

Abstract
Bromodomains (BD) are readers of lysine acetylation marks present in numerous proteins associated with chromatin. Here we describe a dual inhibitor of the bromodomain and PHD finger (BRPF) family member BRPF2 and the TATA box binding protein-associated factors TAF1 and TAF1L. These proteins are found in large chromatin complexes and play important roles in transcription regulation. The substituted benzoisoquinolinedione series was identified by high-throughput screening, and subsequent structure–activity relationship optimization allowed generation of low nanomolar BRPF2 BD inhibitors with strong selectivity against BRPF1 and BRPF3 BDs. In addition, a strong inhibition of TAF1/TAF1L BD2 was measured for most derivatives. The best compound of the series was BAY-299, which is a very potent, dual inhibitor with an IC50 of 67 nM for BRPF2 BD, 8 nM for TAF1 BD2, and 106 nM for TAF1L BD2. Importantly, no activity was measured for BRD4 BDs. Furthermore, cellular activity was evidenced using a BRPF2– or TAF1–histone H3.3 or H4 interaction assay.
Introduction
Proteins involved in chromatin modulation usually contain several functional regions that serve to recruit and tether binding partners, ultimately leading to the formation of large multiprotein complexes. A clear understanding of the function of these complexes will only be possible through the dissection of the interactions between the individual protein partners and the analysis of the exact role of their interactive modules. Different approaches are currently being used to address this. One method is the selective deletion of gene exons by CRISPR-Cas9 followed by phenotypic analyses.1 Another, complementary approach is the identification of specific domain inhibitors and evaluation of their functional impact in cellular, and in some cases, animal models.2,3 Here, great advances have recently been reported in the area of bromodomains (BDs), the best-characterized readers of acetyl-lysine (KAc) marks which are found on histones and other proteins.2−4 BDs are potentially amenable to small molecule inhibition, as impressively shown in the case of bromo and extra-terminal (BET) BD inhibitors which have been extensively used in recent years to understand the role of this family in normal physiology and pathology, ultimately paving the way for their clinical evaluation in cancer indications.3−6 In the wake of this success, several other BDs have been successfully targeted by inhibitors with very different scaffolds, further highlighting the druggability of this target family.7−14
The bromodomain and PHD finger (BRPF) family encompasses three paralogs, BRPF1, BRPF2, and BRPF3, which are found in histone acetyltransferase (HAT) complexes.15 BRPFs possess different functional modules, including C2H2, plant homeodomain-linked fingers, bromo, and PWWP domains. Mouse studies show that loss of either BRPF1 or BRPF2 leads to embryonic lethality at E9.5 and E15.5, respectively, due to different developmental defects.16,17 In sharp contrast, BRPF3 is not needed for mouse development or survival, and no compensatory expression change is observed for BRPF1 or BRPF2, in line with nonoverlapping functions.18 BRPF1 is involved in hematopoietic stem cell development and BRPF2 in embryonic stem cell differentiation.16,19 Little is known about the implication of BRPFs in disease, but a critical role of BRPF1 in the leukemia subgroup harboring the MOZ-TIF fusion has been reported.20 First BRPF BD inhibitors have been published (Figure 1). Compounds 1 (OF-1)21 and 2 (NI-57)10 are pan-inhibitors recognizing all three BRPF paralogs and derived from two distinct structure classes, benzimidazolones and quinolinones, which make them valuable complementary chemical probes. Two additional inhibitors, 3 (PFI-4) and 4 (close analog of IACS-9571), as well as (R)-N-[1,3-dimethyl-6-(2-methylpiperazin-1-yl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl]-2-methoxybenzamide (GSK6853) and N-[2,3-dihydro-1,3-dimethyl-2-oxo-6-(1-piperidinyl)-1H-benzimidazol-5-yl]-2-methoxybenzamide (GSK5959), are derived from the 1,3-dimethyl-benzimidazolone scaffold and exhibit potent and selective binding to BRPF1.22−24 Compound 4 shows additional strong activity on TRIM24, a nuclear receptor cofactor associated with different cancer types,25 being therefore a dual BRPF1/TRIM24 inhibitor.21,23 Altogether, these findings demonstrate the high usefulness of the 1,3-dimethylbenzimidazolone headgroup as a KAc mimetic binding motif and also suggest that the addition of novel decorations to this scaffold may result in a different selectivity profile.21,22,24 To the best of our knowledge, however, no compounds showing selectivity for BRPF2 BD over its two paralogs have yet been reported.
Figure 1.

Comparison of 5 with previously described BRPF inhibitors.
Several TATA-binding protein-associated factors (TAFs) have been identified in the basal transcription factor TFIID. TAF1 is the largest subunit of the complex and represents the essential scaffold to which the other TAFs and the TATA-binding protein attach.26 Recent structural data indicate that TAF1 makes essential interactions with the core promoter DNA.27 It contains multiple modules, including a kinase motif, a ubiquitin-conjugating activity, a HAT region, and two related BDs. This modular organization is also found in the highly related paralog TAF1L, which functionally replaces TAF1 in male germ cells.28 Mutations in the TAF1 gene are associated with neurological pathologies in affected males.29,30 However, little is known about the implication of individual TAF1 domains in these diseases or about a role of TAF1 in cancer. The recent progress reported in the area of BD inhibitors suggests that addressing this module may be a good starting point for functional studies. Indeed, a first paper describing a BET inhibitor with additional submicromolar TAF1 BD2 activity, named UMB-32, has recently been presented.31 This compound is derived from an imidazo-pyrazine scaffold bearing a dimethyl-isoxazole;32−36 however, its usefulness is restricted by its limited on-target activity and cross-reactivity with BET BDs, which are known to exert multiple cellular effects.2,13,37,38 Very recently, TAF1 inhibitors derived from a methyl pyrrolopyridone fragment were described.39,40 The most selective example interacts mainly with TAF1 and TAF1L BD1, but also with BRD9, BRD7, and BET BDs.
Here, we present the results of a screening and optimization strategy which allowed discovery of the 1,3-dimethylbenzimidazolone-substituted benzoisoquinolinedione 5 (BAY-299),41 which exhibits potent dual inhibitory activity for both BRPF2 BD and TAF1/TAF1L BD2 in biochemical and cellular assays. This compound is the first reported BRPF2 inhibitor with high selectivity against the BRPF1 and BRPF3 paralogs. It represents a powerful chemical tool suited for investigation of the role of two little-explored BD-containing proteins. Importantly, 5 is inactive against BET BDs, so that the generated cellular results will not be blurred by residual interference with BRD4 or its paralogs, which are known to possess numerous biological functions.38
Results and Discussion
Screening and Hit Identification
Our original aim was to identify inhibitors of BRPF BDs with cellular activity and without any activity on BET BDs. High-throughput screening (HTS) of a 3.5-million compound library was performed using a BRPF1 time-resolved fluorescence resonance energy transfer (TR-FRET) assay (Figure 2A). Confirmed hits were evaluated by dose–response analysis (IC50 determination) in BRPF1, BRPF2, and BRPF3 TR-FRET (Figure 2A). BRD4 TR-FRET was used as counter screen. This exercise yielded several overlapping hit clusters with nanomolar pan-BRPF BD activities and, surprisingly, allowed the identification of a single hit (6, Figure 2A) containing a 1,3-dimethylbenzimidazolone scaffold22 with nanomolar activity for BRPF2 while being inactive up to concentrations of 20 μM for BRPF1 (Figure 2B), BRPF3 (Figure 2C), and BRD4 (Figure 2D). The binding specificity and selectivity profile of the compound were confirmed by thermal shift assays (TSA), in which hit 6 stabilized BRPF2 with a ΔTm of 0.8 K (Figure 2E), whereas only a ΔTm of 0.2 K was measured for BRPF1 (not shown). After confirmation of sample identity and based on its favorable binding efficiency index and lipophilic ligand efficiency (BEI: 17.3; LLE: 3.7), hit 6 was selected as a starting point for further optimization cycles using additional assays (Figure 2A).
Figure 2.

Screening strategy for the discovery of 6 and identification of 5. (A) Overview of the HTS campaign leading to 6, and of the optimization process which culminated with the identification of the probe. (B–D) Potency correlation plots of HTS hits showing the BRPF2 selectivity of 5: TR-FRET IC50 values for BRPF2 (x-axis) compared with BRPF1 (B), BRPF3 (C), and BRD4 (D) IC50 values. Symbols corresponding to 6 are shown in black. (E) TSA melting curve of BRPF2 in the presence of 6 compared to vehicle.
Chemistry
One structural feature of 6 is the tricyclic decoration of the benzimidazolone at position 5 (naphthalimide substituent). Surprisingly, the first analyses revealed that 6 had a logD of 2.1 and an excellent permeability profile in Caco2 cells; furthermore, a low solubility (3.7 mg/L) was measured, which was not unexpected. Docking studies revealed that the naphthalimide moiety was responsible for the strong BRPF2 selectivity, as it forms key interactions which are not possible with BRPF1 (see Figure 3 cocrystal structure discussion). That this bulky substituent is indeed essential was confirmed by our early attempts to reduce its molecular size or aromatic architecture, which always led to less potent and less selective compounds (data not shown). Based on this binding mode hypothesis, we initiated an optimization program around the BRPF2-selective HTS hit 6.
Figure 3.

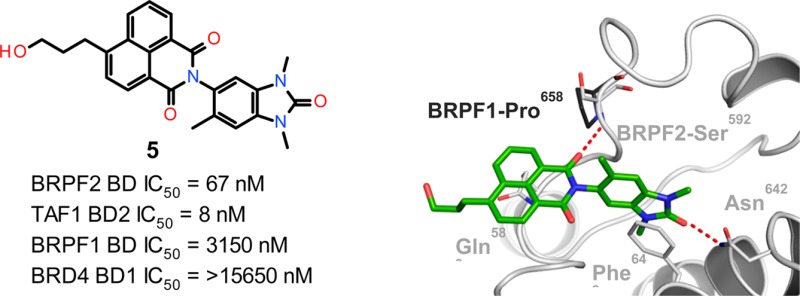
Binding mode of 7 in BRPF2 BD and basis for selectivity. (A) Cartoon representation of BRPF2 BD domain (white) with key interacting side-chains shown in stick representation with C, N, and O atoms colored white, blue, and red, respectively. Compound 7 is also shown in stick representation with C, N, and O atoms colored green, blue, and red, respectively. Hydrogen-bonding interactions between 7 and the protein are shown as red dotted lines. (B) Overlay of the BRPF2–7 complex structure with selected residues from the BRPF1–apostructure (PDB 4LC2) (C, N, and O atoms colored dark gray, blue, and red, respectively). BRPF1 BD Pro658 is unable to form a key hydrogen bond with 7. (C) Overlay of the BRPF2–7 complex structure with selected residues from the BRD4–apostructure (PDB 2OSS) (C, N, and O atoms colored dark gray, blue, and red, respectively).
Structure–Activity Relationships
We first focused on improving in vitro BRPF2 activity. Therefore, the influence of substituents at position 6 of the 1,3-dimethylbenzimidazolone core was studied (Table 1). A significant, 4-fold improvement of the BRPF2 activity was achieved by introducing small groups which lock the central dihedral angle in its bioactive conformation; however, this was also accompanied by an increased interaction with BRPF1. The best results were obtained for the methyl- and methoxy-derivatives (compounds 7 and 8) which offered the highest selectivity for BRPF2 over BRPF1. Compounds 9 and 10 were also potent BRPF2 inhibitors but less selective toward BRPF1. Addition of the larger methylpiperazine moiety (compound 11) did not lead to an activity improvement. Importantly, all compounds demonstrated a high selectivity against BRD4 BDs. Furthermore, a strong activity was also observed for TAF1 BD2.
Table 1. SAR of Position 6 for BRPF2 BD, BRPF1 BD, TAF1 BD2, and BRD4 BD1 Activity.
