

Abstract
Introduction
Transient receptor potential (TRP) channels are broadly expressed cation channels that mediate diverse physiological stimuli and include canonical (TRPC), melastatin (TRPM) and vanilloid (TRPV) subtypes. Recent studies have implicated a role for TRPC6 channels as an important component of signaling via the cytokine, transforming growth factor beta 1 (TGFb1) in right (RV) or left ventricular (LV) failure. Endoglin is a transmembrane glycoprotein that promotes TRPC6 expression and TGFb1 activity. No studies have defined biventricular expression of all TRP channel family members in heart failure.
Hypothesis
We hypothesized that heart failure is associated with distinct patterns of TRP channel expression in the LV and RV.
Methods
Paired viable left (LV) and right (RV) ventricular free wall tissue was obtained from human subjects with end-stage heart failure (n=12) referred for cardiac transplantation or biventricular assist device implantation. Paired LV and RV samples from human subjects without heart failure served as controls (n=3). To explore a functional role for endoglin (Eng) as a regulator of TRP expression in response to RV or LV pressure overload, wild-type (Eng+/+) and Eng haploinsufficient (Eng+/−) mice were exposed to thoracic aortic (TAC) or pulmonary arterial (PAC) constriction for 8 weeks. Biventricular tissue was analyzed by real-time polymerase chain reaction.
Results
Compared to non-failing human LV and RV samples, mRNA levels of TRPC1, 3, 4, 6 and TRPV-2 were increased and TRPM2, 3, and 8 were decreased in failing LV and RV samples. TRPC1 and 6 levels were higher in failing RV compared to failing LV samples. After TAC, murine LV levels of TPRC1 and 6 were increased in both Eng +/+ and Eng +/− mice compared to sham controls. LV levels of TRPC4; TRPM3 and 7; TRPV2 and 4 were increased in Eng +/+, not Eng +/− mice after TAC. After PAC, all TRP channel family members were increased in the RV, but not LV, of Eng +/+ compared to sham controls. In contrast to Eng+/+, PAC did not increase RV or LV levels of TRP channels in Eng +/− mice.
Conclusions
This is the first study to demonstrate that TRP channels exhibit distinct profiles of expression in the LV and RV of patients with heart failure and in murine models of univentricular pressure overload. We further introduce that the TGFb1 co-receptor endoglin selectively regulates expression of multiple TRP channels in the setting of LV or RV pressure overload.
Keywords: Heart failure, Transient receptor potential channels, Right ventricular failure, Endoglin
1. Introduction
Heart failure is a major cause of morbidity and mortality for nearly 24 million individuals worldwide (Bui et al., 2011). While much attention has focused on signaling mechanisms regulating left ventricular (LV) remodeling, the negative impact of right ventricular (RV) dysfunction on survival remains a significant problem for patients with left heart failure or lung disease (Aronson et al., 2013; Ghio et al., 2001; Gulati et al., 2013; Iglesias-Garriz et al., 2012; McLaughlin et al., 2009). However, several lines of evidence suggest that the RV and LV have distinct profiles of response to injury including: 1) the developmental origin of the RV from a heart field distinct from the LV; 2) a thin RV free wall with susceptibility to increased wall stress; 3) a greater dependence of RV stroke volume on afterload; and 4) enhanced RV contractile resilience to pressure overload (Bogaard et al., 2009; Bristow et al., 1998; Rockman et al., 1994; Urashima et al., 2008; Zaffran et al., 2004). Our understanding of the mechanisms governing RV remodeling stem primarily from data generated in models of LV failure. The identification of molecular targets that improve LV and RV function remains a significant unmet need for patients suffering from heart failure and lung disease.
Transient receptor potential (TRP) channels are broadly expressed cation channels that mediate diverse physiological stimuli and include canonical (TRPC), melastatin (TRPM) and vanilloid (TRPV) subtypes (Clapham, 2003). TRP channels are emerging as key mediators of cardiac hypertrophy and fibrosis, however little is known about TRP channel family expression in HF. Several recent studies implicate TRPC-6 as a central mediator of signaling via transforming growth factor beta 1 (TGFb1). In these studies, TGFb1 promotes expression of calcineurin, which increases levels of TRPC-6, which triggers calcium influx and subsequent calcineurin activation, thereby setting up a self-propagating mechanism for pathologic LV hypertrophy, fibrosis, and increased mortality in heart failure (Eder and Molkentin, 2011; Koitabashi et al., 2010; Kuwahara et al., 2006; Patel et al., 2010). Expression of TRP channel family members in heart failure remains poorly understood.
