

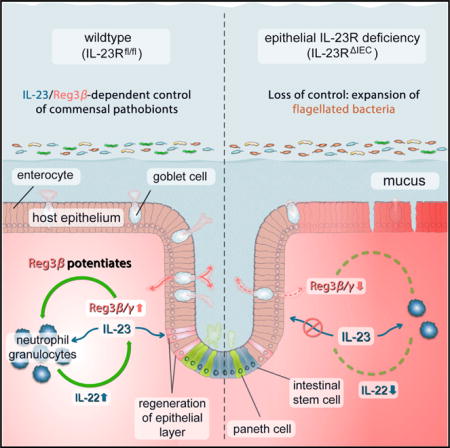
SUMMARY
A plethora of functional and genetic studies have suggested a key role for the IL-23 pathway in chronic intestinal inflammation. Currently, pathogenic actions of IL-23 have been ascribed to specific effects on immune cells. Herein, we unveil a protective role of IL-23R signaling. Mice deficient in IL-23R expression in intestinal epithelial cells (Il23RΔIEC) have reduced Reg3b expression, show a disturbed colonic microflora with an expansion of flagellated bacteria, and succumb to DSS colitis. Surprisingly, Il23RΔIEC mice show impaired mucosal IL-22 induction in response to IL-23. αThy-1 treatment significantly deteriorates colitis in Il23RΔIEC animals, which can be rescued by IL-22 application. Importantly, exogenous Reg3b administration rescues DSS-treated Il23RΔIEC mice by recruiting neutrophils as IL-22-producing cells, thereby restoring mucosal IL-22 levels. The study identifies a critical barrier-protective immune pathway that originates from, and is orchestrated by, IL-23R signaling in intestinal epithelial cells.
Graphical abstract
INTRODUCTION
Interleukin-23 (IL-23) is a heterodimeric cytokine of the IL-12 superfamily, consisting of two subunits, IL-23p19 and IL-12p40, and exerts important functions on T cell-driven immune responses in several organ systems (Parham et al., 2002). Overwhelming genetic and functional evidence has pointed to a pro-inflammatory role of the IL-23/IL-23 receptor (IL-23R) signaling pathway in inflammatory bowel disease (IBD) and other immune diseases (Croxford et al., 2012). Inhibition of IL-23 signaling has been shown to ameliorate several murine models of intestinal inflammation (Elson et al., 2007). So far, the pro-inflammatory effects of IL-23 have been ascribed to perpetuating pathogenic TH17 responses, with increased production of the TH17-associated cytokines IL-17A and IL-17F (Aggarwal et al., 2003; Leppkes et al., 2009). Several genome-wide association studies have identified a number of genetic variants in the genes encoding IL23R, IL12A (IL-12p40), JAK2, and STAT3 as independent risk (or protective) factors for both ulcerative colitis (UC) and Crohn’s disease (CD) (Barrett et al., 2008). So far, a decreased responsiveness of T cell populations to IL-23 is conferred by the presence of a single rare coding variant (R381Q, rs11209026) in the IL23R gene that is associated with decreased susceptibility for IBD (Sarin et al., 2011). Although the link between disease protection and a loss-of-function variant in IL-23 signaling is compelling, it is worth noting that the exact functional role of IL-23R signaling in intestinal homeostasis has not been defined yet, and several conundrums remain.
First, IL-23R activation in mucosal immune cells is a pivotal inducer of the cytokine IL-22, a member of the IL-10 family. IL-22 has been shown to exert protection against intestinal inflammation in either adoptive transfer, chemically induced, or infectious models of colitis (Sugimoto et al., 2008). Therefore, therapeutic inhibition of pro-inflammatory IL-23R signaling may also lead to downregulation of IL-22-dependent signals with unclear consequences.
Second, high susceptibility to experimental colitis was demonstrated in a mouse model with genetic ablation of IL23p19 (Becker et al., 2006). The paradoxical finding has been attributed to a direct cross-regulation of IL12p35/p40. Importantly, all findings reported so far have concentrated on the role of direct IL-23 signaling in leukocytes, although recent evidence points to a role of IL-23 signaling in epithelial cells types (Hasnain et al., 2014). In a previous study, we found IL-23R to be expressed in the intestinal epithelium of normal subjects and IBD patients (Raelson et al., 2007). Here we report that intestinal epithelium-specific deletion of the Il23r gene renders mice susceptible to experimental colitis by impairing IL-22-driven protective mucosal immunity against flagellated commensals.
RESULTS
Il23RΔIEC Mice Show Exacerbated Morbidity and Associated Mortality in Experimental Colitis
To initially test the ability of IL-23 to elicit a signal in intestinal epithelial cells in vitro, ModeK cells were stimulated with recombinant Il-23 (100 ng/ml), and STAT3 phosphorylation and target gene induction were assessed by western blot and real-time PCR, respectively. The experiments revealed a moderate increase in phosphorylation of STAT3 after 30 min as well as a slight (compared with the effect of Il-22, which was tested in parallel) but significant induction of the S100a9 mRNA after 6 hr. Under baseline conditions, Il23r mRNA is present at very low levels in purified murine intestinal epithelial cells, which were upregulated ~5-fold by induction of colonic inflammation (2% dextran sodium sulfate [DSS] for 3 days; Figure S1D). Using a lacZ knockout (KO) reporter mouse, which expresses β-galactosidase contained within the deletion cassette under the endogenous promoter, we further demonstrated that the Il23r promoter is active throughout the colonic epithelium (Figure S1E).
We generated a conditional allele of the Il23r gene (Il23rflox mice) (Figure S2A) and crossed Il23rfl/fl mice to C57Bl/6VillinCre mice to obtain a specific deletion of Il23r in the intestinal epithelium (termed Il23RΔIEC hereafter) and corresponding IL-23Rfl/fl (Il23Rfl) littermates, as depicted in the breeding scheme (Figure S2C). Continuous genotyping revealed no evidence for aberrant germline deletion (Figure S2B). Genotypes were confirmed using tail, tissue, and fecal DNA genotyping (Figure S2D). Although Il23r mRNA in purified intestinal epithelial cell fractions from Il23RΔIEC mice was below the detection threshold (Figure S2E), it was present in Il23Rfl animals at low levels (~30-fold lower compared with lamina propria mononuclear cells). Furthermore, to test the impaired signaling downstream of epithelial IL-23R in vivo, we injected 1.5 μg of Il-23 (or PBS as sham) intraperitoneally (i.p.) and assessed epithelial STAT3 phosphorylation 1 hr later. Only in IL-23-injected colonic epithelial cells in Il23Rfl, but not in Il23RΔIEC, mice could a significant increase of nuclear pSTAT3 immunoreactivity be observed, thereby providing further evidence for direct epithelial IL-23R activity, which is inactivated in Il23RΔIEC mice (Figures S1F and S1G).