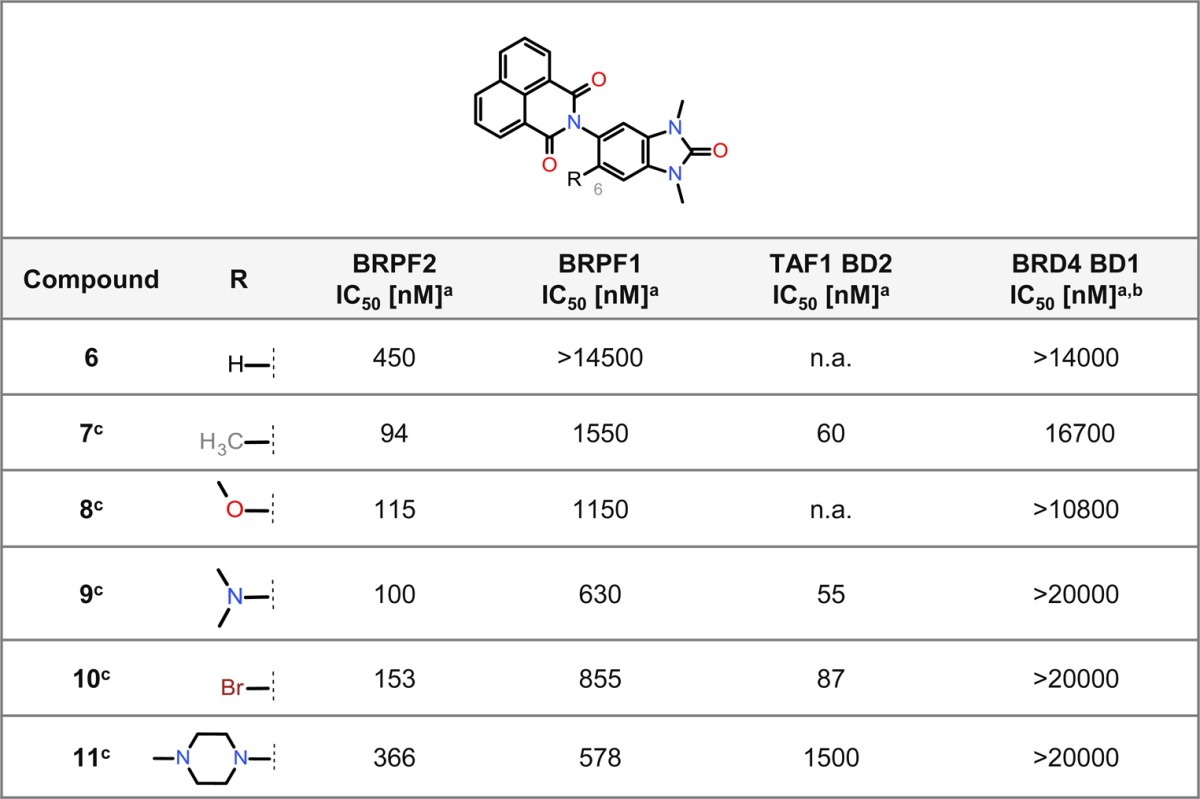
Determined using a TR-FRET assay.
All compounds show an IC50 > 20000 nM for BRD4 BD2; n.a. = not available.
Compounds are assumed to be racemates.
The cellular activity of selected compounds was then determined using the NanoBRET assay42 to measure the interaction between the BRPF BD fused to the NanoLuc luciferase and full-length histone H4 fused to the HaloTag (Supporting Information Table S1). Compound 7 inhibited the interaction of BRPF2 BD and histone H4 with an IC50 of 470 nM, whereas no effect on the BRPF1 BD–histone H4 interaction was observed (IC50 > 10000 nM). Compound 8 was also highly selective for BRPF2 BD (IC50 = 1500 nM), compared to BRPF1 BD (IC50 > 10000 nM). In addition, inhibition of the TAF1 BD2–histone H4 interaction was also measured (Supporting Information Table S1).
The cocrystal structure of 7 with BRPF2 BD revealed the binding mode within the KAc binding pocket (Figure 3A). The expected hydrogen bond is formed between the carbonyl moiety of the benzimidazolone backbone of 7 and residue Asn642 as well as the π-stacking with the Phe648. As predicted by our in silico analyses, a second hydrogen bond of the carbonyl moiety of the naphthalimide moiety to the backbone nitrogen of Ser592 is formed, which is key for the BRPF1 vs BRPF2 selectivity profile. A proline residue (Pro658) at the equivalent position in BRPF1 is unable to form this hydrogen bond (Figure 3B). The crystal structure also revealed that the Gln589 side-chain in BRPF2 unexpectedly adopted an alternative conformation in the presence of 7, forming an additional Van-der-Waals-type interaction by stacking underneath the tricyclic ring system (Figure 3A). Overlay with the BRD4 apostructure revealed a number of key residue differences that contribute to the strong selectivity of our compounds (Figure 3C).
In order to enrich our structure–activity relationship (SAR) knowledge, we then focused on the naphthalimide moiety and explored two different positions (5′ and 6′) (Table 2). For compounds bearing an electron-withdrawing group at position 6′ (compounds 12 and 13), a low, two-digit nanomolar biochemical potency for BRPF2 was reached. Aside from 15, which displayed an IC50 of 63 nM for BRPF2, analogues with a modification at position 5′ (compounds 14, 16–18) displayed no significant improvement over 7. Except for 14, all modified compounds possessed strong additional TAF1 BD2 in vitro activity and unchanged selectivity against BRPF1. The best TAF1 BD2 IC50 values were obtained for 12 and 13, which possess an electron-withdrawing group at position 6′.
Table 2. SAR of Positions 5′ and 6′ for BRPF2 BD, BRPF1 BD, TAF1 BD2, and BRD4 BD1 Activity.
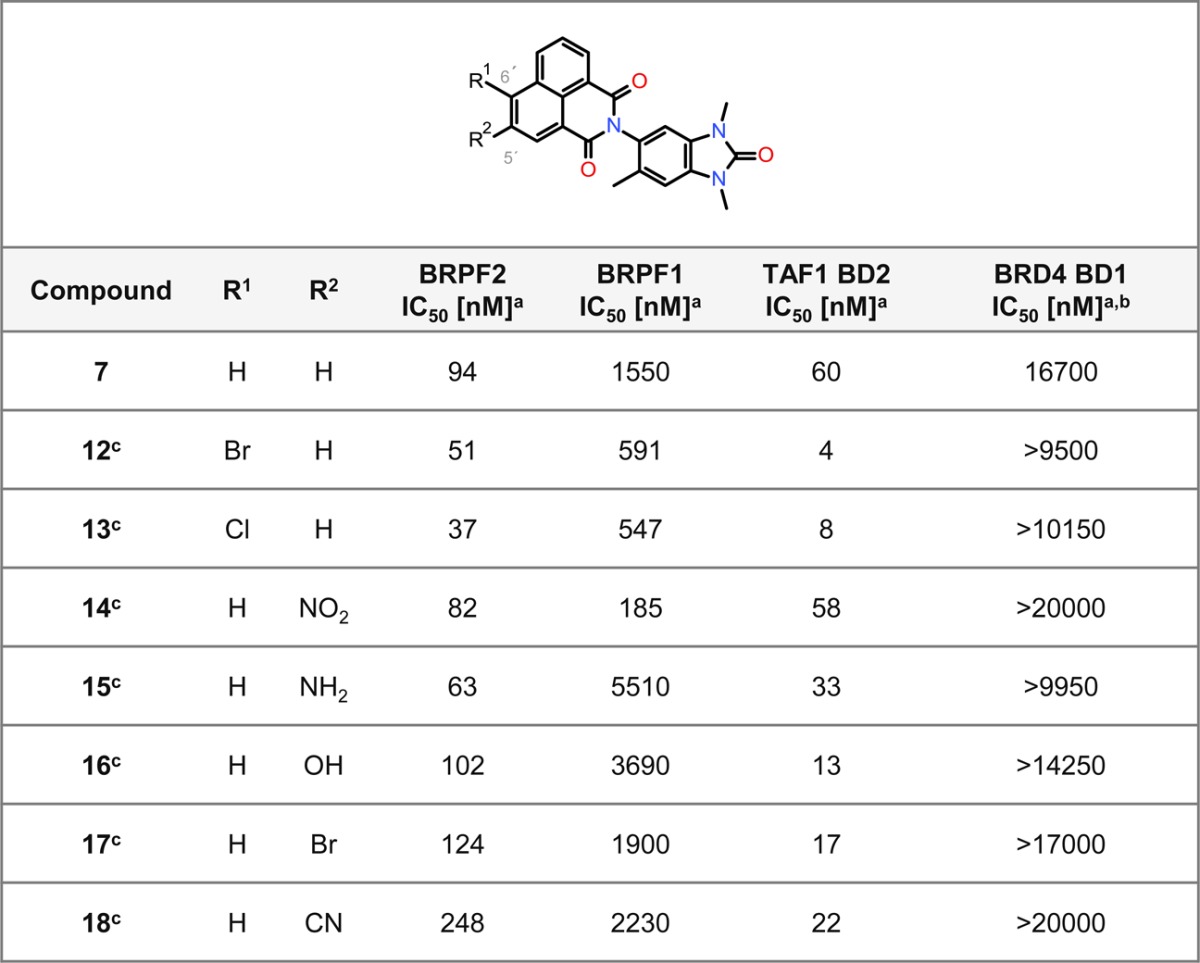
Determined using a TR-FRET assay.
All compounds show an IC50 > 20000 nM for BRD4 BD2.
Compounds are racemates.
Encouraged by these data, we characterized 13 in more detail. While cellular activity in the NanoBRET assay was determined as IC50 675 nM for BRPF2, there was no activity for BRPF1 (Supporting Information Table S1). Inhibition of the TAF1 BD2–histone H4 interaction was furthermore observed (Supporting Information Table S1). In addition, the selectivity of 13 was assessed using TSA with a panel of 48 BDs (Supporting Information Table S2). The strongest stabilization was observed for BRPF2 (ΔTm = 6.2 K), and weaker stabilization was seen for TAF1 BD2, TAF1L BD2, CECR2 BD, and adenoviral E1A binding protein (EP300) BD. As the low solubility of 13 limited further characterization, we elected to optimize this parameter by tuning the polarity via modifications at position 6′ of the naphthalimide moiety (Table 3). A range of amines with diverse degrees of basicity were introduced by nucleophilic aromatic substitution (SNAr) or Buchwald–Hartwig coupling to provide 19–28 (Table 3). A significant increase in the thermodynamic solubility from DMSO solution with maintenance of the selectivity was indeed observed for 21, 22, and 26–28. One acyclic amine displaying high selectivity for BRPF2 over BRPF1 BD (118-fold) and strong TAF1 BD2 activity could also be synthesized (compound 26). Interestingly, synthesis of its corresponding C-substituted terminal alkoxy analogues (5 and 29) led to more selective compounds. We opted for further investigation of 5 (BEI: 16.72, LLE: 4.72), since it showed the strongest selectivity and cellular activity for BRPF2 with a reasonable solubility and a logD of 2.0. The physicochemical profile and characteristics of 5 are summarized in the Supporting Information (Table S3). Overall, these SAR investigations revealed a high tolerability to amines with respect to the BRPF2 BD or TAF1 BD2 in vitro activity.
Table 3. Selected SAR: Optimizing Solubility.