Endoglin is a 180kDA trans-membrane glycoprotein that promotes TGFb1 signaling in heart failure (Kapur et al., 2010; Kapur et al., 2012). Endoglin null mice die at embryonic day 10.5 due to impaired cardiovascular development and extra-embryonic angiogenesis (Li et al., 1999). In contrast, endoglin heterozygous mice (Eng+/−) are viable and have reduced total body levels of endoglin. We recently reported that loss of the TGFb1 co-receptor, endoglin, attenuates increased TRPC-6 expression, reduces RV fibrosis and improves survival in murine models of RV failure (Kapur et al., 2014). Whether endoglin regulates expression of other TRP channel family members is not known. Based on these background data, the purpose of this study was to determine TRP channel expression in the LV and RV from patients with end-stage HF and to explore whether endoglin regulates expression of TRP channels in response to LV or RV pressure overload.
2. Methods
2.1 Human samples
Paired viable left (LV) and right (RV) ventricular free wall tissue was obtained from human subjects with end-stage HF (n=12) referred for orthotopic heart transplantation (OHT) or biventricular assist device implantation (BIVAD). Non-failing LV and RV tissue obtained from the National Disease Research Interchange (NDRI) served as controls (n=3). All tissue was immediately frozen in liquid nitrogen and stored at −80°C until further processing as described below. All surgical procedures and tissue harvesting were performed in concordance with the National Institutes of Health and Tufts University Institutional Review Board guidelines.
2.2 Surgical models of heart failure
Animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals (National Academy of Science). Animal protocols were approved by the Tufts Medical Center Institutional Animal Care and Use Committee. As described previously, adult, male, 12–14 week old C57BL/6 wild-type (Eng+/+) and Eng+/− mice underwent constriction of the thoracic aorta (TAC) or pulmonary artery (PAC) for 8 weeks to generate models of left HF or right HF, respectively (Kapur et al., 2013; Kapur et al., 2012). Sham-operated mice served as controls (n=6). Ten weeks after TAC or PAC, terminal hemodynamics were recorded using biventricular conductance catheters as previously described ()(Kapur et al., 2013) and both RV and LV tissue obtained for further analysis by real-time polymerase chain reaction (RT-PCR).
2.3 PCR
Total RNA was extracted from the LV and RV with Trizol (Life Technologies) and converted to cDNA with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed in triplicate using 40 cycles at 94° C for 15 seconds, 60° C for 30 seconds and 72° C for 30 seconds with an ABI Prism 7900 Sequence Detection System. The primers used for detection are shown in Table 2.
Table 2.
