/jat.27839. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Lee CG. MicroRNA and cancer-focus on apoptosis. J Cell Mol Med. 2009;13:12–23. doi: 10.1111/j.1582-4934.2008.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou DH, Wang X, Feng Q. EGCG enhances the efficacy of cisplatin by downregulating hsa-miR-98-5p in NSCLC A549 cells. Nutr Cancer. 2014;66:636–644. doi: 10.1080/01635581.2014.894101. [DOI] [PubMed] [Google Scholar]
- 17.Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell Mol Life Sci. 2013;70:2859–2872. doi: 10.1007/s00018-012-1194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawamura T, Wakabayashi I, Okamura T. LOX-1 in atherosclerotic disease. Clin Chim Acta. 2015;440:157–163. doi: 10.1016/j.cca.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 19.Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): A critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 2006;69:36–45. doi: 10.1016/j.cardiores.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Li XX, Ma Q, Cui J. The variability of oxLDL-induced cytotoxicity on different types of cell lines. Cell Biochem Biophys. 2013;67:635–644. doi: 10.1007/s12013-013-9552-5. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces and atherogenesis. Ann N Y Acad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 23.Grover-Páez F, Zavalza-Gómez AB. Endothelial dysfunction and cardiovascular risk factors. Diabetes Res Clin Pract. 2009;84:1–10. doi: 10.1016/j.diabres.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Chen CH, Jiang W, Via DP, Luo S, Li TR, Lee YT, Henry PD. Oxidized low-density lipoproteins inhibit endothelial cell proliferation by suppressing basic fibroblast growth factor expression. Circulation. 2000;101:171–177. doi: 10.1161/01.CIR.101.2.171. [DOI] [PubMed] [Google Scholar]
- 25.Yin G, Yang X, Li B, Yang M, Ren M. Connexin43 siRNA promotes HUVEC proliferation and inhibits apoptosis induced by ox-LDL: An involvement of ERK signaling pathway. Mol Cell Biochem. 2014;394:101–107. doi: 10.1007/s11010-014-2085-4. [DOI] [PubMed] [Google Scholar]
- 26.Dong Q, Xiang R, Zhang DY, Qin S. Ox-LDL increases OX40L in endothelial cells through a LOX-1-dependent mechanism. Braz J Med Biol Res. 2013;46:765–770. doi: 10.1590/1414-431X20132733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H, Hristov M, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. 2014;20:368–376. doi: 10.1038/nm.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pirillo A, Catapano AL. Soluble lectin-like oxidized low density lipoprotein receptor-1 as a biochemical marker for atherosclerosis-related diseases. Dis Markers. 2013;35:413–418. doi: 10.1155/2013/716325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw DJ, Seese R, Ponnambalam S, Ajjan R. The role of lectin-like oxidised low-density lipoprotein receptor-1 in vascular pathology. Diab Vasc Dis Res. 2014;11:410–418. doi: 10.1177/1479164114547704. [DOI] [PubMed] [Google Scholar]
- 30.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, Masaki T. An endothelial receptor for oxidized low-density lipoprotein. Nature. 1997;386:73–77. doi: 10.1038/386073a0. [DOI] [PubMed] [Google Scholar]
- 31.Akhmedov A, Rozenberg I, Paneni F, Camici GG, Shi Y, Doerries C, Sledzinska A, Mocharla P, Breitenstein A, Lohmann C, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J. 2014;35:2839–2848. doi: 10.1093/eurheartj/eht532. [DOI] [PubMed] [Google Scholar]
- 32.Mehta JL, Li DY. Identification and autoregulation of receptor for OX-LDL in cultured human coronary artery endothelial cells. Biochem Biophys Res Commun. 1998;248:511–514. doi: 10.1006/bbrc.1998.9004. [DOI] [PubMed] [Google Scholar]
- 33.Hermonat PL, Zhu H, Cao M, Mehta JL. LOX-1 transcription. Cardiovasc Drugs Ther. 2011;25:393–400. doi: 10.1007/s10557-011-6322-8. [DOI] [PubMed] [Google Scholar]
- 34.Lu J, Yang JH, Burns AR, Chen HH, Tang D, Walterscheid JP, Suzuki S, Yang CY, Sawamura T, Chen CH. Mediation of electronegative low-density lipoprotein signaling by LOX-1: a possible mechanism of endothelial apoptosis. Circ Res. 2009;104:619–627. doi: 10.1161/CIRCRESAHA.108.190116. [DOI] [PubMed] [Google Scholar]
- 35.Mollace V, Gliozzi M, Musolino V, Carresi C, Muscoli S, Mollace R, Tavernese A, Gratteri S, Palma E, Morabito C, et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int J Cardiol. 2015;184:152–158. doi: 10.1016/j.ijcard.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Imanishi T, Hano T, Sawamura T, Takarada S, Nishio I. Oxidized low density lipoprotein potentiation of Fas-induced apoptosis through lectin-like oxidized-low density lipoprotein receptor-1 in human umbilical vascular endothelial cells. Circ J. 2002;66:1060–1064. doi: 10.1253/circj.66.1060. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Mehta JL, Haider N, Zhang X, Narula J, Li D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res. 2004;94:370–376. doi: 10.1161/01.RES.0000113782.07824.BE. [DOI] [PubMed] [Google Scholar]
- 38.Hong D, Bai YP, Gao HC, Wang X, Li LF, Zhang GG, Hu CP. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis. 2014;235:310–317. doi: 10.1016/j.atherosclerosis.2014.04.028. [DOI] [PubMed] [Google Scholar]
- 39.Sugawara T, Fujimura M, Noshita N, Kim GW, Saito A, Hayashi T, Narasimhan P, Maier CM, Chan PH. Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx. 2004;1:17–25. doi: 10.1602/neurorx.1.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han Z, Hendrickson EA, Bremner TA, Wyche JH. A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J Biol Chem. 1997;272:13–432. doi: 10.1074/jbc.272.20.13432. 13,436. [DOI] [PubMed] [Google Scholar]
- 41.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 42.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kataoka H, Kume N, Miyamoto S, Minami M, Morimoto M, Hayashida K, Hashimoto N, Kita T. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:955–960. doi: 10.1161/01.ATV.21.6.955. [DOI] [PubMed] [Google Scholar]
- 44.Li DY, Chen HJ, Mehta JL. Statins inhibit oxidized-LDL-mediated LOX-1 expression, uptake of oxidized-LDL and reduction in PKB phosphorylation. Cardiovasc Res. 2001;52:130–135. doi: 10.1016/S0008-6363(01)00371-6. [DOI] [PubMed] [Google Scholar]