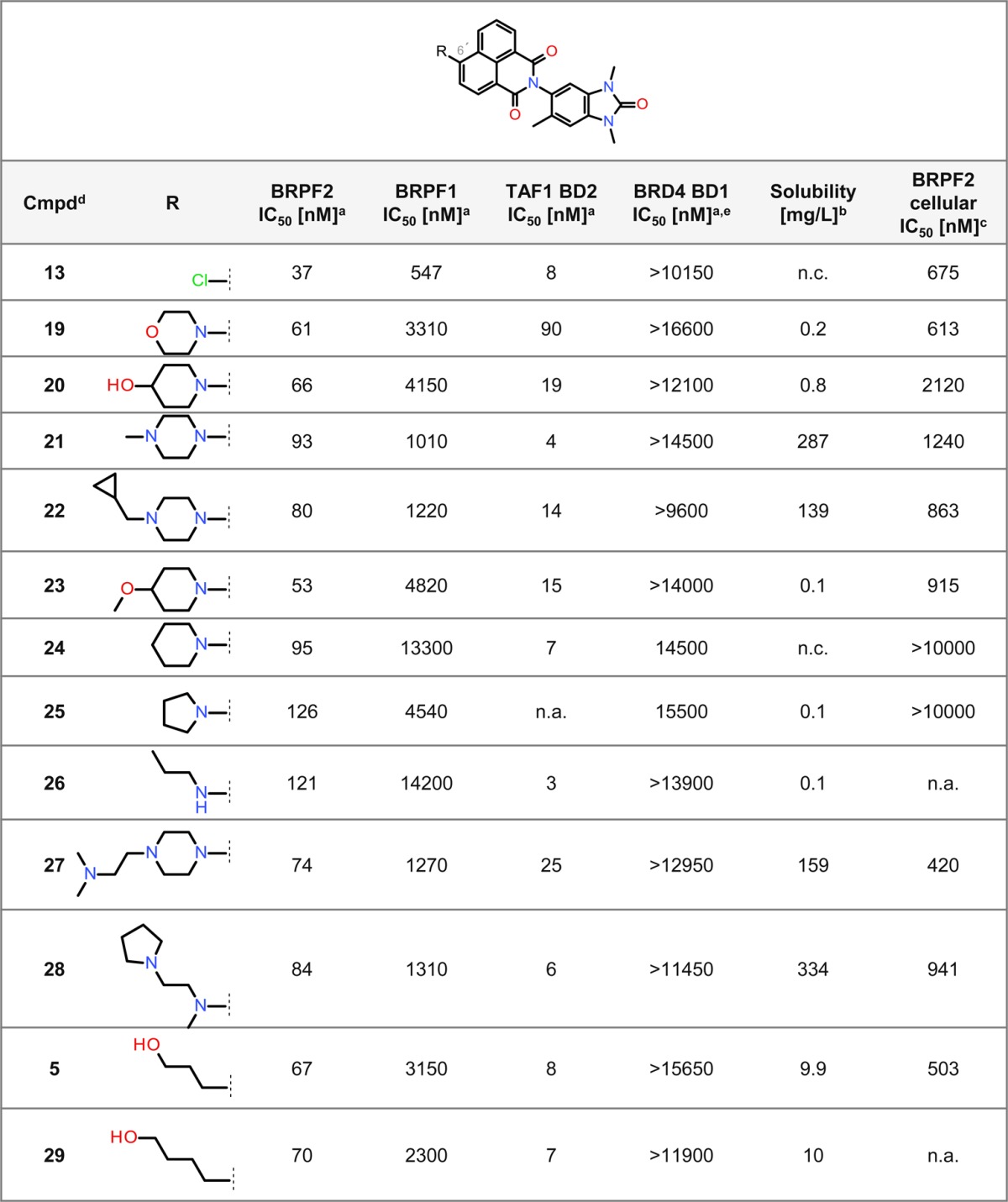
Determined using a TR-FRET assay.
Solubility from DMSO solution.
Determined using NanoBRET.
All compounds are racemates.
All compounds show an IC50 > 15000 nM for BRD4 BD2; n.a. = not available; n.c. = no calibration possible.
General Synthesis of 1,3,6-Trimethylbenzimidazolone-Substituted Naphthalimides
The 1,3,6-trimethylbenzimidazolone-substituted naphthalimides were synthesized via a linear and straightforward reaction pathway, as outlined in Scheme 1. Condensation of amine 30 with various commercially available 1,8-naphthalic anhydrides resulted in 7, 12–14, and 16–18. Subsequent reaction of aryl bromide 12 with the appropriate amines via nucleophilic aromatic substitution provided 19–25, 27, and 28. Compound 26 was synthesized from aryl bromide 12 and propylamine via Buchwald–Hartwig amination. Sonogashira coupling of aryl bromide 12 with propargyl alcohol or but-3-yn-1-ol, followed by hydrogenation of the resulting coupled alkynes with palladium on carbon, resulted in 5 and its analogue 29.
Scheme 1. General Route for the Synthesis of 1,3,6-Trimethylbenzimidazolone-Substituted Naphthalimides.

a) 1,8-naphtalic substituted anhydrids, CH3COOH, rfx; b) amine, potassium carbonate, DMSO, 110 °C; c) amine, cesium carbonate, tris(dibenzylideneacetone)dipalladium, Xantphos, toluene, 60 °C d) alkyl-alcohol, Et3N, bis(triphenylphosphine)palladium(II) chloride, CuI, THF, 65 °C; e) H2, Pd/C, EtOAc, rt.
Atropisomerism
Introduction of the methyl group at position 6 of the benzimidazolone core of the substituted naphthalimides hinders the rotation around the central torsion (axial chirality).43 Compound 5 is therefore a racemic mixture of the two atropisomers shown in Figure 4A. Several compounds containing C–N-coupled heterobiaryls with related atropisomerism have already been described.44−46 The calculated rotational energy barrier of 25 kcal/mol for a representative fragment (Figure 4B) classifies the atropisomers of 5 as “class two”.47 For this class of atropisomers, interconversion might occur depending on the medium the compound is exposed to.47 In the case of 5; the atropisomers 5a and 5b could be separated using chiral high-performance liquid chromatography (HPLC) and individually characterized (Table 4). Compound 5 and its individual atropisomers showed similar biochemical potency, selectivity, and cellular NanoBRET activity for BRPF BD, TAF1 BD2, and BRD4 BDs. Furthermore, the isothermal titration calorimetry (ITC) and Caco2 cell permeability profiles were comparable. Stirring experiments with the individual atropisomers revealed slight racemization in acetonitrile and DMSO after 1 week at 37 °C, and 30% racemization after 2 days at 80 °C (data not shown). The racemate 5 was therefore selected as the chemical probe.
Figure 4.

Atropisomerism. (A) Atropisomers 5a and 5b of 5. (B) Energy profile of the central torsion for a 6′-methyl- (solid lines) and unsubstituted (dashed lines) phenylnaphthalimide representative fragment. Optimization was performed at the B3LYP/6-31G* level followed by single-point LMP2/cc-pVTZ(-f) calculations.54
Table 4. Comparison of 5 with Its Atropisomersa.
| Compound 5 | atropisomer 1 | atropisomer 2 | |
|---|---|---|---|
| IC50 BRPF1 (TR-FRET) [nM] | 3150 | 5860 | 5490 |
| IC50 BRPF2 (TR-FRET) [nM] | 67 | 62 | 74 |
| IC50 TAF1 BD2 (TR-FRET) [nM] | 8 | 17 | 11 |
| IC50 BRD4 BD2 (TR-FRET) [nM] | >20000 | >20000 | >20000 |
| KD BRPF2 (ITC) [nM] | 45 | 36 | 37 |
| KD TAF1 BD2 (ITC) [nM] | 17 | n.a. | n.a. |
| IC50 BRPF2 cellular [nM] | 575 | 354 | 565 |
| BRPF2 cellular inhibition [%] | 82 | 84 | 84 |
| TAF1 BD2 cellular inhibition [%] | 64 | 63 | 63 |
| Caco2 Papp(A-B) [nm/s]/Papp (B-A) [nm/s]/ratio | 163/191/1.2 | 181/183/1.0 | 151/206/1.4 |
n.a. = not available.
Biochemical Potency, Binding, and Selectivity
TR-FRET assays showed that 5 was a potent inhibitor of BRPF2 BD with an IC50 of 67 nM, and a selectivity of 47- and 83-fold over BRPF1 and BRPF3 BDs (Table 5). The profile of 5 was further confirmed by AlphaScreen assay, where an IC50 of 97 nM and a selectivity of 23- and 25-fold over BRPF1 and BRPF3 BDs were measured (Table 5). Expanded selectivity studies using TSA with a panel of 48 BDs (Figure 5A) confirmed the selectivity of 5 within the BRPF family with a thermal shift for BRPF2 BD more than five degrees higher than for BRPF1 BD and BRPF3 BD. Results from the BROMOscan panel further confirmed the high selectivity with the BD family (Figure 5B). TR-FRET assays also disclosed a potent inhibition of TAF1 BD2 and the highly related TAF1L BD2 (Table 5). Finally, ITC was performed and KD values of 45 nM and 17 nM were determined for BRPF2 and TAF1 BD2, respectively (Figure 6A–B). A cocrystal structure of 5 in complex with TAF1 BD2 revealed a highly conserved binding mode reminiscent of the one obtained for the precursor 7 in BRPF2 BD (Supporting Information Figure S1). In addition, interactions of 5 with the BDs of cyclic-AMP response element binding protein (CREBBP) and the related EP300 were also detected by ITC (Figure 6A–B). However, with the BROMOscanTM panel and in the AlphaScreen assays, only a weak activity was recorded (30% and 41% inhibition in the presence of 100 nM compound; IC50 (CREBBP) = 1710 nM) (Supporting Information Table S4 and Table 5). ITC also revealed only modest interaction of 5 with CREBBP BD (KD = 1390 nM) (Figure 6B). Compound 5 was also tested at 10 μM in a panel containing over 300 kinases, and in all cases, inhibition was less than 50% (Table 5). Finally, 5 was evaluated in the LeadProfilingScreen, which contains 68 potential targets, and no inhibition or stimulation exceeding 25% was noted in the presence of 10 μM compound (Table 5).
Table 5. Biochemical Characterization of 5.
| Compound 5 | |
|---|---|
| IC50 BRPF2 (TR-FRET/Alpha Screen) [nM] | 67/97 |
| IC50 TAF1 BD2 (TR-FRET) [nM] | 8 |
| IC50 TAF1L BD2 (TR-FRET) [nM] | 106 |
| IC50 BRPF1 (TR-FRET/Alpha Screen) [nM] | 3150/2280 |
| IC50 BRPF3 (TR-FRET/Alpha Screen) [nM] | 5550/2450 |
| IC50 BRD4 BD1/BD2 (TR-FRET) [nM] | >15650/>20000 |
| IC50 CREBBP (Alpha Screen) [nM] | 1710 |
| Kd BRPF2 (ITC) [nM] | 45 |
| Kd TAF1 BD2 (ITC) [nM] | 17 |
| IC50 BRPF2 BD/H4 cellular [nM]a | 575 |
| IC50 TAF1 BD2/H4 cellular [nM]a | 970 |
| Selectivity Kinases (300 entries) | >10000 |
| Selectivity LeadProfilingScreen (68 entries) | >10000 |
Determined using NanoBRET.
Figure 5.

Biophysical selectivity profile of 5. (A) TSA results for 48 BDs at 10 μM compound concentration. (B) BROMOscan panel results from DiscoverX at 100 nM compound concentration.
Figure 6.

ITC results for 5. (A) ITC binding study: data show raw injection heats for titration of protein into compound. The inset shows the normalized binding enthalpies corrected for the heat of protein dilution as a function of binding site saturation. Solid lines represent a nonlinear least-squares fit using a single-site binding model. (a) BRPF2 BD–5; (b) TAF1 BD2–5; (c) TAF1L BD2–5; (d) CREBBP BD–5. (B) Summary of ITC data.
Cellular Activity
NanoBRET experiments (Figure 7A–C) showed that the interaction of BRPF2 BD with histones H4 and H3.3 was blocked by 5 with IC50 values of 575 and 825 nM, respectively. For TAF1 BD2, the IC50 values were 970 and 1400 nM, respectively. No inhibitory effect was observed for the interaction between BRPF1 or BRD4 and histone H4 up to 10 μM for 5.
Figure 7.

Cellular activity of 5 and 31 in NanoBRET assays. (A) Dose-dependent activity of 5. (B) Dose-dependent activity of 31. (C) Overview of IC50 values determined for 5 and 31.