Primers used for real time PCR analysis References
| Mouse | Forward primer | Reverse primer |
|---|---|---|
| Endoglin | ctgccaatgctgtgcgtgaa | gctggagtcgtaggccaagt |
| TRPC1 | ctgcgaacagcaaagcaatg | gaagatgtaccagaacagagc |
| TRPC3 | ggtggtcgttttactcaacatgc | catcgaagtaggagagccaaag |
| TRPC4 | gactatgacttaagccccacgg | gatttcaaacattttgcctgcc |
| TRPC6 | cacagaagacctagcagagctca | ataatatggcttcaagtggagaaat |
| TRPV2 | cggaccagcaagtacctcactg | ccggaatccctgtcaatctg |
| TRPV4 | ccctggcaagagtgaaatctacc | catctgcgcttgagttcttgttc |
| TRPM3 | atcgcttcaattcgtccaacg | tgctagccggatgtccacg |
| TRPM4 | ccaacactgttctcgagtcctgac | gcaaacacctagacatccaccag |
| TRPM6 | ctgacctcttggccttcacttac | ggaaaaggtgcttagactgtctaag |
| TRPM7 | gaagagcagtgtgttgagatgtac | tatgtagttgacacgatctccaac |
| Human | Forward primer | Reverse primer |
| TRPC1 | tatggatgttgcacctgtcattt | actgggagacaaactctttctgg |
| TRPC3 | acgacttctacgcttacgacgag | cttaatggcaagtttgacacgac |
| TRPC4 | gaggtactctgcctactcccttca | gagccattgcttatgttatgtcttt |
| TRPC5 | caatgtgaaagccagacacgaat | tctatttcccaagaggtcaagca |
| TRPC6 | ttgacgaaagtaacattgggagac | accagattgaagggtacaggaag |
| TRPC7 | ttgtggaacctgctagatttcgg | ggttgtatttggcacctcggtag |
| TRPM1 | aaaactttcggaccctttacaac | aagaatccccatgataaccttca |
| TRPM2 | cctcatcgccatgttcaactacac | ctcctccgtcttcttcctgcctc |
| TRPM3 | agaaggaggcagaagaaccagag | ccaccagcataatgatgacaaag |
| TRPM4 | ggcggagaccctggaagaca | tgcggatgagcgagttgg |
| TRPM5 | ctggacgagattgatgaagcc | acgagcaccgagcagtagtt |
| TRPM6 | gggcaagtatggaaatgaaatga | atggtaagaaagcgatggaggtt |
| TRPM7 | tgtcccatatcccacaatctcaa | atctagcaaacgcacataccaaa |
| TRPM8 | gcaatgccatctcctacgctcta | gagttctatgtccatctcgtccc |
| TRPV1 | gattgaagacgggaagaatgact | tctgcctgaaactctgcttgacc |
| TRPV2 | acatcgccattgagaagaggagt | ccacagaagccaggtcatacagc |
| TRPV3 | atgcctggttccactttgtcttt | gaatctgctcctcagccattctg |
| TRPV4 | aaagtcttcaaccggcctatcct | gcagcaggtcgtacatcttggta |
| TRPV5 | ctggctgccttgtacctgctcta | gactggttgggtcctctgtctgg |
| TRPV6 | accttcgagctgttccttaccat | gttgattatcccacgcaggtctc |
2.4 Statistics
All statistical analyses were performed using Graph Pad Prism v6 (Graph Pad Software, Inc.). Comparison between two experimental groups was performed with the unpaired student’s T test and for three groups or more with a one-way ANOVA. α values less than 0.05 were accepted as statistically significant.
3. Results
3.1 TRP channel expression in human heart failure
To determine whether TRP channel expression is altered in patients with heart failure, LV and RV samples were obtained from patients with advanced heart failure referred for OHT (n=9) or BIVAD support (n=3; Table 1). Non-ischemic cardiomyopathy was the primary etiology for heart failure in 11 subjects. Compared to non-failing control LV and RV samples, levels of TRPC1, 3, 4 and 6 mRNA were increased in the failing LV and RV respectively (Figure 1). Levels of TRPC1 and 6 mRNA were higher in the failing RV than the failing LV. TRPV1 and 3 were not detectable in the human samples. TRPV2 levels were similarly increased in the failing LV and RV. Levels of TRPM 1,6, and 7 mRNA were not detectable in the LV or RV. Levels of TRPM2, 3 and 8 were reduced to a similar extent in the failing LV and RV compared to non-failing control LV and RV samples. TRPC2 is a pseudo-gene in humans and was not analyzed (Clapham, 2003). TRPC5 and 7 were not detectable in the human LV or RV. Compared to non-failing human LV and RV samples, Endoglin mRNA levels were increased in the LV and RV (Figure 2).
Table 1.
Clinical characteristics of patients with advanced heart failure
| Patient | Age | Gender | Cardiomyopathy | Indication for surgery |
|---|---|---|---|---|
| 1 | 20 | F | NICM | OHT for post-partum cardiomyopathy |
| 2 | 52 | F | NICM | OHT for NICM post-LVAD |
| 3 | 64 | M | NICM | OHT for HCM |
| 4 | 53 | F | NICM | BIVAD for myocarditis |
| 5 | 45 | M | NICM | BIVAD for myocarditis |
| 6 | 34 | M | NICM | OHT for NICM post-LVAD |
| 7 | 50 | F | NICM | OHT for NICM |
| 8 | 53 | F | NICM | OHT for chemotherapy related NICM post-LVAD |
| 9 | 44 | F | NICM | OHT for giant cell myocarditis |
| 10 | 64 | M | ICM | OHT post-LVAD |
| 11 | 56 | M | NICM | BIVAD for myocarditis |
| 12 | 67 | F | NICM | OHT for chemotherapy related NICM |
Figure 1. Distinct expression of RV and LV TRP channels in human subjects with advanced heart failure.