Macromorphological and histological analysis of the small and large intestine of Il23RΔIEC mice did not reveal any spontaneous abnormalities apart from a slight reduction in goblet cells (Figure S3), as reported earlier for the constitutive knockout (Benham et al., 2014).
When Il23RΔIEC mice were exposed to chronic DSS (Wirtz et al., 2007), fulminant colitis was observed in DSS-treated Il23rΔIEC animals based on clinical and histological assessment (Figure 1). The survival rate of mutant animals declined to 50% in Il23RΔIEC mice compared with 100% survival of Il23Rfl mice (Figure 1B). Lower numbers of proliferating epithelial cells shown by bromodeoxyuridine (BrdU) labeling (Figures 1E and 1F) and reduced expression of the Reg3g and Reg3b transcripts of proliferation-associated markers of STAT3 activation in intestinal epithelial cells (IECs) (Figure 1I) were present in inflamed colon tissue of Il23RΔIEC mice compared with Il23Rfl littermates. The results were phenocopied in an additional DSS protocol (Wirtz et al., 2007) with longer treatment cycles (Figure S4), together demonstrating an unexpected protective role of epithelial IL-23R signaling against intestinal inflammation.
Figure 1. Increased Lethality and Disease Activity in Il23RΔIEC Mice in Chronic Intestinal Inflammation.
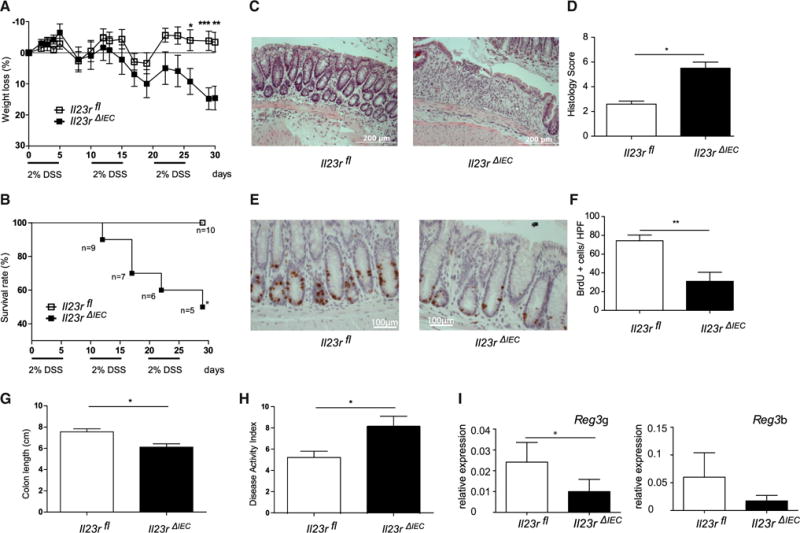
Colitis was induced by cyclic administration of 2% DSS (n = 10 male animals/group).
(A and B) Weight loss (A) and overall survival (B) were monitored every other day until day 30.
(C) Representative histologies (H&E) of DSS-treated Il23rΔIEC and Il23rfl mice on day 30.
(D) The histological score displays the combined score of inflammatory cell infiltration and tissue damage.
(E) Colonic sections from Il23rΔIEC and Il23rfl mice were immunostained with antibodies for BrdU.
(F) Positively stained cells were counted per high-power field (HPF, 4003).
(G) The columns represent the median of the colon length obtained from 10 animals/group postmortem on day 30.
(H) Overall disease activity index on day 30.
(I) Colon transcript levels of STAT3-dependent proliferation-associated Reg3b and Reg3g mRNA in Il23rΔIEC (n = 10 males) and IL23Rfl (n = 10 males) mice (qRT-PCR).
Significance was determined using two-tailed Student’s t test and is expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Epithelial IL-23R Signaling Controls the Abundance of Flagellated Intestinal Bacteria
To determine the effect of epithelial Il23R signaling on gut microbiome composition, we performed 16S rDNA profiling of Il23RΔIEC and Il23Rfl feces before and after DSS treatment. Principle coordinate analysis (PcoA) on Bray Curtis distances revealed four distinct clusters (Figure 2A) based on genotype and treatment. At higher taxonomical levels, the major bacterial phylum Firmicutes (Figure S5) showed a significantly increased abundance in naive (p = 0.010) and DSS-treated (p = 0.041) Il23RΔIEC mice compared with control littermates. Interestingly, overall mostly flagellated bacterial groups (e.g., Lachnospiraceae, Helicobacter, Escherichia/Shigella, Clostridium groups) were more prevalent in Il23RΔIEC mice with further expansion upon DSS treatment (Figure 2B). Functional metagenomic prediction using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) algorithm (Langille et al., 2013) inferred a significant increase of genes assigned to flagellar assembly pathways in Il23RΔIEC compared with Il23Rfl mice at baseline and after DSS treatment (Figure 2C). Using a HEK-Blue Toll-like receptor (TLR)-5 reporter assay (Cullender et al., 2013), we indeed confirmed an increased abundance of biologically active flagellin in Il23rΔIEC compared with Il23rfl feces in response to DSS treatment (Figure 2D). Importantly, co-housing Il23RΔIEC mice with an excess of Il23Rfl littermates could correct disease activity and flagellar expansion, indicating that altered microbiota significantly contributes to the increased disease risk of Il23RΔIEC mice (Figures S5B and S5C).
Figure 2. Epithelial IL-23R Controls Microbial Expansion of Flagellated Bacteria in DSS Colitis.
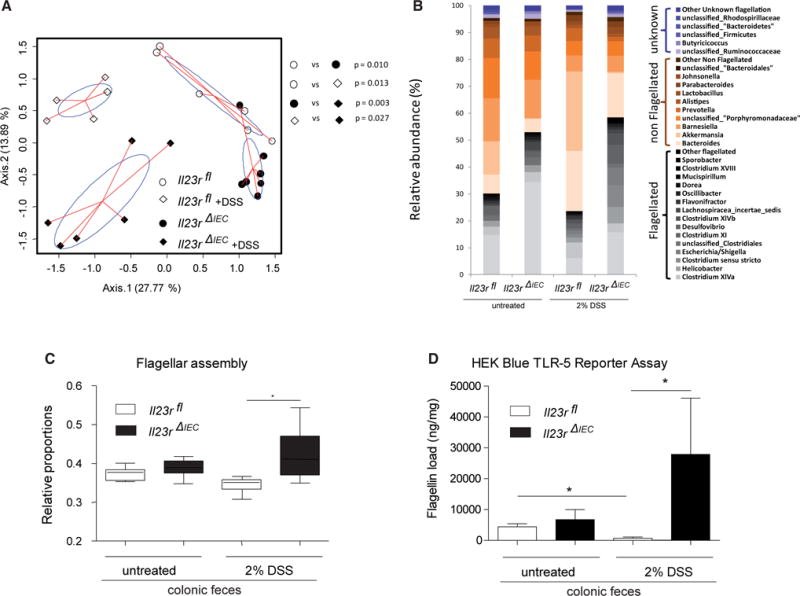
(A) PcoA of 16S rDNA sequences was performed on Bray Curtis distance matrices calculated on log-transformed species level (97% similarity) operational taxonomic unit (OUT) abundance data. p Values were generated by NPMANOVA.