Antiproliferative Activity
Cell viability experiments were performed with 5 to determine a potential link between BRPF2 or TAF1 with proliferative diseases. Altogether little or no effect was observed. A weak inhibition was seen in cell lines originating from hematological tumors (Supporting Information Table S5). Interestingly, a newly described quinoline-2-one inhibitor targeting mainly BRPF1 and BRPF2 BDs also shows low micromolar activity in leukemia cell lines.48
In Vitro and in Vivo Pharmacokinetics
The metabolic stability of 5 was high after incubation with mouse, rat, dog, or human liver microsomes (Table 6A). In contrast, metabolic stability was low after incubation with rat or dog hepatocytes. This difference may result from phase II metabolism, and indeed, corresponding metabolites have been identified in vitro. Permeability through Caco2 cell monolayers was high, with no hint of active transport. Plasma protein binding was low in mouse and moderate in human.
Table 6. Pharmacokinetic Properties of 5.
| Part A | |
|---|---|
| Compound 5 | |
| Metabolic Stability after Incubation with Liver Microsomes | |
| Mouse CLblood [L/h/kg]/Fmax [%] | 0.19/96 |
| Rat CLblood [L/h/kg]/Fmax [%] | 0.15/96 |
| Dog CLblood [L/h/kg]/Fmax [%] | <0.001/100 |
| Human CLblood [L/h/kg]/Fmax [%] | <0.001/100 |
| Metabolic Stability after Incubation with Hepatocytes | |
| Rat CLblood [L/h/kg]/Fmax [%] | 3.8/9.4 |
| Dog CLblood [L/h/kg]/Fmax [%] | 1.5/28 |
| Permeability in Caco2 Monolayer Cells | |
| Caco2 Papp(A-B) [nm/s]/Papp (B-A) [nm/s]/ratio | 106/204/1.9 |
| Plasma Protein Binding (Fraction Unbound) | |
| NMRI mouse female [%] | 0.58 |
| Caucasian human (mixed gender) [%] | 4.3 |
| Part B | |||||||
|---|---|---|---|---|---|---|---|
| species | CLblood[L/h/kg] | Vss [L/kg] | t1/2[h] i.v. | AUC [mg·h/L]i.v. | AUC [mg·h/L] p.o. | F [%] p.o. | Cb/Cp |
| rat | 0.73 | 1.5 | 10 | 1.0 | 1.5 | 73 | 0.52 |
Studies of the in vivo pharmacokinetic properties of 5 in rat revealed that blood clearance was low (ca. 17% of hepatic blood flow), volume of distribution in steady-state high, terminal half-life long to very long, and bioavailability high (F = 73%) (Table 6B). In vivo blood clearance was as anticipated based on rat liver microsome values but lower than expected based on hepatocyte data. Bioavailability matched blood clearance.
Negative Control
In order to deliver a valuable tool, synthesis of an inactive, structurally close analogue of a chemical probe, as a direct comparator from the same chemotype in biochemical and cellular assays, is recommended.13 For this purpose, the key potency drivers of candidate 5 were disrupted. As a first strategy, we opted for inducing steric hindrance at the 1,3-dimethyl-part of the benzimidazolone moiety. The corresponding diverse set of compounds had, however, remaining BRPF2 or TAF1 activity. Then a different strategy aiming at reducing the size of the molecule was followed. Previous results suggested that a significant loss of potency could be achieved upon removing the methyl group at position 6 of the benzimidazolone. Additional removal of one or both methyl groups from the 1,3-dimethylimidazolone led to a complete loss of activity on BRPF2 BD, low residual activity on TAF1 BD2, and no activity on TAF1L BD2. 31 (BAY-364)41 was selected among several candidates due to its favorable permeability profile (Figure 8). Compound 31 showed no activity for BRPF3 or BRD4 BDs in the AlphaScreen assay. Also, no cellular effect was seen in the NanoBRET assay for BRPF2 BD or TAF1 BD2, or for BRPF1 or BRD4 BDs (Figure 7A–C). Furthermore, 31 did not show any activity up to 10 μM in a panel of 22 kinases (data not shown). Thus, 31 fulfills the requirements of an inactive, structurally close analogue of probe 5.
Figure 8.

Biochemical and cellular data for the negative control 31.
Conclusion
An HTS approach followed by medicinal chemistry optimization led to the identification of 5, a highly potent and first selective dual inhibitor of BRPF2 BD and TAF1/TAF1L BD2, which differs from previously reported BRPF inhibitors. Our straightforward three-step synthesis ensures 5 is an easily accessible tool. A cocrystal structure of the close congener 7 in complex with BRPF2 BD revealed four key interactions, including a hydrogen bond between a carbonyl group and Ser592 which was essential for selectivity over BRPF1 BD. Importantly 5 was demonstrated to engage BRPF2 and TAF1 in cells at submicromolar concentrations. Antiproliferative activity was generally low and mainly observed in leukemia cell lines. The strong biochemical and cellular activities of 5 suggest it to be a high-quality chemical probe for the in-depth analysis of the physiological function and pathological relevance of BRPF2 and TAF1/TAF1L, which has not been possible to date. This novel tool enlarges the repertoire of available chemical probes for epigenetic players such that new knowledge about an implication of these two BD-containing proteins in diverse pathologies can be expected forthwith.49−53
Experimental Procedures
Chemistry
General Comments
Chemical names and numbers of the compounds are given in italics and in bold, respectively. Chemical names were generated using ACD/Name Batch or Autonom 2000, following IUPAC nomenclature.
Stereochemistry
In certain cases (e.g., where the substituent at position 6 is not a hydrogen atom) atropisomerism could be induced, and the compounds are assumed to be obtained as racemic mixtures of atropisomers. In the cases where the atropisomeric ratio was investigated, the ratio and the retention time of the corresponding atropisomers are given in the analytical data part of the corresponding racemates. In some cases, the atropisomers were separated using chiral HPLC. The HPLC methods used for separation are described in the individual cases, along with the corresponding analytical data. The atropisomers with the shorter retention time (LCMS) were defined as atropisomer 1 (atrop1) and those with the longer retention time as atropisomer 2 (atrop2). The exact configuration of the obtained atropisomers was not determined.
Analytical Methods
All NMR spectroscopy results were recorded on Bruker Avance III HD spectrometers. 1H spectra were obtained at 400 MHz and referenced to residual solvent signals (2.50 ppm for [D]6DMSO). All spectra were obtained at ambient temperature (22 ± 1 °C). Peak forms and multiplicities are specified as apparent in the spectra; potential higher-order effects have not been considered. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) in hertz (Hz). Spin multiplicities are reported as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, and mc = centered multiplet. Optical rotations were measured on a P2000 JASCO polarimeter using a 1 mL microcell (10 cm thickness, 3 mm diameter) and the alpha-D-line of sodium at 20 °C.
Chromatographic Methods
Analytical liquid chromatography–mass spectrometry (LC-MS) methods were performed using UPLCMS on a Waters Acquity UPLCMS SingleQuad instrument (with an Acquity UPLC BEH C18 1.7 μm, 50 × 2.1 mm column or a Kinetex 2.6 μm 50 × 2.1 mm column) or on an Agilent 1290 UPLCMS 6230 time-of-flight instrument with a BEH C18 1.7 μm, 50 × 2.1 mm column. The different methods are described below.
Analytical methods with the Waters instruments:
Method I: column: Acquity UPLC BEH C18 1.7 μm, 50 × 2.1 mm; eluent A: water + 0.1 vol % formic acid (99%), eluent B: acetonitrile (ACN); gradient: 0–1.6 min 1–99% B, 1.6–2.0 min 99% B; flow: 0.8 mL/min; temperature: 60 °C; diode array detector (DAD) scan: 210–400 nm.
Method II: column: Acquity UPLC BEH C18 1.7 μm, 50 × 2.1 mm; eluent A: water + 0.2 vol % aqueous ammonia (32%), eluent B: ACN; gradient: 0–1.6 min 1–99% B, 1.6–2.0 min 99% B; flow: 0.8 mL/min; temperature: 60 °C; DAD scan: 210–400 nm.
Method III: column: BEH C18 1.7 μm 50 × 2.1 mm; eluent A: water + 0.05% formic acid (99%), eluent B: ACN + 0.05% formic acid (99%); gradient: 0–0.2 min 2% B, 0.2–1.7 min 2–90% B, 1.7–1.9 min 90% B; 1.9–2.0 min 90–2% B, 2.0–2.5 min 2% B, flow: 1.3 mL/min; temperature: 60 °C; DAD scan: 200–400 nm.
Method IV: column: Kinetex 2.6 μm, 50 × 2.1 mm; eluent A: water + 0.05% formic acid (99%), eluent B: ACN + 0.05% formic acid (99%); gradient: 0–1.9 min 1–99% B, 1.9–2.1 min 99% B; flow: 1.3 mL/min; temperature: 60 °C; DAD scan: 200–400 nm.
Analytical methods with the Agilent instruments:
Method V: eluent A: water + 0.05% formic acid (99%), eluent B: ACN + 0.05% formic acid (99%); gradient: 0–1.7 min 2–90% B, 1.7–2.0 min 90% B; flow: 1.2 mL/min; temperature: 60 °C; DAD scan: 190–400 nm.
Reagents
All reagents for which the synthesis is not described below are either commercially available or were synthesized according to literature procedures. All final products were at least 95% pure, as determined by analytical HPLC, apart from the two cases otherwise indicated.
General Procedure for the Condensation of Naphthalic Anhydrides under Acidic Conditions (GP1)
In a pressure vessel, or a round-bottom flask, the aniline (1.0 equiv) was suspended in acetic acid (3.8 mL/mmol) under argon atmosphere. Then, the commercially available anhydride partner (0.9–1.5 equiv) was added. The reaction mixture was heated to reflux for 3–16 h. After cooling, the mixture was concentrated under reduced pressure. The residue was suspended in methanol (10 mL/mmol), and 4 N sodium hydroxide solution (177 μL/mmol) was added. After stirring at room temperature (RT) for 1 h, water (74 mL/mmol) was added and the mixture was further stirred for 10 min. The precipitate was separated by filtration, washed three times with water and methanol, and dried overnight in a vacuum oven at 40 °C. If necessary, the precipitate was purified by HPLC or flash column chromatography to give the desired product.
General Procedure for the Nucleophilic Substitution of Amines (GP2)
In a pressure vessel, the aryl bromide (1.0 equiv) was dissolved in DMSO (7.5 mL/mmol) under argon atmosphere. The commercially available amine partner (1.1 equiv) and potassium carbonate (2.5 equiv) were added ,and the mixture was heated to 110 °C overnight. After cooling, the precipitate was separated by filtration and purified by HPLC.
General Procedure for the Sonogashira Coupling (GP3)
In a pressure vessel, the aryl bromide (1.0 equiv) was dissolved in tetrahydrofuran (0.4 mL/mmol). The commercially available catalyst bis(triphenylphosphine)palladium(II) dichloride (0.02 equiv), copper(I) iodide (0.02 equiv), triethylamine (7.4 equiv), and the coupling partner (2.0 equiv) were added to the solution, and the mixture was degassed with argon for 5 min. The mixture was heated to 65 °C for 5–16 h. After cooling, the mixture was diluted with aqueous ammonia (6 mL/mmol) and was stirred again for 5 min. Dichloromethane (60 mL/mmol) and water (15 mL/mmol) were added, and the layers were separated. The aqueous layer was extracted with dichloromethane (2 × 40 mL/mmol). The combined organic layers were washed with brine (60 mL/mmol) and dried over sodium sulfate. After filtration, the solvent was removed under reduced pressure. The residue was purified by HPLC to give the desired product. In some cases, the crude product was directly used in the next step without any further purification.