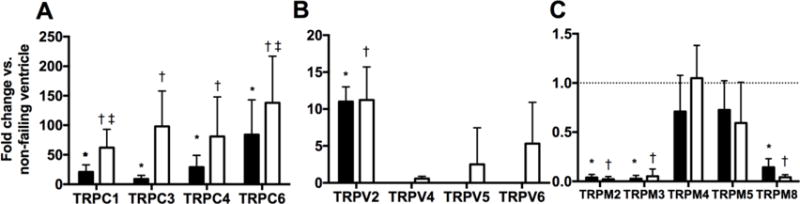
mRNA expression levels of TRPC (A), TRPV (B) and TRPM (C) in human LV (closed bars) and RV (open bars) samples. Data are expressed as fold change compared to normal ventricle. p<0.05: *, vs Normal LV; †, vs Normal RV; ‡, vs Failing LV. ND=not detected.
Figure 2. Endoglin mRNA expression following 8 weeks of LV or RV pressure overload.
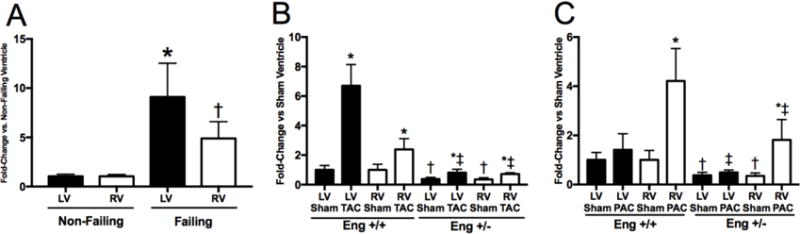
mRNA expression levels of Endoglin in human LV and RV (A). Data are expressed as fold change compared to non-failing ventricle. p<0.05: *, vs Non-Failing LV; †, vs Non-Failing RV. mRNA expression levels of Endoglin in the LV (closed bars) and RV (open bars) following 8 weeks of TAC (B) or PAC (C).*, vs. Sham operated corresponding ventricle; †, vs. Eng +/+ Sham operated corresponding ventricle; ‡, vs. Eng +/+ TAC or PAC corresponding ventricle
3.2 TRP channel expression in LV pressure overload
To explore whether isolated LV failure altered TRP channel expression and the effect of endoglin haploinsufficiency on TRP channel expression, we employed the well-established model of LV pressure overload induced by trans aortic constriction (TAC) in WT and endoglin haploinsufficient mice (Eng+/−). Compared to sham controls and WT mice after TAC, both RV and LV mRNA levels of endoglin are lower in Eng+/− mice (Figure 2). In sham-operated controls, TRPC-1, 3, 4 and 6; TRPV-2, and 4; and TRPM-3, 4, 6 and 7 are expressed in the LV and RV. Compared to sham-operated controls, LV pressure overload increased LV mRNA levels of TRPC-1, 4, and 6; TRPM-3, 7; and TRPV-2 and 4 levels (Figure 3). TRPM-4 and TRPM-6 levels were unchanged with TAC in the LV and RV (data not shown). LV pressure overload only increased RV mRNA levels of TRPC-4 (Figure 4). Compared to sham-operated controls, LV pressure overload increased LV mRNA levels of TRPC1 and TRPC 6 only in Eng+/− mice (Figure 3). No change in RV mRNA levels of any TRP channels were observed after LV pressure overload in Eng+/− mice. Compared to WT mice subjected to TAC, LV mRNA levels of TRPC-4, TRPM-3, TRPV-2, and TRPV-4 were lower in Eng+/− mice after TAC. We observed increased LV peak systolic pressure in Eng +/− mice, not Eng +/+ mice, following TAC (Figure 3D). LV end diastolic pressure was similarly increased in Eng +/+ and Eng +/− mice after TAC (Figure 3E). LV and RV stroke volume was decreased in Eng +/+ mice and preserved in Eng +/− mice (Figure 3F). These findings indicate LV function was improved in Eng +/− mice compared to WT mice following TAC.