(B) Abundances of dominant bacterial groups (genus level) before and after DSS treatment in the feces of Il23rfl and Il23rΔIEC mice according to inferred flagellation status (flagellated, non-flagellated, or unknown; see the Supplemental Experimental Procedures for details) in response to DSS.
(C) Bar plot showing the relative proportion of genes assigned to flagellar assembly pathways in Il23rfl and Il23rΔIEC animals at baseline and after DSS treatment (PICRUst algorithm). The median, lower, and upper quartiles are shown. Asterisks indicate significant differences (one-way ANOVA followed by Tukey-Kramer multiple comparison test and Welch’s test for two groups (*p < 0.05). See the Supplemental Experimental Procedures for details regarding phylogenomic analyses.
(D) Load of fecal flagellin measured in Il23rfl and Il23rΔIEC mice before and after induction of colitis as measured by TLR5 reporter assay (HEK Blue cells) (Cullender et al., 2013). Data are representative of minimum of n = 3 male animals per group.
Epithelial IL-23 Signaling Licenses Mucosal IL-22 Production
The intestinal epithelium interacts with immune cells that critically contribute to tissue regeneration by producing IL-22 (Sanos et al., 2011). We thus hypothesized that altered mucosal IL-22 levels may contribute to the disease risk in Il23RΔIEC mice. Indeed, ex vivo stimulation with IL-23 led to strong IL-22 mRNA upregulation (Figure 3A) in crude small intestinal crypts prepared from Il23Rfl animals, which was significantly attenuated in crypts from Il23RΔIEC mice. Known IL-22-expressing cells in the gut consist of a heterogeneous cell population comprising Th17 cells, innate-type immune cells (e.g., natural killer [NK] cells, innate lymphoid cells) (Buonocore et al., 2010; Cox et al., 2012) and neutrophilic granulocytes (Zindl et al., 2013). Because isolated splenocytes showed indistinguishable IL-22 induction between Il23Rfl and Il23RΔIEC animals (Figure 3C), we asked whether extravasation of IL-22-producing cells may contribute to different IL-22 levels in Il23Rfl and Il23RΔIEC mice. Because treatment with the αThy-1 antibody inhibits the extravasation of leukocytes (including T cells, innate lymphoid cells [ILCs], and neutrophils) into the lamina propria (Wetzel et al., 2006; Cox et al., 2012), Il23Rfl and IL23RΔIEC mice were treated for 2 consecutive days with 100 or 200 μg of a Thy-1 antibody (clone T24/31), and a significant reduction of CD3+ lymphocytes (Figure 3D) as well as neutrophils (Gr1+, data not shown) was observable. IL-23-induced Il-22 expression was blunted in preparations derived from animals that underwent αThy-1 administration at a high dose, regardless of their genotype (Figure 3E), demonstrating that epithelial cells are not a direct source of IL-22 in response to IL-23.
Figure 3. Epithelial Signaling Licenses IL-22 Production.
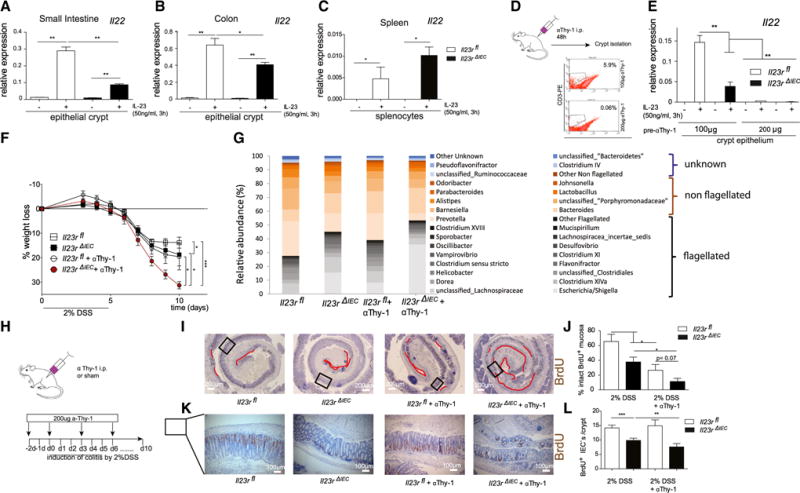
(A–C) Crude epithelial crypt preparations from the small intestine (A), colon (B), and primary splenocytes (C) from untreated mice were isolated and stimulated for 3 hr with IL-23 (50 ng/ml), and IL-22 mRNA levels were investigated by real-time PCR. Data are representative of a minimum of 3 animals/genotype and experiment.
(D) Depletion of leukocytes using anti Thy-1 antibody treatment of 100 or 200 μg (T24/31) per mouse 2 days before crypt isolation. Note that such preparations contain associated leukocytes. The effect of the Thy-1 antibody was confirmed by FACS staining of small intestine lamina propria CD3+ cells.
(E) Isolated cell fractions were stimulated for 3 hr with IL-23 (50 ng/ml), and gene expression of IL-22 was assessed.
(F) Weight loss was monitored from day 3 every day until day 10.
(G) Increased abundance of flagellated bacteria upon aThy-1 administration in Il23RΔIEC mice.
(H) Colitis was induced by administration of 2% DSS in the following groups: Il23Rfl (n = 6; 3 male [m], 3 female [f]), Il23RΔIEC (n = 7; 4 m, 3 f), Il23Rfl +aThy1 (n = 5; 3 m, 2 f), and Il23RΔIEC + aThy1 (n = 5; 4 m, 1 f). Anti-Thy-1 administration occurred 48 hr prior to DSS induction, on day 0, and then every third day by injection (i.p.) of 200 μg αThy-1 or IgG control.
(I and K) Colon Swiss rolls from Il23Rfl, Il23RΔIEC, Il23Rfl +aThy1, and Il23RΔIEC + aThy1 mice were immunostained with antibodies for BrdU. Representative pictures are shown with red lines indicating the area of BrdU absent cells in the colon mucosa (I) or the number of BrdU+ cells per crypt (L).