General Procedure for Hydrogenation with Palladium on Carbon (GP4)
In a hydrogenation flask, the alkyne (1.0 equiv) was dissolved in ethyl acetate (450–1000 mL/mmol) and Pd/C (0.02 equiv) was added. The suspension was flushed with argon and vigorously shaken under a 1 bar hydrogen atmosphere for 2–4 h. After reaction completion, the suspension was flushed with argon again, the catalyst was removed by filtration through a pad of Celite, and the solvent was removed under reduced pressure. The residue was purified by HPLC or flash column chromatography to give the desired product.
6-(3-Hydroxypropyl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (5)
According to GP3, aryl bromide 12 (125 mg, 0.278 mmol), propargyl alcohol (32 μL, 0.555 mmol), triethylamine (288 μL, 2.07 mmol), CuI (1.1 mg, 0.006 mmol), and bis(triphenylphosphine)palladium(II) dichloride (3.9 mg, 0.006 mmol) in tetrahydrofuran (1.2 mL) gave the corresponding alkyne, which, after purification, was obtained as a yellow solid (83 mg, 67%). According to GP4, the alkyne (72 mg, 0.169 mmol) and Pd/C (36 mg, 0.034 mmol) in EtOAc (56 mL) gave 5 as a colorless solid (49 mg, 64%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.88 (mc, 2 H), 2.08, 3.29 (2 s, 3 H each), 3.30 (mc, 2 H), 3.38 (s, 3 H), 3.54 (mc, 2 H), 4.67 (t, J = 5.2 Hz, 1 H), 7.17 (mc, 2 H), 7.78 (d, J = 7.6 Hz, 1 H), 7.94 (dd, J = 7.4, 8.6 Hz, 1 H), 8.46 (d, J = 7.6 Hz, 1 H), 8.55 (dd, J = 1.0, 7.4 Hz, 1 H), 8.69 (dd, J = 1.0, 8.4 Hz, 1 H) ppm; LCMS (method V): tR = 0.87 min; m/z: [M + H]+ = 430; atropisomeric ratio: atrop 1/atrop 2 = 1:1; tR (atrop1) = 2.64 min; tR (atrop2) = 3.44 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IA 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); eluent B: ACN; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
The atropisomers were separated using the following chiral HPLC method: instrument: Labomatic HD5000, Labocord-5000; Gilson GX-241, Labcol Vario 4000, column: Chiralpak IA 5 μm 250 × 30 mm; eluent A: methanol + 0.1 vol % diethylamine (99%); eluent B: 2-methoxy-2-methylpropane + 0.1% diethylamine (99%); isocratic: 50% A + 50% B; flow: 50.0 mL/min; UV 254 nm.
(−)-6-(3-Hydroxypropyl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (5a)
[α]20D = −1.8 (c = 1.00, DMSO); 1H NMR (400 MHz, [D]6DMSO): δ = 1.88 (mc, 2 H), 2.08, 3.29 (2 s, 3 H each), 3.30 (mc, 2 H), 3.38 (s, 3 H), 3.54 (mc, 2 H), 4.67 (t, J = 5.2 Hz, 1 H), 7.17 (mc, 2 H), 7.78 (d, J = 7.6 Hz, 1 H), 7.94 (dd, J = 7.4, 8.6 Hz, 1 H), 8.46 (d, J = 7.6 Hz, 1 H), 8.55 (dd, J = 1.0, 7.4 Hz, 1 H), 8.69 (dd, J = 1.0, 8.4 Hz 1 H) ppm; LCMS (method V): tR = 0.87 min; m/z: [M + H]+ = 430.
(+)-6-(3-Hydroxypropyl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (5b)
[α]20D = +4.8 (c = 1.00, DMSO); 1H NMR (400 MHz, [D]6DMSO): δ = 1.88 (mc, 2 H), 2.08, 3.29 (2 s, 3 H each), 3.30 (mc, 2 H), 3.38 (s, 3 H), 3.54 (mc, 2 H), 4.67 (t, J = 5.2 Hz, 1 H), 7.17 (mc, 2 H), 7.78 (d, J = 7.6 Hz, 1 H), 7.94 (dd, J = 7.4, 8.6 Hz, 1 H), 8.46 (d, J = 7.6 Hz, 1 H), 8.55 (dd, J = 1.0, 7.4 Hz, 1 H), 8.69 (dd, J = 1.0, 8.4 Hz 1 H) ppm; LCMS (method V): tR = 0.87 min; m/z: [M + H]+ = 430.
2-(1,3,6-Trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (7)
According to GP1, 30 (150 mg, 0.784 mmol) and 1,8-naphthalic anhydride (233 mg, 1.18 mmol) in acetic acid (3 mL) gave 7 as a colorless solid (40 mg, 13%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.09, 3.29, 3.38 (3 s, 3 H each), 7.18 (mc, 2 H), 7.93 (dd, J = 7.8 Hz, 2 H), 8.53–8.56 (m, 4 H) ppm; LCMS (method III): tR = 1.04 min; m/z: [M + H]+ = 372. Compound 7 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
2-(6-Methoxy-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (8)
According to GP1, 5-amino-6-methoxy-1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-one (159 mg, 0.769 mmol) and 1,8-naphthalic anhydride (229 mg, 1.15 mmol) in acetic acid (3 mL) gave 8 as a colorless solid (4.2 mg, 1%). 1H NMR (400 MHz, [D]6DMSO): δ = 3.29, 3.41, 3.72 (3 s, 3 H each), 7.13, 7.21 (2 mc, 1 H each), 7.92 (t, J ≈ 7.8 Hz, 2 H), 8.53 (t, J ≈ 8.4 Hz, 4 H) ppm; LCMS (method III): tR = 0.98 min; m/z: [M + H]+ = 388. Compound 8 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
2-[6-(Dimethylamino)-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl]-1H-benzo[de]isoquinoline-1,3(2H)-dione (9)
According to GP1, 5-amino-6-(dimethylamino)-1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-one dihydrochloride (244 mg, 0.833 mmol) and 1,8-naphthalic anhydride (248 mg, 1.15 mmol) in acetic acid (3 mL) gave 9 as a colorless solid (20 mg, 6%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.47, 3.29, 3.40 (3 s, 6 H, 3 H, 3 H), 7.18, 7.26 (2 mc, 1 H each), 7.92 (t, J ≈ 7.7 Hz, 2 H), 8.53, 8.55 (2 mc, 2 H each) ppm; LCMS (method V): tR = 0.78 min; m/z: [M + H]+ = 401. Compound 9 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
2-(6-Bromo-1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (10)
According to GP1, 5-amino-6-bromo-1,3-dimethyl-1,3-dihydro-2H-benzimidazol-2-one (150 mg, 0.586 mmol) and 1,8-naphthalic anhydride (174 mg, 0.879 mmol) in acetic acid (3 mL) gave 10 as a colorless solid (9 mg, 3%). 1H NMR (400 MHz, [D]6DMSO): δ = 3.32, 3.40 (2 s, 3 H each), 7.46, 7.66 (2 mc, 1 H each), 7.96 (t, J ≈ 7.8 Hz, 2 H), 8.55–8.58 (m, 4 H) ppm; LCMS (method V): tR = 1.03 min; m/z: [M + H]+ = 436, [(M + 2) + H]+ = 438 (Br isotopic pattern). Compound 10 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
2-[1,3-Dimethyl-6-(4-methylpiperazin-1-yl)-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl]-1H-benzo[de]isoquinoline-1,3(2H)-dione (11)
Commercially available 5-chloro-1,3-dimethyl-6-nitro-1,3-dihydro-2H-benzimidazol-2-one (250 mg, 1.03 mmol) was suspended under an atmosphere of argon in N,N-dimethylformamide (10 mL), and sodium carbonate (286 mg, 2.07 mmol) and 1-methylpiperazine (170 μL, 1.6 mmol) were added. The reaction mixture was heated to 120 °C overnight. After cooling, the reaction mixture was filtered and the solvent was removed under reduced pressure. The obtained residue was then suspended in ethanol (19.2 mL), and Pd/C (57.9 mg, 0.054 mmol) was added under argon. The suspension was flushed with argon and vigorously shaken under an atmosphere of hydrogen (1 bar) for 4 h. After reaction completion, the suspension was flushed with argon again, the catalyst was removed by filtration through a pad of Celite, and the solvent was removed under reduced pressure. The crude product was directly used in the next step without any further purification. LCMS (method I): tR = 0.48 min; m/z: [M + H]+ = 275. According to GP1, crude product (200 mg, 0.726 mmol) and 1,8-naphthalic anhydride (144 mg, 0.269 mmol) in acetic acid (2.6 mL) gave 11 was obtained as a colorless solid (20 mg, 6%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.87 (s, 3 H), 2.72 (mc, 4 H), 3.30, 3.39 (2 s, 3 H each), 7.22, 7.27 (2 mc, 1 H each), 7.93 (t, J ≈ 7.7 Hz, 2 H), 8.53, 8.55 (2 mc, 2 H each) ppm; four proton signals could not be detected; LCMS (method V): tR = 0.61 min; m/z: [M + H]+ = 456. Compound 11 was assumed to be a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
6-Bromo-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (12)
According to GP1, 30 (60 mg, 0.314 mmol) and 4-bromo-1,8-naphthalic anhydride (96 mg, 0.345 mmol) in acetic acid (1.2 mL) gave 12 as a yellow solid (90 mg, 61%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.08, 3.28, 3.37 (3 s, 3 H each), 7.17* (mc, 2 H), 8.06 (dd, J = 7.4, 8.1 Hz, 1 H), 8.29, 8.37 (2 d, J = 7.9 Hz, 1 H each), 8.63–8.67 (m, 2 H) ppm; *overlapping signals; LCMS (method V): tR = 1.12 min; m/z: [M + H]+ = 450, [(M + 2) + H]+ = 452 (Br isotopic pattern). Compound 12 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
6-Chloro-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (13)
According to GP1, 30 (60 mg, 0.314 mmol) and 4-chloro-1,8-naphthalic anhydride (80 mg, 0.345 mmol) in acetic acid (1.2 mL) gave 13 as a yellow solid (57 mg, 43%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.09, 3.28, 3.38 (3 s, 3 H), 7.18* (mc, 2 H), 8.07 (dd, J = 7.4, 8.4 Hz, 1 H), 8.11, 8.48 (2 d, J = 8.0 Hz, 1 H each), 8.63 (dd, J = 7.0 Hz, 1 H), 8.71 (dd, J = 8.6 Hz, 1 H) ppm; *overlapping signals; LCMS (method V): tR = 1.16 min; m/z: [M + H]+ = 406. Compound 13 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
5-Nitro-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (14)
According to GP1, 30 (60 mg, 0.314 mmol) and 3-nitro-1,8-naphthalic anhydride (84 mg, 0.345 mmol) in acetic acid (1.2 mL) gave 14 as a yellow solid (113 mg, 78%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.11, 3.29, 3.38 (3 s, 3 H each), 7.19* (mc, 2 H), 8.12 (t, J ≈ 7.7 Hz, 1 H), 8.72 (dd, J = 7.4 Hz, 1 H), 8.87 (dd, J = 8.4 H, 1 H), 9.00 (d, J = 2.3 Hz), 9.57 (d, J = 2.0 Hz, 1 H) ppm; *overlapping signals; LCMS (method V): tR = 1.02 min; m/z: [M + H]+ = 417. Compound 14 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
5-Amino-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinolone-1,3(2H)-dione (15)
Compound 14 (78 mg, 0.187 mmol) was suspended in methanol (110 mL), and Pd/C (40 mg, 0.037 mmol) was added. The suspension was flushed with argon and vigorously shaken under an atmosphere of hydrogen (1 bar) for 3 h. After reaction completion, the suspension was flushed with argon again, the catalyst was removed by filtration through a pad of Celite, and the solvent was removed carefully under reduced pressure. The residue (23 mg) was purified by HPLC (basic) and lyophilized to give the desired aniline 15 as a yellow solid (10 mg, 12%), which was stored under argon in the fridge. 1H NMR (400 MHz, [D]6DMSO): δ = 2.06, 3.29, 3.37 (3 s, 3 H each), 6.05 (bs, 2 H), 7.16* (mc, 2 H), 7.35 (d, J = 2.3 Hz, 1 H), 7.66 (dd, J = 7.4, 8.1 Hz, 1 H), 8.00 (d, J = 2.3 Hz, 1 H), 8.09 (mc, 2 H) ppm; *overlapping signals; LCMS (method V): tR = 0.86 min; m/z: [M + H]+ = 387. Compound 15 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