Figure 3. Biventricular TRPC expression and hemodynamics following 8 weeks of LV pressure overload.
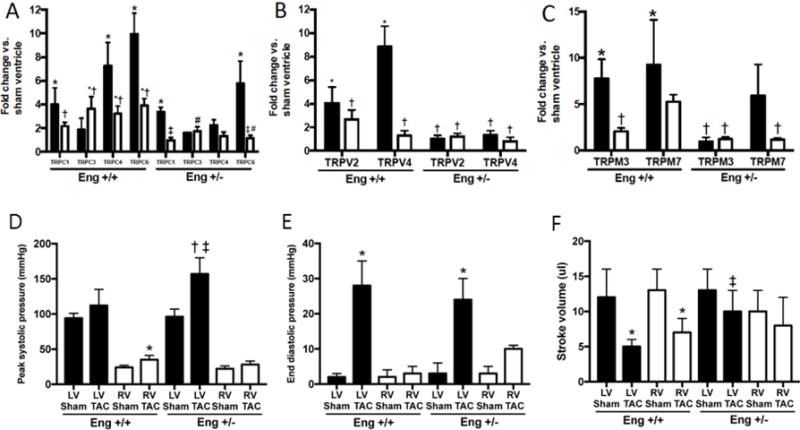
mRNA expression levels of TRPC (A), TRPV (B) and TRPM (C) in the LV (closed bars) and RV (open bars) following 8 weeks of TAC. Data are expressed as fold change compared to sham operated ventricle. p<0.05: *, vs. Sham operated corresponding ventricle; †, vs. Eng +/+ TAC LV; ‡, vs. Eng +/− TAC LV; #, vs. Eng +/+ TAC RV; %, vs. Eng +/− TAC RV.Peak systolic pressure (D), end diastolic pressure (E) and stroke volume (F) were measured under steady state conditions. p<0.05: *, vs. Sham operated corresponding ventricle; †, vs. Eng +/+ Sham operated corresponding ventricle; ‡, vs. Eng +/+ TAC corresponding ventricle
Figure 4. Biventricular TRP expression and hemodynamics following 8 weeks of RV pressure overload.
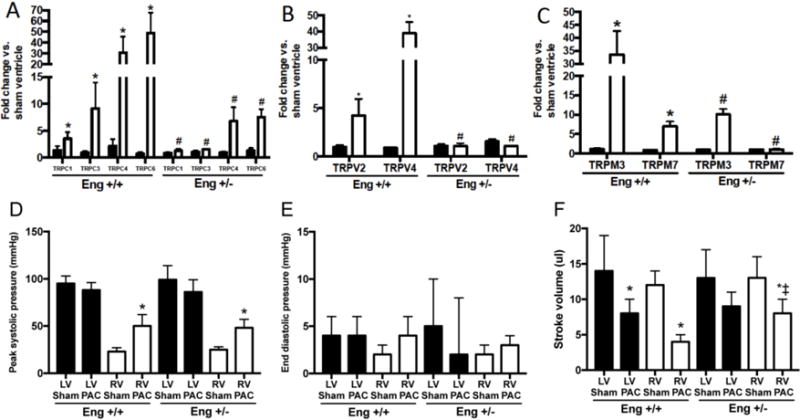
mRNA expression levels of TRPC (A), TRPV (B) and TRPM (C) in the LV (closed bars) and RV (open bars) following 8 weeks of PAC. Data are expressed as fold change compared to sham operated ventricle. p<0.05: *, vs. Sham operated corresponding ventricle; †, vs. Eng +/+ PAC LV; ‡, vs. Eng +/− PAC LV; #, vs. Eng +/+ PAC RV; %, vs. Eng +/− PAC RV. Peak systolic pressure (D), end diastolic pressure (E) and stroke volume (F) were measured under steady state conditions. p<0.05: *, vs. Sham operated corresponding ventricle; †, vs. Eng +/+ Sham operated corresponding ventricle; ‡, vs. Eng +/+ TAC corresponding ventricle
3.3 TRP channel expression in RV pressure overload
To explore whether isolated RV failure altered TRP channel expression, we employed the well-established model of RV pressure overload induced by pulmonary artery constriction (PAC). Compared to sham controls and WT mice after PAC, both RV and LV mRNA levels of endoglin are lower in Eng+/− mice (Figure 2). Compared to sham controls, RV pressure overload increased RV mRNA levels of all TRP channels studied (TRPC-1, 3, 4, 6; TRPM-3, 7; and TRPV-2, and 4). TRPM-4 and TRPM-6 levels were unchanged with PAC in the LV and RV (data not shown). RV pressure overload did not induce expression of any TRP channel studied in the LV (Figure 4). Next, to explore whether endoglin regulates TRP channel expression in RV pressure overload, TRP mRNA expression was studied in Eng+/− mice subjected to PAC. Compared to sham-operated controls, RV pressure overload did not increase RV or LV mRNA levels of any TRP channel in Eng +/− mice (Figure 4). Compared to WT mice subjected to TAC, RV mRNA levels of all TRP channels were higher after PAC and LV mRNA levels were unchanged. We observed increased RV systolic pressure (RVSP) and no change in end-diastolic pressure in both Eng +/+ and Eng +/− mice (Figure 4D–E). Despite equally increased RVSP in both Eng +/+ and +/− mice, LV and RV stroke volume was decreased in Eng +/+, not Eng +/−, mice (Figure 3F). Compared to WT mice, these findings suggest RV function was improved in Eng +/− mice following PAC.