(J and L) Statistical assessment of intact BrdU+ mucosa (see Experimental Procedures) (K) and BrdU+ cells per crypt (M) in colon Swiss rolls reveals a dramatic reduction of proliferation in Il23RΔIEC and Il23RΔIEC + aThy1 mice
Significance was determined using two-tailed Student’s t test and is expressed as the mean ±± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Subsequently, we analyzed the effect of αThy-1 treatment as an in vivo model to reduce the mucosal IL-22 level in DSS-induced colitis (Cox et al., 2012). Il23Rfl and Il23RΔIEC mice were treated prior to induction of DSS colitis (2%) and then every 3 days with 200 μg αThy-1 (Il23Rfl +αThy1; Il23RΔIEC + αThy1) or an immunoglobulin G (IgG) (Il23Rfl, Il23RΔIEC) control (clone LTF-2) (Figure 3H). Il23RΔIEC animals treated with αThy1 antibody became moribund shortly after induction with the 2% DSS regimen, as determined by a dramatic weight loss over the first 10 days, compared with αThy1-treated Il23Rfl mice (Figure 3F). This was accompanied by impaired intestinal proliferation because the length of the BrdU+ epithelial lining (Figures 3I and 3J) and the number of BrdU+ cells per crypt (Figures 3K and 3L) were both reduced in Il23RΔIEC mice, which was further deteriorated by αThy-1 treatment. In line with previous reports on IL-22-deficient mice (Kreymborg et al., 2007; Zheng et al., 2008), αThy-1 treatment in Il23RΔIEC mice increased the abundance of flagellated (54.38% in Il23RΔIEC and only 39.41% in Il23Rfl mice) bacterial groups (Figure 3G).
To demonstrate the direct role of IL-22 in the setting, mice were pretreated with αThy-1 prior to DSS administration as described above. αThy-1-treated Il23RΔIEC mice were then either supplemented with PBS (+PBS) or recombinant (rec.) IL-22 (+IL-22) every other day and sacrificed on day 10. Notably, exogenous IL-22 completely rescued DSS-induced weight loss (Figure 4A), increased colon length (Figure 4D), reduced the abundance of flagellated bacteria (data not shown), and improved wound healing (as measured by BrdU staining and statistical assessment of the BrdU+ area) (Figures 4B and 4C), indicating that lack of IL-22 is indeed a decisive element for the observed phenotype in Il23RΔIEC mice. The findings indicate that coordination of both the epithelial renewal and the composition of the microbiota relies on the presence of functional IL-23R in intestinal epithelial cells, which influences optimal mucosal IL-22 secretion.
Figure 4. IL-22 Rescues Thy-1-Treated Il23RΔIEC Mice through Restoration of Epithelial Regeneration.
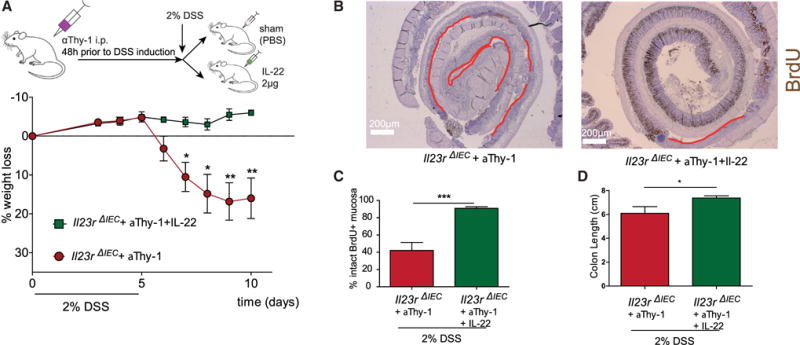
DSS colitis and aThy-1 administration were induced in Il23RΔIEC + IL-22 (n = 5; 3 m, 2 f) and Il23RΔIEC + PBS (n = 5; 3 m, 2 f) mice as described in Figure 4. Mice received 2μg rec. IL-22 on day 0 and every second day or an equivalent volume of PBS.
(A) Weight loss was monitored from day 3 every day until day 10. Colon Swiss rolls from Il23RΔIEC + aThy1+ IL-22 and Il23RΔIEC + aThy1 + PBS mice were immunostained with antibodies for BrdU.
(B and C) Representative pictures with red lines indicating the BrdU-negative areas in the colon mucosa (B) and corresponding statistical assessment of BrdU+ areas (proliferative area) (C).
Significance was determined using two-tailed Student’s t test and is expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Intestinal Epithelial IL-23R Activation Induces Reg3b Expression
We next addressed the potential mechanism that mediates the influence of epithelial IL-23R deficiency on IL22 production by leukocytes. From other studies, which showed direct activation effects on myelomonocytic cells (Lörchner et al., 2015), we postulated that the C-type lectin Reg3b, a known STAT3 target gene in intestinal epithelial cells could be a decisive epithelium-derived co-factor to modulate IL-22 expression.
Reg3b expression was already significantly lower in small intestinal and colonic crypts from Il23RΔIEC mice compared with Il23Rfl animals at baseline (Figure 5A) and was further induced in epithelial cells upon DSS colitis in an IL-23-dependent manner in Il23Rfl but not in Il23RΔIEC animals (Figure 5B). Reg3b could be directly induced in epithelial cells upon i.p. injection with recombinant IL-23 in Il23Rfl but not Il23RΔIEC mice (Figure 5C), and induction was mediated by epithelial STAT3, because STAT3ΔIEC mice failed to upregulate Reg3b in the colon epithelium compared with their STAT3fl littermates (Figure 5D). As an endogenous strong inducer of IL-23 (Kinnebrew et al., 2012), we further employed i.p. injection of the TLR5 ligand flagellin, which also led to upregulation of epithelial Reg3b in Il23Rfl animals (Figures S6A and S6C) but not in Il23RΔIEC mice. Notably, this regulation pattern induced by the endogenous flagellin/IL-23 model also coincided with differential IL22 mRNA and protein induction (Figures S6A and S6B). The failure to upregulate IL22 upon flagellin stimulation was not observable in mixed splenocyte cultures from Il23RΔIEC mice, thus rendering a systemic TLR5 signaling defect unlikely (data not shown).
Figure 5. Epithelial IL-23R Orchestrates Reg3b Expression.
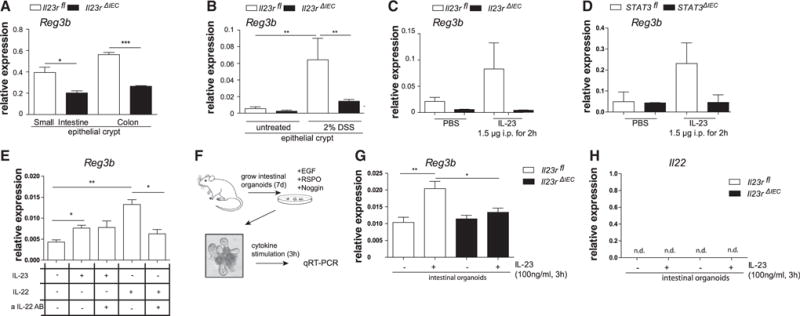
(A) Small intestinal and colon crypts were isolated, and gene expression of Reg3b was assessed. Data are representative of at least 3 animals/genotype.