5-Hydroxy-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (16)
According to GP1, 30 (60 mg, 0.314 mmol) and 3-hydroxy-1,8-naphthalic anhydride (74 mg, 0.345 mmol) in acetic acid (1.2 mL) gave 16 as a bright yellow solid (80 mg, 60%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.07, 3.29, 3.27 (3 s, 3 H each), 7.16*, 7.17* (2 mc, 1 H each), 7.72 (d, J = 2.5 Hz, 1 H), 7.79 (dd, J = 8.1, 8.4 Hz, 1 H), 8.06 (d, J = 2.5 Hz, 1 H), 8.27–8.32 (m, 2 H), 10.63 (bs, 1 H) ppm; *overlapping signals; LCMS (method IV): tR = 0.85 min; m/z: [M + H]+ = 388. Compound 16 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
5-Bromo-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (17)
According to GP1, 30 (100 mg, 0.522 mmol) and 3-bromo-1,8-naphthalic anhydride (159 mg, 0.575 mmol) in acetic acid (2 mL) gave 16 as a bright yellow solid (110 mg, 44%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.08, 3.27, 3.37 (3 s, 3 H each), 7.16, 7.17 (2 mc, 1 H each), 7.95 (t, J ≈ 7.6 Hz, 1 H), 8.47–8.54 (m, 3 H), 8.84 (mc, 1 H) ppm; LCMS (method V): tR = 1.17 min; m/z: [M + H]+ = 450, [(M + 2) + H]+ = 452 (Br isotopic pattern). Compound 17 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
1,3-Dioxo-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-2,3-dihydro-1H-benzo[de]isoquinoline-5-carbonitrile (18)
According to GP1, 30 (58 mg, 0.305 mmol) and 3-cyano-1,8-naphthalic anhydride (75 mg, 0.336 mmol) in acetic acid (1.2 mL) gave 18 as a yellow solid (30 mg, 22%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.09, 3.28, 3.38 (3 s, 3 H each), 7.18* (mc, 2 H), 8.07 (t, J ≈ 7.7 Hz, 1 H), 8.62 (d, J ≈ 8.4 Hz, 1 H), 8.68* (d, J ≈ 7.4 Hz, 1 H), 8.70*, 9.18 (2 mc, 1 H each) ppm; *overlapping signals; LCMS (method V): tR = 0.95 min; m/z: [M + H]+ = 397. Compound 18 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
6-(Morpholin-4-yl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (19)
According to GP2, aryl bromide 12 (150 mg, 0.333 mmol), morpholine (32 μL, 0.366 mmol), and potassium carbonate (115 mg, 0.833 mmol) in DMSO (2 mL) gave 19 as a yellow solid (58 mg, 34%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.06 (s, 3 H), 3.26 (mc, 4 H), 3.29, 3.37 (2 s, 3 H each), 3.94 (t, J ≈ 5.0 Hz, 4 H), 7.14, 7.17 (2 s, 1 H each), 7.41 (d, J = 8.1 Hz, 1 H), 7.87 (dd, J = 7.4, 8.3 Hz, 1 H), 8.45 (d, J ≈ 8.5 Hz, 1 H), 8.52 (dd, J = 1.1, 7.2 Hz, 1 H), 8.58 (dd, J = 1.1, 8.4 Hz, 1 H) ppm; LCMS (method III): tR = 1.12 min; m/z: [M + H]+ = 457; atropisomeric ratio: atrop 1/atrop 2 = 50:50; tR (atrop1) = 2.74 min; tR (atrop2) = 3.28 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IB 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); Eluent B: methanol; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-(4-Hydroxypiperidin-1-yl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (20)
According to GP2, aryl bromide 12 (150 mg, 0.333 mmol), 4-hydroxypiperidine (37 mg, 0.366 mmol), and potassium carbonate (115 mg, 0.833 mmol) in DMSO (2 mL) gave 20 as a yellow solid (48 mg, 28%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.77, 2.02 (2 mc, 2 H each), 2.06 (s, 3 H), 3.07 (mc, 2 H), 3.29, 3.37 (2 s, 3 H each), 3.49, 3.79 (2 mc, 2 H, 1 H), 4.94 (bs, 1 H), 7.13, 7.17 (2 mc, 1 H each), 7.37 (d, J = 8.3 Hz, 1 H), 7.86 (dd, J = 7.5, 8.2 Hz, 1 H), 8.4 (d, J = 8.0 Hz, 1 H), 8.49 (mc, 2 H) ppm; LCMS (method V): tR = 0.90 min; m/z: [M + H]+ = 471; atropisomeric ratio: atrop 1/atrop 2 = 47:53; tR (atrop1) = 4.86 min; tR (atrop2) = 7.23 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IA 3 μm 100 × 4.6 mm; eluent A: methanol + 0.1 vol % diethylamine (99%); eluent B: ethanol; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-(4-Methylpiperazin-1-yl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (21)
According to GP2, aryl bromide 12 (150 mg, 0.333 mmol), 1-methylpiperazine (41 μL, 0.366 mmol), and potassium carbonate (115 mg, 0.833 mmol) in DMSO (2 mL) gave 21 as a yellow solid (39 mg, 22%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.06, 2.32 (2 s, 3 H each), 2.67, 3.29 (2 mc, 4 H, 7 H), 3.37 (s, 3 H), 7.14, 7.17 (2 mc, 1 H each), 7.39 (d, J = 8.4 Hz, 1 H), 7.86 (dd, J = 7.5, 8.3 Hz, 1 H), 8.43 (d, J = 8.1 Hz, 1 H), 8.50, 8.52 (2 mc, 1 H each) ppm; LCMS (method V): tR = 0.62 min; m/z: [M + H]+ = 470; atropisomeric ratio: atrop1/atrop2 = 41:42 (17% impurities); tR (atrop1) = 2.72 min; tR (atrop2) = 4.11 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IB 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); eluent B: methanol; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-[4-(Cyclopropylmethyl)piperazin-1-yl]-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (22)
According to GP2, aryl bromide 12 (100 mg, 0.222 mmol), 1-(cyclopropylmethyl)piperazine (36 μL, 0.244 mmol) and potassium carbonate (77 mg, 0.555 mmol) in DMSO (2 mL) gave 22 as a yellow solid (16 mg, 14%). 1H NMR (400 MHz, [D]6DMSO): δ = 0.15, 0.52, 0.91 (3 mc, 2 H, 2 H, 1 H), 2.06 (s, 3 H), 2.33 (mc, 2 H), 2.80 (mc, 4 H), 3.29 (s, 3 H), 3.30 (mc, 4 H), 3.37 (s, 3 H), 7.14, 7.17 (2 mc, 1 H each), 7.39 (d, J = 8.1 Hz, 1 H), 7.86 (dd, J = 7.4, 8.4 Hz, 1 H), 8.43 (d, J = 8.2 Hz, 1 H), 8.50–8.53 (m, 2 H) ppm; LCMS (method V): tR = 0.70 min; m/z: [M + H]+ = 510; atropisomeric ratio: atrop1/atrop2 = 50:50; tR (atrop1) = 2.59 min; tR (atrop2) = 3.83 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IB 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); eluent B: Methanol; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-(4-Methoxypiperidin-1-yl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (23)
According to GP2, aryl bromide 12 (100 mg, 0.222 mmol), 4-methoxypiperidine (29 μL, 0.244 mmol), and potassium carbonate (77 mg, 0.555 mmol) in DMSO (4 mL) gave 23 as a yellow solid (30 mg, 26%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.82 (mc, 2 H), 2.06 (s, 3 H), 2.13, 3.09 (2 mc, 2 H each), 3.28, 3.37 (2 s, 3 H each), 3.48 (mc, 3 H), 7.13, 7.16 (2 mc, 1 H each), 7.38 (d, J = 8.1 Hz, 1 H), 7.85 (t, J = 7.7 Hz, 1 H), 8.41 (d, J = 8.4 Hz, 1 H), 8.50 (mc, 2 H) ppm; LCMS (method III): tR = 1.25 min; m/z: [M + H]+ = 485; atropisomeric ratio: atrop1/atrop2 = 47:47 (6% impurities); tR (atrop1) = 2.70 min; tR (atrop2) = 3.17 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IB 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); eluent B: methanol; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-(Piperidin-1-yl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (24)
Aryl bromide 12 (100 mg, 0.222 mmol), piperidine (29 μL, 0.244 mmol), and potassium carbonate (77 mg, 0.555 mmol) in DMSO (4 mL) gave 24 as a yellow solid (25 mg, 24%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.68, 1.85 (2 mc, 2 H, 4 H), 2.06 (s, 3 H), 3.24 (mc, 4 H), 3.28, 3.37 (2 s, 3 H), 7.13, 7.16 (2 mc, 1 H each), 7.36 (2 s, 1 H each), 7.85 (dd, J = 7.4, 8.4 Hz, 1 H), 8.41 (d, J = 8.1 Hz, 1 H), 8.49 (mc, 2 H each) ppm; LCMS (method III): tR = 1.42 min; m/z: [M + H]+ = 455; atropisomeric ratio: atrop1/atrop2 = 50:50; tR (atrop1) = 3.60 min; tR (atrop2) = 4.59 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak ID 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); eluent B: ACN; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-(Pyrrolidin-1-yl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (25)
According to GP2, aryl bromide 12 (50 mg, 0.111 mmol), pyrrolidine (10 μL, 0.122 mmol), and potassium carbonate (38 mg, 0.278 mmol) in DMSO (1 mL) gave 25 as a yellow solid (15 mg, 29%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.04 (mc, 7 H), 3.29, 3.37 (2 s, 3 H each), 3.81 (mc, 4 H), 6.94 (d, J = 8.9 Hz, 1 H), 7.09, 7.15 (2 mc, 1 H each), 7.66 (dd, J = 7.4, 8.6 Hz, 1 H), 8.27 (d, J = 8.6 Hz, 1 H), 8.46 (dd, J = 0.9, 7.2 Hz, 1 H), 8.81 (dd, J = 1.0, 8.6 Hz, 1 H) ppm; LCMS (method V): tR = 1.11 min; m/z: [M + H]+ = 441; atropisomeric ratio: atrop 1/atrop 2 = 50:50; tR (atrop1) = 2.01 min; tR (atrop2) = 2.25 min.
6-(Propylamino)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (26)
In a pressure vessel, aryl bromide 12 (100 mg, 0.222 mmol, 1.0 equiv) was suspended in toluene (2 mL) under argon atmosphere. The commercially available catalyst tris(dibenzylideneacetone)dipalladium (8.1 mg, 0.009 mmol, 0.040 equiv), 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene (Xantphos, 5.1 mg, 0.009 mmol, 0.040 equiv), cesium carbonate (218 mg, 0.671 mmol, 3.0 equiv), and propylamine (56 μL, 0.671 mmol, 2.1 equiv) were added, and the mixture was degassed with argon for 5 min. The reaction mixture was heated to 90 °C for 18 h. After cooling, the solvent was removed under reduced pressure. The residue was purified by HPLC, giving product 26 as a green solid (10 mg, 10%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.00 (t, J = 7.4 Hz, 3 H), 1.74 (mc, 2 H), 2.04, 3.29, 3.37* (3 s, 3 H each), 3.14–3.37* (m, 2 H), 6.83 (d, J = 8.9 Hz, 1 H), 7.09, 7.15 (2 mc, 1 H each), 7.72 (t, J = 7.9 Hz, 1 H), 7.89 (mc, 1 H), 8.28 (d, J = 8.6 Hz, 1 H), 8.45 (d, J = 6.8 Hz, 1 H), 8.79 (d, J = 8.1 Hz, 1 H) ppm; *overlapping signals; LCMS (method III): tR = 1.18 min; m/z: [M + H]+ = 429. Compound 26 was assumed to be obtained as a racemic mixture of atropisomers. The atropisomeric ratio was not investigated at this stage.