4. Discussion
Our central finding is that biventricular TRP channel expression is altered in heart failure. Specifically, we report the following: 1) mRNA levels of multiple TRP channel family members are increased in the LV and RV of patients with advanced heart failure, 2) LV pressure overload increases LV and RV mRNA levels of multiple TRP channels in a murine model of thoracic aortic constriction, 3) RV pressure overload increases RV, not LV, mRNA levels of multiple TRP channels, and 4) reduced endoglin expression attenuates increased mRNA levels of multiple TRP channels after LV or RV pressure overload. Our findings suggest that further exploration of the role of TRP channels in biventricular remodeling may identify novel targets of therapy for heart failure or pulmonary hypertension and further introduces that the TGFb1 co-receptor, endoglin, regulates biventricular TRP channel expression.
The TRPC family has been well described as mediators of pathological cardiac remodeling. TRPC-1,3,4 and 6 are mechanosensitive mediators of calcium influx into cardiomyocytes which activate NFAT/calcineurin signaling and downstream maladaptive signaling as well as promote transformation of fibroblasts into highly secretory myofibroblasts (Seth 2009, Camacho Londono 2015, Seo PNAS 2014). TRPC loss of function studies using genetic and pharmacological approaches have shown suppression of pressure overload induced heart failure in mouse models (Seth 2009, Camacho Londono 2015, Seo PNAS 2014). We observed increased levels of TRPC (1, 3, 4, 6) in failing human samples and mouse models of pressure overload induced heart failure. These data further substantiate that TRPC channels are upregulated in human heart failure and suggest targeting TRPC channel expression or activity may ameliorate heart failure.
The role of TRPV and TRPM channels in human HF is not well defined.. Prior studies have established expression of specific TRPM (3,7) and TRPV (2,4) subtypes in human cardiac fibroblasts and suggest calcium influx mediated by these channels contribute to myofibroblast transformation (Adapala et al., 2013; Du et al., 2010; Iwata et al., 2013). We found increased levels of TRPV2 and reduced levels of TRPM 2,3, and 8 in the failing LV and RV. Functional TRP channels assemble from homomeric and heteromeric oligomerization of TRPC, TRPM and TRPV subunits (Alessandri-Haber et al., 2009; Hofmann et al., 2002; Kobori et al., 2009; Poteser et al., 2006). The subunit composition of TRP channels in HF is not well established and TRPV or TRPM may participate in the formation of functional TRP channels underlying pathological cardiac remodeling. Future studies may focus on the functional significance of TRPV and TRPM transcriptional regulation in HF.