(B) Expression of Reg3b in response to DSS induction was quantified in colonic epithelial cells isolated after 3 days of 2% DSS colitis induction.
(C and D) Il23rfl and Il23rΔIEC (C) and STAT3fl and STAT3ΔIEC (D) animals were injected with IL-23 (1.5 μg) for 2 hr. Colon transcript levels of Reg3b were assessed by real-time PCR.
(E) Freshly isolated epithelial cells from WT animals were stimulated with IL-23 (50 ng/ml) or IL-22 (50 ng/ml) in the presence or absence of neutralizing αIL-22 antibody (1 μg/ml) for 3 hr. Transcript levels of Reg3b were assessed by real-time PCR.
(F) Cultivation of intestinal organoids from Il23rfl and Il23rΔIEC mice. (G and H) Organoids were stimulated for 24 hr with IL-23 (100 ng/ml), and Reg3b (G) and IL-22 (H) mRNA expression was determined by real-time PCR. Significance was determined using two-tailed Student’s t test and is expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To rule out an indirect role of autocrine IL-22 as a Reg3b inducer, freshly isolated epithelial cells from wild-type (WT) animals were stimulated with IL-23 or IL-22 in the presence or absence of a neutralizing αIL-22 antibody for 3 hr. IL-23 and IL-22 both induced expression of Reg3b in intestinal epithelial cells, whereas only IL-22- but not IL-23-induced Reg3b expression was blocked by the neutralizing αIL-22 antibody (Figure 5E). Last, intestinal organoids (Figure 5F) from Il23RΔIEC and Il23Rfl (Sato et al., 2009) mice were stimulated with IL-23 for 24 hr, and mRNA transcript levels of Reg3b showed significant epithelial Reg3b expression in organoids derived from Il23Rfl but not Il23RΔIEC cultures (Figure 5G). IL-22 was not detectable, again excluding autocrine IL-22 effects on Reg3b expression (Figure 5H). These data demonstrate that IL-23 induced Reg3b in intestinal epithelial cells via epithelial cell-expressed IL-23R.
Systemic Reg3b Administration Protects Il23RΔIEC Mice from DSS-Induced Colitis by Restoring IL-22 Production and Recruitment of Neutrophils
We hypothesized that, if indeed Reg3b derived by epithelial cells is important for the observed phenotype in Il23RΔIEC mice, then systemic administration of Reg3b should protect Il23RΔIEC mice from colitis and influence IL-22 production within the inflamed mucosa. Therefore, colitis was induced by 2% DSS, and Il23RΔIEC mice received either PBS or rec. Reg3b intraperitoneally every other day for 10 days at a dose far below the concentrations that have been shown to have a direct effect on microbiota when applied luminally in earlier studies (Zheng et al., 2008). Systemic Reg3b treatment significantly improved DSS-induced body weight loss (Figure 6A), reconstituted epithelial proliferation (Figures 6B and 6C), and increased local production of IL-22 in Reg3b-treated colon explant cultures (Figure 6F). Given that mucosal IL-22 secretion negatively regulates the expansion of flagellated β-proteobacteria, we next analyzed the bacterial composition of the fecal microbiota using 16S rRNA sequencing. A Bray Curtis PcoA plot showed significant (nonparametric multivariate ANOVA [NPMANOVA], p = 0.0089) differentiation between untreated and Reg3b-treated Il23RΔIEC mice (Figure 6G), and systemic Reg3b treatment resulted in a significant decrease of flagellated bacteria in the colonic feces of Il23RΔIEC mice (Figure 6H).
Figure 6. Systemic Reg3b Substitution Rescues Il23RΔIEC Mice by Increasing Local IL-22 Production.
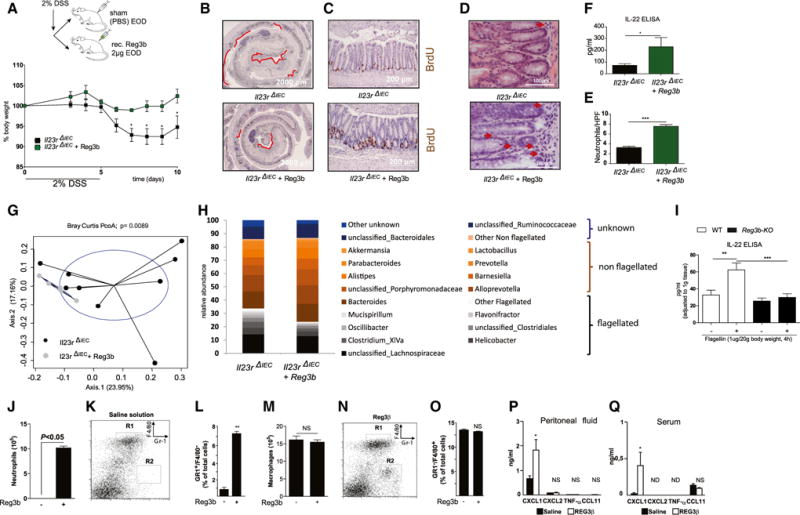
DSS colitis was induced in Il23RΔIEC + Reg3b (n = 5; 3 m, 2 f) and Il23RΔIEC + (n = 9; 5 m, 4 f) mice as described above. Mice received 2 μg rec. Reg3b or an equivalent volume of PBS i.p. on day 1 and every second day.
(A) Weight loss was monitored from day 3 every day until day 10.
(B) Colon Swiss rolls from Il23RΔIEC and Il23RΔIEC + Reg3b mice were immunostained with antibodies for BrdU. Representative pictures are shown, with red lines indicating the BrdU-negative area.
(C) Representative pictures of colonic sections stained with anti-BrdU+ antibody.
(D and E) Representative H&E pictures of colon showing increased neutrophil influx in Reg3b-treated mice (D) and statistical evaluation of counted neutrophils per HPF (E).
(F) IL-22 ELISA of colon explants from Il23RΔIEC+Reg3b versus Il23RΔIEC mice.
(G) Principle coordinate analysis (Bray Curtis distance matrices) of fecal flora obtained by 16S rDNA V4 sequencing from Il23RΔIEC+Reg3b versus Il23RΔIEC animals after DSS treatment (see the Supplemental Experimental Procedures for details).
(H and I) Abundances of dominant bacterial groups (genus level) after DSS treatment in the feces of Il23RΔIEC+ Reg3b versus Il23RΔIEC mice according to the flagellatin status (flagellated, non-flagellated, or unknown) in response to DSS (H), IL-22 ELISA from WT or Reg3b KO explants after stimulation with 1 μg ultrapure flagellin for 2 hr i.p. (I).