6-{4-[2-(Dimethylamino)ethyl]piperazin-1-yl}-2-(1,3,6,-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (27)
According to GP2, aryl bromide 12 (300 mg, 0.666 mmol), 1-(2-dimethylaminoethyl)piperazine (129 mL, 0.733 mmol), and potassium carbonate (230 mg, 1.66 mmol) in DMSO (5 mL) gave 27 as a yellow solid (25 mg, 7%). 1H NMR (400 MHz, [D]6DMSO): δ = 2.06, 2.18 (2 s, 3 H, 6 H), 2.43, 2.52, 2.76, 3.28 (4 mc, 2 H, 2 H, 4 H, 7 H), 3.37 (s, 3 H), 7.13, 7.17 (2 s, 1 H each), 7.38 (d, J = 8.4 Hz, 1 H), 7.85 (dd, J = 7.4, 8.2 Hz, 1 H), 8.42 (d, J = 8.1 Hz, 1 H), 8.50, 8.52 (2 mc, 1 H each) ppm; LCMS (method III): tR = 0.76 min; m/z: [M + H]+ = 527; atropisomeric ratio: atrop1/atrop2 = 50:50; tR (atrop1) = 2.69 min; tR (atrop2) = 4.13 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IB 3 μm 100 × 4.6 mm; eluent A: 2-methoxy-2-methylpropane + 0.1 vol % diethylamine (99%); eluent B: methanol; isocratic: 50% A + 50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD: 254 nm.
6-{Methyl[2-(pyrrolidin-1-yl)ethyl]amino}-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (28)
According to GP2, aryl bromide 12 (50 mg, 0.111 mmol), methyl[2-(pyrrolidin-1-yl)ethyl]amine (17 μL, 0.122 mmol), and potassium carbonate (39 mg, 2.278 mmol) in DMSO (1 mL) gave 27 as a yellow solid (18 mg, 29%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.63 (mc, 4 H), 2.06 (s, 3 H), 2.44, 2.82 (2 mc, 4 H, 2 H), 3.10, 3.29, 3.37 (3 s, 3 H each), 3.51 (mc, 2 H), 7.13, 7.16 (2 mc, 1 H each), 7.34 (d, J = 8.4 Hz, 1 H), 7.80 (dd, J = 7.4, 8.6 Hz, 1 H), 8.38 (d, J = 8.4 Hz, 1 H), 8.49 (dd, J = 1.0, 7.4 Hz, 1 H), 8.69 (dd, J = 1.0, 8.6 Hz, 1 H) ppm; LCMS (method III): tR = 0.78 min; m/z: [M + H]+ = 498; atropisomeric ratio: atrop1/atrop2 = 50:50; tR (atrop1) = 2.30 min; tR (atrop2) = 3.14 min. The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Waters Acquity UPLCMS SingleQuad; column: Acquity UPLC BEH C18 1.7 μm, 50 × 2.1 mm; eluent A: water + 0.1 vol % formic acid (99%), eluent B: ACN; gradient: 0–1.6 min 1–99% B, 1.6–2.0 min 99% B; flow: 0.8 mL/min; temperature: 60 °C; DAD: 210–400 nm.
6-(4-Hydroxybutyl)-2-(1,3,6-trimethyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (29)
According to GP3, aryl bromide 12 (150 mg, 0.333 mmol), but-3-yn-1-ol (50 μL, 0.666 mmol), triethylamine (345 μL, 2.48 mmol), CuI (1.3 mg, 0.007 mmol), and bis(triphenylphosphine)palladium(II) dichloride (4.7 mg, 0.007 mmol) in tetrahydrofuran (1.5 mL) gave, after workup, the violet residue (150 mg, 0.341 mmol) which was directly used in the next step according to GP4 with Pd/C (145 mg, 0.034 mmol) in EtOAc (112 mL). 29 was obtained as a yellow solid (48 mg, 30%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.57, 1.76 (2 mc, 2 H each), 2.08 (s, 3 H), 3.26 (mc, 2 H), 3.29, 3.38 (2 s, 3 H each), 3.46 (mc, 2 H), 4.44 (t, J = 5.2 Hz, 1 H), 7.17 (mc, 2 H), 7.78 (d, J = 7.6 Hz, 1 H), 7.94 (dd, J = 7.2, 8.5 Hz, 1 H), 8.46 (d, J = 7.4 Hz, 1 H), 8.54 (dd, J = 1.0, 7.4 Hz, 1 H), 8.69 (dd, J = 0.9, 8.5 Hz, 1 H) ppm; LCMS (method V): tR = 0.93 min; m/z: [M + H]+ = 444; atropisomeric ratio: atrop 1/atrop 2 = 45:47 (8% impurities); tR (atrop1) = 2.90 min; tR (atrop2) = 3.33 min.
The atropisomeric ratio was determined using the following chiral HPLC method: instrument: Agilent HPLC 1260; column: Chiralpak IE 3 μm 100 × 4.6 mm; eluent A: ACN + 0.1 vol % diethylamine (99%); eluent B: methanol; isocratic: 50% A+50% B; flow: 1.4 mL/min; temperature: 25 °C; DAD 325 nm.
6-(3-Hydroxypropyl)-2-(1-methyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (31)
According to GP1, 6-bromo-2-(1-methyl-2-oxo-2,3-dihydro-1H-benzimidazol-5-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione was prepared from 5-amino-1-methyl-1,3-dihydro-2H-benzimidazol-2-one (3.34 g, 20.5 mmol) and 4-bromo-1,8-naphthalic anhydride (5.96 g, 21.5 mmol) in acetic acid (82 mL). The corresponding aryl bromide was obtained as a yellow solid (6.22 g), part of which (500 mg, 1.18 mmol) was used in the next step, according to GP3, with propargyl alcohol (138 μL, 2.39 mmol), triethylamine (1.3 mL, 8.81 mmol), CuI (4.5 mg, 0.024 mmol), and bis(triphenylphosphine)palladium(II) dichloride (17 mg, 2.37 mmol) in tetrahydrofuran (40 mL). After workup, the residue [200 mg, LCMS (method II): tR = 0.87 min; m/z: [M + H]+ = 397] was used directly in the next step. According to GP4, intermediate alkyne and Pd/C (214 mg, 0.101 mmol) in EtOAc (167 mL) gave 31 as a yellow solid (7 mg, 3%). 1H NMR (400 MHz, [D]6DMSO): δ = 1.87, 3.28 (2 mc, 2 H each), 3.34* (s, 3 H), 3.52, 4.66 (2 mc, 2 H, 1 H), 6.98–7.03 (m, 2 H), 7.18 (d, J = 8.1 Hz, 1 H), 7.76 (d, J = 7.6 Hz, 1 H), 7.92 (dd, J = 7.2, 8.5 Hz, 1 H), 8.42 (d, J = 7.4 Hz, 1 H), 8.51 (dd, J = 1.0, 7.1 Hz, 1 H), 8.65 (dd, J = 1.0, 8.5 Hz, 1 H), 11.00 (bs, 1 H) ppm; *overlapping with water solvent peak of [D]6DMSO; LCMS (method IV): tR = 0.75 min; m/z: [M + H]+ = 402.
Protein Expression and Purification
For screening and biophysical testing cDNAs encoding human BRPF2 BD (residues 563–687, UniProt O95696), human TAF1 BD2 (residues 1501–1635, UniProt P21675), TAF1L (residues 1523–1654, UniProt Q8IZX4), and human CREBBP BD (residues 1081–1197, UniProt Q92793) were cloned into a modified pET28-derived expression vector (pNIC28-Bsa4) using ligation-independent cloning. Alternatively for TR-FRET assays, GST-TAF1 and GST-TAF1L proteins were purchased from BPS Biosciences. Expression constructs encoding the respective target proteins were then transformed into competent E. coli BL21 (DE3)-R3-pRARE2 bacteria (phage-resistant derivative with a pRARE plasmid encoding rare codon tRNAs). Bacteria were then grown at 37 °C in Terrific Broth from overnight cultures until the OD600 reached between 0.6 and 0.8, followed by cooling and addition of 0.2 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) to allow protein expression at 18 °C for 16 h. Cells were lysed in loading buffer (20 mM Hepes pH 7.5, 500 mM NaCl, 20 mM imidazole, 1 mM TCEP, 5% glycerol) in the presence of Protease Inhibitor Cocktail EDTA-free using an EmulsiFlex-C5 high-pressure homogenizer. Lysates were cleared by centrifugation at 14000g for 45 min at 4 °C, after which the supernatant was loaded onto a nickel affinity column (HisTrap Chelating FF 5 mL), followed by washing and elution with a linear imidazole gradient. The eluted proteins were collected and treated overnight with Tobacco Etch Virus protease at 4 °C, followed by a second affinity purification step. Proteins were further purified by size exclusion chromatography (HiLoad 16/600 Superdex 75 GE Healthcare Life Sciences). Protein identity was confirmed by intact mass analysis.
The construct for cocrystallization of human BRPF2 encoded the residues 556–688 with two mutations P566E and V569R integrated in the pNIC28-Bsa4 vector (Addgene, Teddington, U.K.). The expression construct was introduced into competent E. coli BL21 (DE3) bacteria, which were then grown in buffered LB medium until the OD550 reached 1.0. This was followed by cooling and addition of 0.1 mM IPTG to allow protein expression at 17 °C for 24 h. Cells were resuspended in base buffer (10 mM Hepes pH 7.5, 500 mM NaCl, 10 mM imidazole, 0.5 mM TCEP, 5% glycerol) supplemented with 0.1% Triton X100 and 0.025% NP40 in the presence of Protease Inhibitor Cocktail EDTA-free, and passed through a Microfluidizer (3 × 900 bar) for lysis. Lysates were cleared by centrifugation at 30000g at 4 °C for 60 min, after which the supernatant was loaded onto a nickel affinity column (HisTrap Chelating HF 5 × 5 mL), followed by a washing step with 25 mM and subsequent elution step with 250 mM imidazole in base buffer. The eluted proteins were collected, dialyzed against base buffer, and treated with Tobacco Etch Virus protease at 4 °C overnight, followed by a second affinity purification step. Proteins were further purified by size exclusion chromatography using base buffer without imidazole (HiLoad 35/600 Superdex 75 GE Healthcare Life Sciences). Monomeric peak fractions were pooled and concentrated to 13 mg/mL. These samples were directly used for crystallization trials. Protein identity was confirmed by intact mass analysis. For cocrystallization of human TAF1 BD2, the same plasmid construct, expression, and purification conditions as above were used.
Computational Methods
Coordinate Scans
Optimization was performed at the B3LYP/6-31G* level followed by single-point LMP2/cc-pVTZ(-f) calculations.54
Predictions and Calculation Method
BEI and LLE were determined by a proprietary software tool using the following equations at physiological pH: LLE = −log(IC50) – clogD and BEI = [−log(IC50)/MWcorr]*1000. clogD (pH 7.5) as estimated from HPLC retention times was calculated by using a highly predictive method.55−57
Crystallography
BRPF2
Apo BRPF2 crystals were grown at 22 °C using the sitting-drop method by mixing 200 nL of protein (13 mg/mL/0.825 mM) with 100 nL of well solution (100 mM bis-tris pH 6.5, 30% (w/v) PEG 3350). For complex formation, a single crystal was transferred into a soaked drop containing mother liquor supplemented with 10 mM compound (prepared as a 100 mM stock solution in 100% ethylene glycol). After soaking for 48 h, the crystal was briefly immersed in cryoprotection solution consisting of mother liquor supplemented with 10 mM compound and 30% ethylene glycol and then flash-frozen in liquid nitrogen. X-ray data were collected on the Helmholtz-Zentrum Berlin beamline 14–1 at a wavelength of 0.91814 Å and using a PILATUS detector. Data were integrated, scaled, and merged using the programs XDS and SCALA.58,59 The structure was solved by molecular replacement using the program Phaser.60 Initial electron density maps clearly indicated binding of the compound. The model was refined through iterative manual and maximum-likelihood refined using the programs COOT and REFMAC5.58,61 Statistics for the final model are given in the Supporting Information Table S6. Coordinates and structure factors have been submitted to the PDB database and are accessible with the code 5N49.