RV failure is an unmet clinical need without specific medical therapy. All of the examined TRPC channels were upregulated in the human failing LV and RV. We observed profound upregulation of the examined TRPC channels in the pressure-overloaded RV and severely impaired RV systolic function. Our findings indicate TRPC channel upregulation may be a molecular signature of the failing RV and a potential therapeutic target. Development of pharmacological inhibitors targeting TRP subtypes in prior pre-clinical studies of pressure overload and post-myocardial infarction HF has shown limited progress to date, in part due to the inherent difficulty of modulating multiple TRP channels with a single agent (Camacho Londono et al., 2015; Makarewich et al., 2014; Seo et al., 2014). Endoglin deficiency attenuated TRP channel upregulation among all subtypes in the pressure overloaded RV and improved RV function.We have previously shown reduction of endoglin activity limits TRPC6 mediated calcineurin activation and fibrosis in the failing RV (Kapur et al., 2014). Further studies are necessary to establish if the salutatory functional effects of endoglin blockade in the pressure overloaded RV are attributable to modulation of TRP channel expression and/or non-TRP mediated signaling pathways.
Prior studies of TRP channel expression in the failing heart have reported disparate findings. TRPV2 and TRPC6 have been alternately reported as not detectable, unchanged or increased in the LV of patients with dilated cardiomyopathy (Iwata et al., 2013; Kuwahara et al., 2006; Watanabe et al., 2009). In part the lack of agreement may be explained by methodological differences including sample location (atria vs. ventricle), patient characteristics (non-ischemic vs. ischemic cardiomyopathy), and sample type (isolated cell preparations vs. tissue homogenates). We sought to examine differential mRNA expression of the TRPC, TRPV and TRPM families in the failing LV and RV.
Our study is limited by the availability of commercially available antibodies with sufficient specificity to differentiate between TRP channel subtypes in mice and humans and therefore we employed real time PCR as performed in contemporaneous studies (Makarewich et al., 2014; Seo et al., 2014). Without measurement of TRP channel protein levels, we cannot determine if the observed differences are manifest at the post-transcriptional level. As reliable and specific antibodies for the TRP channel subtypes are developed, future studies may permit detailed cell and TRP subunit specific analysis of the functional role of TRP channels in adverse cardiac remodeling.
5. Conclusions
In conclusion, heart failure is a major cause of global mortality. Limited studies have explored the expression profile of TRP channels in heart failure. Recent reports have suggested a link between TGFb1, endoglin, and TRPC-6 as mediators of cardiac fibrosis in right and left heart failure. We now introduce that biventricular expression of multiple TRP channels is altered in patients with heart failure and that LV or RV pressure overload generate distinct profiles of TRP expression. Furthermore, we show that intact reduced endoglin expression prevents upregulation of TRP channels in murine models of right or left ventricular pressure overload. Our studies support that TRP channels may represent novel targets of therapy for heart failure.
Highlights.
-
-
A distinct TRP expression profile was observed in advanced human heart failure
-
-
Select TRP channel expression is higher in the failing RV than LV
-
-
TRP channel upregulation requires full endoglin activity in the failing RV
Acknowledgments
This work was supported by a grant from the National Institutes of Health (K08HL094909-03 and R56HL118113-01A1) to N.K. and a Heart Failure Society of America Research Fellowship to K.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
There are no relevant financial relationships to disclose.