(J–Q) Peritoneal exudates of five wild-type mice were harvested 6 hr after intraperitoneal injection of either sterile saline solution (K) or 100 μg/kg of purified, endotoxin-free Reg3b (N), and relative numbers of neutrophils or macrophages were assessed by cytospin (J and M), or Gr-1 and F4/80 staining was quantified by FACS analysis (K, L, N, and O). Representative flow cytometric analyses (K and N) of peritoneal cell populations and statistical analysis (L and O) show that Reg3b triggers the influx of neutrophils. Also shown is an ELISA on the indicated cytokines on peritoneal fluid (P) and sera (Q) after Reg3b stimulation as described above. Significance was determined using two-tailed Student’s t test and is expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Because we had excluded the possibility of a direct synergistic effect of Reg3b stimulation on IL-23-induced IL22 expression by co-stimulation experiments in crude crypt preparations (data not shown), we next tested the hypothesis whether endogenous Reg3b could serve as a chemoattractant principle for IL-22-producing cells also in the intestine because expression of Reg3b was recently described to control the accumulation of activated immune cells in myocardial ischemia (Lörchner et al., 2015). Indeed, Il23RΔIEC mice exhibited reduced numbers of Thy-1+, CD3+, and CD4+ cells in the small intestinal lamina propria compared with their Il23Rfl littermates under baseline conditions (Figures S7A–S7C). We specifically observed diminished numbers of neutrophilic granulocytes in the colonic lamina propria, as indicated in fluorescence-activated cell sorting (FACS) staining (Gr-1, Figure S7D) and colon histology of untreated IL23RΔIEC compared with Il23Rfl mice (Figures S7E and S7F). Neutrophil numbers were notably increased in the lamina propria of Reg3b-treated Il23RΔIEC animals despite the clear anti-inflammatory effect of Reg3b treatment (Figures 6D and 6E).
To further test the chemotactic role of Reg3b for immune cells, WT mice were injected i.p. with PBS or recombinant endotoxin-free Reg3b, and peritoneal cells were collected. Cytospin and FACS analysis of peritoneal cell composition showed a significant influx of neutrophils 6 hr after Reg3b administration, whereas the level of macrophages, eosinophils, and basophils remained unchanged (Figures 6J–6O and data not shown). In line with a positive influence of Reg3b on chemotaxis, Cxcl1 production was also increased in peritoneal fluids (Figure 6P) and sera (Figure 6Q) after Reg3b treatment. Finally, we could show that Reg3b KO mice produce less IL-22 in response to flagellin compared with similarly treated WT littermate controls (Figure 6I).
Taken together, these data provide evidence for a mechanism involving IL-23R-dependent epithelial Reg3b expression, which is needed for adequate attraction of IL-22 secreting cells, most notably neutrophils. In addition to a direct regenerative effect, this mechanism may help to control colonic microbiota composition.
DISCUSSION
Intestinal homeostasis requires a balanced interplay of a plethora of cell types. A growing body of evidence highlights the key role of transducing tonic signals from commensal bacteria into cytokine-mediated epithelial regenerative programs (Veldhoen et al., 2008; Martin et al., 2009; Mielke et al., 2013; Leung et al., 2014). So far, the epithelium has been mainly thought to “passively” receive cytokine signals and transduce them into an antimicrobial and regenerative response that is known to be dependent on STAT3 (Pickert et al., 2009).
Here we demonstrate that IL-23-dependent activation of the intestinal epithelium serves as a crucial modulatory factor toward an orchestrated immune response from epithelial cells that subsequently control antimicrobial and regenerative processes in the inflamed mucosa.
It must be noted that the expression levels of IL-23R in intestinal epithelial cells are low compared with immune cells. Although we provide evidence for direct signaling and IL-23R-dependent target gene expression in IECs and Il23r mRNA upregulation during intestinal inflammation, the different expression patterns between the epithelial compartment and immune cells warrant an adequate discussion of the potential pitfalls of the model. We have controlled for the precision of the Cre excision by the villin-Cre line in Il23RΔIEC mice both by demonstrating the genomic deletion, which is only present in IECs but not in other cellular compartments, as well as by showing that mixed immune cells (splenocytes) from Il23RΔIEC mice respond functionally normal to IL-23 stimulation. In an independent cross for which we used the lysM-Cre line, deletion of the Il23r gene in the myeloid compartment caused a clear impairment of IL-23-dependent target gene expression in CD11c+ cells (unpublished data), demonstrating that cre-mediated excision is principally also able to impair IL-23R-dependent signals in another genetic model. The employed villin-cre line has been characterized extensively in many studies, making an inadequate activation of the promoter in specific immune cells extremely unlikely as the underlying cause of the phenotype. We thus think that, despite the low level of expression, it is justified to assume that the observed protective effects are directly linked to an activation of IL-23R on intestinal epithelial cells.
We report that Il23RΔIEC mice rapidly succumb to chronic DSS-induced colitis because of reduced wound healing and impaired IL-22-mediated protective immune responses. Il23RΔIEC animals significantly differ in their composition of the gut microbiota with expansion of flagellated bacterial genera. Interestingly, the expansion of flagellated bacterial taxa became even more pronounced upon DSS challenge or inhibition of leukocyte extravasation and correlated with the severity of colitis. These observations agree with findings that the abundance of flagellated intestinal bacteria is a critical factor for the severity of experimental colitis (Rhee et al., 2005; Vijay-Kumar et al., 2007; Ivison et al., 2010). One may therefore presume that the expansion of flagellated taxa in Il23RΔIEC mice may result from the lack of flagellin-induced IL22 secretion, which is known to require endogenous IL-23 from dendritic cells (Kinnebrew et al., 2012). Recent studies in Il22−/− mice (Zenewicz et al., 2013; Behnsen et al., 2014) and our own data reconstituting Il23RΔIEC mice with exogenous IL-22 corroborate this hypothesis. As in many other DSS-driven models, the altered microbiota augments the inflammatory phenotype observed in Il23RΔIEC mice, whereas co-housing with an excess of floxed littermates rescues Il23RΔIEC mice from disease risk.
Our data indicate that epithelial IL-23R signaling exerts pivotal control over mucosal IL-22 secretion through a newly described mechanism. Interestingly, our screening for potential chemokines secreted by the epithelium revealed that IL-23-dependent secretion of the c-type lectin Reg3b is likely to act as a paracrine factor amplifying IL-22 production in the lamina propria. The few studies on the effect of Reg3 proteins on intestinal mucosal immunity describe mainly a direct antimicrobial activity because mice deficient in Reg3g display a different mucus architecture (Vaishnava et al., 2011), and luminal application of high doses of recombinant Reg3g seems to reduce colonization with Salmonella (van Ampting et al., 2012). However, the exclusive direct antimicrobial effect is much less clear for Reg3b. The fact that genetic ablation of Reg3b in mice leads to prolonged colonization with Y. pseudotuberculosis in ileal Peyer’s patches without affecting luminal bacterial levels indicates a potential effect of Reg3b beyond its presence in the intestinal mucus (Dessein et al., 2009; van Ampting et al., 2012). A very recent report in the ischemic heart points toward a role of Reg3b in mediating macrophage trafficking to control protective immune cell infiltration into the affected heart tissue (Lörchner et al., 2015). Furthermore, Reg3b is skewing macrophage polarization through an unknown mechanism (Gironella et al., 2013).
Although we could not demonstrate a direct synergistic effect of Reg3b on IL-23-mediated induction of IL22 expression in vitro, we show by intraperitoneal injection that the protein acts as a chemoattractant for neutrophils. Interestingly, neutrophils have recently been reported to serve as a source for mucosal IL-22 in DSS-induced inflammation (Zindl et al., 2013). In line with the idea that epithelium-derived Reg3b could control mucosal levels of IL22-producing neutrophils, intraperitoneal Reg3b administration attenuated the increased clinical susceptibility to DSS-induced inflammation in Il23RΔIEC mice. Under such conditions, Reg3b injection clearly upregulated neutrophil numbers in the lamina propria and restored IL-22-secretion in Il23RΔIEC animals.
Thereby we demonstrate that IL-23R-dependent Reg3b triggers an influx of IL-22-producing immune cells. It remains to be determined whether the alarmin Reg3b may dictate the recruitment of only one or also other IL-22-producing cell types; however, our data clearly point to neutrophils as a plausible candidate. It also remains to be determined whether Reg3b is actively secreted on the basolateral side in the physiological setting or whether it may only enter the lamina propria via epithelial barrier defects. Taken together, we could show that the epithelial IL-23R licenses mucosal IL-22 production and serves as a critical factor orchestrating protective mucosal immune responses in the context of microbiota-fueled inflammation. These findings have important implications for future therapeutic developments in chronic inflammatory diseases because the rationale of IL-23 neutralization is the dampening of TH17 cell activity. However, in clinical settings like IBD, where IL-22 secretion might be an important protective factor, proregenerative functions of IL-23 on epithelial cells should be kept in mind.
EXPERIMENTAL PROCEDURES
Mice
Weight- and gender-matched mice (genetic background C57Bl6JxSv129, backcrossed for at least six generations) were used at an age of 8–12 weeks for all experiments. For DSS experiments, the indicated numbers (minimum n = 5 per genotype) of animals were employed. Il23Rfl/fl mice were generated by targeted insertion of LoxP sites flanking exon 4 of the IL23R gene (Genoway). Villin-Cre animals (strain B6.SJL-Tg(Vil-cre)997Gum/J) were purchased from Jackson Laboratory. Conditional knockout of Il23R in the intestinal epithelium was established by crossing Villin-Cre with Il23Rfl/fl mice, resulting in Il23RΔIEC or IL23Rfl. It has been well established that this knockout, apart from some expression of the transgene in renal tubuli, is specific for the intestinal epithelium and does not involve immune cells in the intestine (El Marjou et al., 2004). STAT3fl/fl and STAT3ΔIEC mice were provided by Christine Watson (Cambridge University). Reg3b WT and Reg3b KO mice were kindly provided by Stephen P. Hunt (University College London). The β-galactosidase reporter mouse (Il23R knockin/knockout mouse [(il23rtm1a (EUCOMM)Wtsi mouse with lacZ cassette]) was kindly provided by the EUCOMM Consortium. Mice were maintained in a 12-hr light-dark cycle under standard conditions and were provided with food and water ad libitum. Procedures involving animal care were conducted according to national and international laws and policies and with appropriate permission. Appropriate permission was given by the local animal safety review board of the federal ministry of Schleswig Holstein.
Antibodies and Reagents
Murine αThy-1 (clone T24/31) was purchased from BioXcell. Flagellin (FLA-ST ultrapure) was purchased from InvivoGen. Recombinant murine Reg3b was purchased from R&D Systems. Recombinant murine IL-22 was purchased from PeproTech, and recombinant murine IL-23 was purchased from BioLegend.
FACS
Flow cytometry was performed on a FACSCalibur (BD Biosciences), and data were analyzed using CellQuest software (BD Biosciences) using standard parameters and antibodies (details can be found in the Supplemental Experimental Procedures).
Isolation of Primary Cells and Cultivation of Intestinal Organoids
Murine intestinal epithelial cells and lamina propria cells were isolated using the Hank’s balanced salt solution (HBSS)-EDTA method followed by enzymatic digestion. Mouse intestinal organoids were cultivated as described before (Sato et al., 2009). For details, see the Supplemental Experimental Procedures.
Induction of Colitis, Determination of Clinical Scores, and Histology
For chronic colitis induction, mice were supplied with 2% of DSS (molecular mass, 40 kDa; TdB Consultancy) dissolved in drinking water for 5 days, followed by 5 days of regular drinking water. The disease activity index (DAI) was obtained as described previously (Siegmund et al., 2001). Consumption of drinking water was measured daily. A high-resolution mouse video endoscopic system was used on day 30 of the experiment (Karl Storz AIDA VET), and endoscopic scores were obtained as described previously (Wirtz et al., 2007). For induction of short colitis, mice were supplied with 2% DSS dissolved in drinking water for 3 days and sacrificed immediately for intestinal epithelial cell isolation as described elsewhere (Pickert et al., 2009). Histopathological analyses were performed according to standard methods (see the Supplemental Experimental Procedures for details).
Colon Culture
Colons were washed in cold PBS supplemented with penicillin and streptomycin (Gibco) and cut longitudinally. Four segments of 0.5-cm length were cultured in 24-well flat-bottom culture plates in RPMI 1640 medium (Gibco) for 24 hr at 37°C. Supernatants were analyzed for IL-22 concentration using an IL-22 ELISA kit according to the manufacturer’s protocol(PeproTech).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (version 4.0) for Windows software (GraphPad). Statistical significance was evaluated by Mann-Whitney U test for nonparametric data or Student’s t test for parametric data unless indicated otherwise. p values less than 0.05 were considered statistically significant.
Supplementary Material
In Brief.
Aden et al. show that epithelial IL-23R signaling initiates a Reg3b-dependent chemoattraction of IL-22-producing neutrophil granulocytes into the intestinal lamina propria, limiting flagellated bacteria content and intestinal inflammation.
Highlights.