References
- Adapala RK, Thoppil RJ, Luther DJ, et al. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. doi: 10.1016/j.yjmcc.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Chen X, et al. TRPC1 and TRPC6 channels cooperate with TRPV4 to mediate mechanical hyperalgesia and nociceptor sensitization. J Neurosci. 2009;29:6217–6228. doi: 10.1523/JNEUROSCI.0893-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson D, Darawsha W, Atamna A, et al. Pulmonary hypertension, right ventricular function, and clinical outcome in acute decompensated heart failure. J Card Fail. 2013;19:665–671. doi: 10.1016/j.cardfail.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Bogaard HJ, Natarajan R, Henderson SC, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Zisman LS, Lowes BD, et al. The pressure-overloaded right ventricle in pulmonary hypertension. Chest. 1998;114:101S–106S. doi: 10.1378/chest.114.1_supplement.101s. [DOI] [PubMed] [Google Scholar]
- Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. doi: 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho Londono JE, Tian Q, Hammer K, et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J. 2015;36:2257–2266. doi: 10.1093/eurheartj/ehv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Du J, Xie J, Zhang Z, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. 2011;108:265–272. doi: 10.1161/CIRCRESAHA.110.225888. [DOI] [PubMed] [Google Scholar]
- Ghio S, Gavazzi A, Campana C, et al. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol. 2001;37:183–188. doi: 10.1016/s0735-1097(00)01102-5. [DOI] [PubMed] [Google Scholar]
- Gulati A, Ismail TF, Jabbour A, et al. The prevalence and prognostic significance of right ventricular systolic dysfunction in nonischemic dilated cardiomyopathy. Circulation. 2013;128:1623–1633. doi: 10.1161/CIRCULATIONAHA.113.002518. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, et al. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias-Garriz I, Olalla-Gomez C, Garrote C, et al. Contribution of right ventricular dysfunction to heart failure mortality: a meta-analysis. Rev Cardiovasc Med. 2012;13:e62–69. doi: 10.3909/ricm0602. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Ohtake H, Suzuki O, et al. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc Res. 2013;99:760–768. doi: 10.1093/cvr/cvt163. [DOI] [PubMed] [Google Scholar]
- Kapur NK, Heffernan KS, Yunis AA, et al. Usefulness of soluble endoglin as a noninvasive measure of left ventricular filling pressure in heart failure. Am J Cardiol. 2010;106:1770–1776. doi: 10.1016/j.amjcard.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur NK, Paruchuri V, Aronovitz MJ, et al. Biventricular remodeling in murine models of right ventricular pressure overload. PLoS One. 2013;8:e70802. doi: 10.1371/journal.pone.0070802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur NK, Qiao X, Paruchuri V, et al. Reducing endoglin activity limits calcineurin and TRPC-6 expression and improves survival in a mouse model of right ventricular pressure overload. J Am Heart Assoc. 2014;3 doi: 10.1161/JAHA.114.000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur NK, Wilson S, Yunis AA, et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 2012;125:2728–2738. doi: 10.1161/CIRCULATIONAHA.111.080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori T, Smith GD, Sandford R, et al. The transient receptor potential channels TRPP2 and TRPC1 form a heterotetramer with a 2:2 stoichiometry and an alternating subunit arrangement. J Biol Chem. 2009;284:35507–35513. doi: 10.1074/jbc.M109.060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koitabashi N, Aiba T, Hesketh GG, et al. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K, Wang Y, McAnally J, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DY, Sorensen LK, Brooke BS, et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- Makarewich CA, Zhang H, Davis J, et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res. 2014;115:567–580. doi: 10.1161/CIRCRESAHA.115.303831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–1619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Patel A, Sharif-Naeini R, Folgering JR, et al. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch. 2010;460:571–581. doi: 10.1007/s00424-010-0847-8. [DOI] [PubMed] [Google Scholar]
- Poteser M, Graziani A, Rosker C, et al. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Ono S, Ross RS, et al. Molecular and physiological alterations in murine ventricular dysfunction. Proc Natl Acad Sci U S A. 1994;91:2694–2698. doi: 10.1073/pnas.91.7.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo K, Rainer PP, Shalkey Hahn V, et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A. 2014;111:1551–1556. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urashima T, Zhao M, Wagner R, et al. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol. 2008;295:H1351–H1368. doi: 10.1152/ajpheart.91526.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Murakami M, Ohba T, et al. The pathological role of transient receptor potential channels in heart disease. Circ J. 2009;73:419–427. doi: 10.1253/circj.cj-08-1153. [DOI] [PubMed] [Google Scholar]
- Zaffran S, Kelly RG, Meilhac SM, et al. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004;95:261–268. doi: 10.1161/01.RES.0000136815.73623.BE. [DOI] [PubMed] [Google Scholar]