IL-23R transduces signals into the intestinal epithelium
Il23RΔIEC mice are susceptible to DSS colitis and have a disturbed gut microflora
Epithelial IL-23R is required for optimal secretion of the c-type lectin Reg3b
The c-type lectin Reg3b promotes recruitment of IL-22-producing cells as an alarmin
Systemic substitution of Reg3b rescues the gut barrier defect of Il23RΔIEC mice
Acknowledgments
We gratefully appreciate the technical assistance of Maren Reffelmann, Tanja Klostermeier, Manuela Kramp, Katharina Schwarzer, Sabine Kock, and Tatjana Schmidtke. We appreciate the design of the graphical abstract by Lennart Lenk. This work was supported by DFG Excellence Cluster Inflammation at Interfaces; the SFB877 B9, the SFB 1182 C2 project, and the BMBF IHEC DEEP project TP2.3 and 5.2 (to P.R.); the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement 260961 (to A.K.); the National Institute for Health Research Cambridge Biomedical Research Centre, ERC CoG GA 648889, and WTIA 106260-Z-14-Z (to A.K.); NIH DK53056, DK44319, and DK088199 (to R.S.B.); and the Fondation pour la Recherche Medicale (to M.C.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.054.
AUTHOR CONTRIBUTIONS
K.A. and P.R. conceived and designed the study; acquired, analyzed, and interpreted the data; performed statistical analysis; and wrote the manuscript. A.R., M.F.P., J.K., F.T., S.P., A.B., A.L., M.J., R.H., R.S.T., S.B.B., O.W., S.L., T.A., J.I., T.S., and S.L.K. performed data acquisition, analysis, and interpretation. K.A, R.S.B., B.B., M.C., A.K., S.S., and P.R. contributed to the interpretation of data and writing of the manuscript. A.R., M.F-P. and T.S. contributed equally.
References
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Dornhoff H, Neufert C, Fantini MC, Wirtz S, Huebner S, Nikolaev A, Lehr HA, Murphy AJ, Valenzuela DM, et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 2006;177:2760–2764. doi: 10.4049/jimmunol.177.5.2760. [DOI] [PubMed] [Google Scholar]
- Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, Raffatellu M. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40:262–273. doi: 10.1016/j.immuni.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham H, Rehaume LM, Hasnain SZ, Velasco J, Baillet AC, Ruutu M, Kikly K, Wang R, Tseng HW, Thomas GP, et al. Interleukin-23 mediates the intestinal response to microbial β-1,3-glucan and the development of spondyloarthritis pathology in SKG mice. Arthritis Rheum. 2014;66:1755–1767. doi: 10.1002/art.38638. [DOI] [PubMed] [Google Scholar]
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JH, Kljavin NM, Ota N, Leonard J, Roose-Girma M, Diehl L, Ouyang W, Ghilardi N. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012;5:99–109. doi: 10.1038/mi.2011.54. [DOI] [PubMed] [Google Scholar]
- Croxford AL, Mair F, Becher B. IL-23: one cytokine in control of autoimmunity. Eur J Immunol. 2012;42:2263–2273. doi: 10.1002/eji.201242598. [DOI] [PubMed] [Google Scholar]
- Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessein R, Gironella M, Vignal C, Peyrin-Biroulet L, Sokol H, Secher T, Lacas-Gervais S, Gratadoux JJ, Lafont F, Dagorn JC, et al. Toll-like receptor 2 is critical for induction of Reg3 β expression and intestinal clearance of Yersinia pseudotuberculosis. Gut. 2009;58:771–776. doi: 10.1136/gut.2008.168443. [DOI] [PubMed] [Google Scholar]
- El Marjou F, Janssen KP, et al. Tissue-specific and inducible Cremediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- Gironella M, Calvo C, Fernández A, Closa D, Iovanna JL, Rosello-Catafau J, Folch-Puy E. Reg3β deficiency impairs pancreatic tumor growth by skewing macrophage polarization. Cancer Res. 2013;73:5682–5694. doi: 10.1158/0008-5472.CAN-12-3057. [DOI] [PubMed] [Google Scholar]
- Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen AC, Loudovaris T, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. 2014;20:1417–1426. doi: 10.1038/nm.3705. [DOI] [PubMed] [Google Scholar]
- Ivison SM, Himmel ME, Hardenberg G, Wark PA, Kifayet A, Levings MK, Steiner TS. TLR5 is not required for flagellin-mediated exacerbation of DSS colitis. Inflamm Bowel Dis. 2010;16:401–409. doi: 10.1002/ibd.21097. [DOI] [PubMed] [Google Scholar]
- Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, Flavell RA, Littman DR, Pamer EG. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–8104. doi: 10.4049/jimmunol.179.12.8098. [DOI] [PubMed] [Google Scholar]
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Leung JM, Davenport M, Wolff MJ, Wiens KE, Abidi WM, Poles MA, Cho I, Ullman T, Mayer L, Loke P. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 2014;7:124–133. doi: 10.1038/mi.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lörchner H, Pöling J, Gajawada P, Hou Y, Polyakova V, Kostin S, Adrian-Segarra JM, Boettger T, Wietelmann A, Warnecke H, et al. Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat Med. 2015;21:353–362. doi: 10.1038/nm.3816. [DOI] [PubMed] [Google Scholar]
- Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117–1124. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raelson JV, Little RD, Ruether A, Fournier H, Paquin B, Van Eerdewegh P, Bradley WE, Croteau P, Nguyen-Huu Q, Segal J, et al. Genome-wide association study for Crohn’s disease in the Quebec Founder Population identifies multiple validated disease loci. Proc Natl Acad Sci USA. 2007;104:14747–14752. doi: 10.1073/pnas.0706645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH, Im E, Riegler M, Kokkotou E, O’brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci USA. 2005;102:13610–13615. doi: 10.1073/pnas.0502174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanos SL, Vonarbourg C, Mortha A, Diefenbach A. Control of epithelial cell function by interleukin-22-producing RORγt+ innate lymphoid cells. Immunology. 2011;132:453–465. doi: 10.1111/j.1365-2567.2011.03410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci USA. 2011;108:9560–9565. doi: 10.1073/pnas.1017854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 β-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci USA. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ampting MTJ, Loonen LMP, Schonewille AJ, Konings I, Vink C, Iovanna J, Chamaillard M, Dekker J, van der Meer R, Wells JM, Bovee-Oudenhoven IM. Intestinally secreted C-type lectin Reg3b attenuates salmonellosis but not listeriosis in mice. Infect Immun. 2012;80:1115–1120. doi: 10.1128/IAI.06165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, Gewirtz AT. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel A, Wetzig T, Haustein UF, Sticherling M, Anderegg U, Simon JC, Saalbach A. Increased neutrophil adherence in psoriasis: role of the human endothelial cell receptor Thy-1 (CD90) J Invest Dermatol. 2006;126:441–452. doi: 10.1038/sj.jid.5700072. [DOI] [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, Flavell RA. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol. 2013;190:5306–5312. doi: 10.4049/jimmunol.1300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. IL-22–producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA. 2013;110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.