TAF1
Aliquots of purified TAF1 BD2 were setup for crystallization using a mosquito crystallization robot (TTP Labtech, Royston U.K.). Coarse screens were typically set up in Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (200 + 100 nL, 150 + 150 nL, and 100 + 200 nL). All crystallizations were carried out using the sitting-drop vapor diffusion method at 4 °C. TAF1 crystals with 5 were grown by mixing 100 nL of protein (11.3 mg/mL mixed with 1:3 molar ratio of ligand) with 200 nL of reservoir solution containing 20% PEG 10K, 0.20 M NaCl, and 0.1 M citrate/phosphate pH 4.2. TAF1 complex crystals were cryoprotected using well solution supplemented with additional 20% ethylene glycol for flash-freezing in liquid nitrogen. Data were collected at Diamond beamline I02 at a wavelength of 0.9794 Å. Indexing, integration, and scaling were carried out using autoPROC62 (Global Phasing Limited - GPhL); this package uses XDS,59 POINTLESS,63 AIMLESS,64 and CCP4.65 Initial phases were calculated by molecular replacement with PHASER60 using the apo TAF1 structure (PDB ID: 3UV4). Initial models were built by ARP/wARP66 followed by manual building in COOT.67 Refinement was carried out in REFMAC5.68 GRADE69 (GPhL) was used to generate compound coordinates and cif files. All model validations were carried out using MolProbity.70 Data collection and refinement statistics can be found in Supporting Information Table S6. The model and structure factors have been deposited with PDB accession code 5MG2.
Time-Resolved Fluorescence Resonance Energy Transfer Assays
TR-FRET binding competition assays were performed at RT in 1536- or 384-well black microliter plates in a final volume of 5 μL. Compounds were diluted in DMSO at a single concentration (10 μM for primary screening, 5 and 25 μM for confirmation tests) or in a titration series (3.5-fold, 11-points, maximal test concentration: 20 μM) and dispensed 100× concentrated onto the plates. Then, 25 nM His-tagged BRPF1 (a.a. 626–740), BRPF2 (a.a. 563–688), or BRPF3 (a.a. 592–711), 5 nM GST-tagged TAF1 (a.a. 1519–1651), 5 nM GST-tagged TAF1L (a.a. 1517–1649) or 10 nM BRD4 BD1 (a.a. 67–152) in assay buffer [50 mM Hepes pH 7.5, 50 mM NaCl, 50 mM KF, 0.01% NP40, 0.05% bovine serum albumin (BSA) and 1 mM dithiothreitol (DTT)] was added, and the plates were incubated for 15 min. Finally, 50 nM (25 nM for TAF assays) C-terminal biotinylated synthetic acetylated peptides derived from histone H4 a.a.1–20 (K5,12 diacetylated for BRPF1, 2, 3 or K5,8,12,16 tetra-acetylated for TAF1, TAF1L and BRD4) and detection reagents (10 nM anti-6His-XL665 (Cisbio), 2.5 nM streptavidin Eu (PerkinElmer) or 2.5 nM anti-GST-Tb Cryptate and 3.12 nM streptavidin-XL665 (Cisbio), both in assay buffer) were dispensed and further incubated for at least 1 h. TR-FRET signals corresponding to the number of protein–peptide complexes in equilibrium were acquired either with Viewlux (PerkinElmer) or Pherastar (BMG Lab Technologies) microtiter plate readers. The normalized ratios of the fluorescence emission at 665 nm and at 620–622 nm after excitation at 330–350 nm, at increasing compound concentrations were used to calculate IC50 values by regression analysis based on a four-parameter equation [minimum, maximum, IC50, Hill; Y = Max + (Min – Max)/(1 + (X/IC50)Hill)].
AlphaScreen
Assay conditions: BRD4 BD1, BRPF1B, BRPF2, BRPF3, CECR2, CREBBP, and FALZ/BPTF assays were performed as described previously,71 with minor modifications of the manufacturer’s protocol (PerkinElmer) using the same acetylated H4 peptide. All reagents were diluted in 25 mM Hepes, 100 mM NaCl, 0.1% BSA, pH 7.4 supplemented with 0.05% CHAPS. Plates were filled with 5 μL of the assay buffer followed by 7 μL of biotinylated peptide and His-tagged protein to achieve final assay concentrations of typically 50 nM. Plates were sealed and incubated for a further 30 min, before the addition of 8 μL of the mixture of streptavidin-coated donor beads (12.5 μg/mL) and nickel chelate acceptor beads (12.5 μg/mL) under low light conditions. Plates were foil-sealed for protection from light, and incubated at RT for 60 min before reading.
Isothermal Titration Calorimetry
For BRPF2 and TAF1, experiments were performed in buffer (20 mM Hepes pH 7.5, 100 mM NaCl) on a VP-ITC microcalorimeter (MicroCal) at 20 °C. For CREBBP and TAF1L, experiments were done at 15 °C using a buffer containing 20 mM Hepes pH 7.5, 150 mM NaCl, and 0.5 mM TCEP. Protein concentrations were between 200 and 240 μM, and the compound concentration was set between 18 and 25 μM. The titrations were conducted using an initial injection of 2 μL followed by identical injections of 8 μL (between 24 and 35 injections). The dilution heats were determined in separate experiments and subtracted from the titration data. Thermodynamic parameters were calculated using ΔG = ΔH – TΔS = −RT ln KB, where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively. In all cases, a single binding site model was employed.
Thermal Shift Assay
Thermal melting experiments were carried out using an Mx3005p Real-Time PCR machine (Stratagene), as described.72 Proteins were prepared in 10 mM Hepes pH 7.5, 500 mM NaCl, and assayed in a 96-well plate at a final concentration of 2 μM in 20 μL volume. Compounds were added at a final concentration of 10 μM. SYPRO Orange (Molecular Probes) was added as a fluorescence probe at a dilution of 1:1000. The temperature gradient was 3 °C per min. The melting temperature was calculated by fitting the Boltzmann equation using Prism 5. Thermal shift assays for hit validation were performed with a ViiA Real-Time PCR machine (Thermo Fisher Scientific) in a 384-well plate format. The thermal ramping was 4 °C per minute from 25 to 80 °C. BRPF1 protein was diluted to 2.5 μM in 10 mM HEPES pH 7.5, 150 mM NaCl, and BRPF2 with a protein concentration of 2.7 μM in HEPES pH 7.5 and 300 mM NaCl. Both proteins were measured in the presence or absence of 100 μM of ligand (1% final DMSO concentration) and a 6-fold SYPRO-Orange dye concentration. TSA data were analyzed using a Genedata Assay Analyzer.
NanoBRET Assay
The assay principle has been described.42 Expression vectors were purchased from Promega (Madison, Wisconsin, U.S.A). The NanoLuc luciferase was C-terminally fused to BRPF2 BD (a.a. 560–666), BRPF1 BD (a.a. 626–732), or TAF1 BD2 (a.a. 1528–1640). A BRD4-NanoLuc fusion was used as control. The HaloTag was C-terminally fused to histone H3.3 or H4. The resulting plasmids were transfected into HCT116 cells using standard procedures. Following addition of the HaloTag ligand, the cells were treated with compound (10 nM to 10 μM) for 4 or 24 h, and the IC50 values were calculated.
LeadProfilingScreen and Kinase Panel
The LeadProfilingScreen containing 68 different targets and the kinase panel experiment (300 kinases) were performed by Eurofins, Cerep Panlabs (Taipei, Taiwan).
Cell Proliferation Assay
Cell lines were obtained from ATCC (Manassas, Virginia, USA) or the DSMZ (Braunschweig, Germany), authenticated, and grown following standard procedures. The cells were treated with compound while in the logarithmic growth phase, and their viability was determined by AlamarBlue staining (ThermoFisher, Waltham, Massachusetts, U.S.A.).
Permeability Assay
Caco2 cells purchased from the DSMZ were seeded at a density of 4.5 × 104 cells per well and grown for 15 days in DMEM with typical supplements. Cells were kept at 37 °C in a humidified 5% CO2 atmosphere. Before the permeation assay was run, the culture medium was replaced by a FCS-free Hepes carbonate transport buffer (pH 7.2). For assessment of monolayer integrity, the transepithelial electrical resistance was measured. Test compounds were predissolved in DMSO and added to either the apical or basolateral compartment at a final concentration of 2 μM. Before and after 2 h incubation at 37 °C, samples were taken from both compartments. Analysis of compound content was conducted after precipitation with methanol by LC/MS/MS analysis. Permeability (Papp) was calculated in the apical to basolateral (A → B) and basolateral to apical (B → A) directions. The efflux ratio basolateral (B) to apical (A) was calculated by dividing Papp(B-A) by Papp(A-B). Reference compounds were analyzed in parallel as assay control.
Pharmacokinetics
Metabolic Stability in Hepatocytes
A hepatocyte cell suspension of the respective species was filtered through sterile gauze in 50 mL Falcon tubes and centrifuged at 50g for 3 min at RT. The cell pellet was resuspended in 30 mL of WME and centrifuged through a Percoll gradient twice at 100g. The hepatocytes were washed again with WME and resuspended in medium containing 5% FCS. Liver cells were distributed in WME containing 5% FCS to glass vials at a density of 1.0 × 106 vital cells/mL. Compound 5 was added at a final concentration of 1 μM. During incubation, the hepatocyte suspensions were continuously shaken at 580 rpm and aliquots were taken at 2, 8, 16, 30, 45, and 90 min, to which equal volumes of cold methanol were immediately added. Samples were frozen at −20 °C overnight, and then centrifuged for 15 min at 3000 rpm, and the supernatant was analyzed by LC/MS/MS. The half-life of 5 was determined from the concentration–time plot. The intrinsic clearances were calculated from the half-life.
Metabolic Stability in Liver Microsomes
The in vitro metabolic stability was determined by incubating compounds at 1 μM concentration in a suspension of liver microsomes in 100 mM phosphate buffer, pH 7.4 (NaH2PO4·H2O + Na2HPO4·2H2O) and at a protein concentration of 0.5 mg/mL at 37 °C. The microsomes were activated by adding a cofactor mix containing 8 mM glucose-6-phosphate, 4 mM MgCl2, 0.5 mM NADP, and 1 IU/mL G-6-P-dehydrogenase in phosphate buffer, pH 7.4. The metabolic assay was started shortly afterward by adding 5 to the incubation in a final volume of 1 mL. During incubation, the microsomal suspensions were continuously shaken at 580 rpm and aliquots were taken at 2, 8, 16, 30, 45, and 60 min, to which equal volumes of cold methanol were immediately added. Samples were frozen at −20 °C overnight and then centrifuged for 15 min at 3000 rpm, and the supernatant was analyzed by LC/MS/MS. The half-life of 5 was determined from the concentration–time plot. The intrinsic clearances were calculated from the half-life.
In Vivo Rat Pharmacokinetics
Compound 5 was administered to three male Wistar rats per arm, either intravenously or intragastrally formulated as solutions. 5 was given as i.v. bolus, and blood samples were taken at 2 min, 8 min, 15 min, 30 min, 45 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h after dosing. For pharmacokinetics after intragastral administration, 5 was given intragastrally to fasted rats and blood samples were taken at 5 min, 15 min, 30 min, 45 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h after dosing. Blood was collected into lithium-heparin tubes (Monovettes, Sarstedt) and centrifuged for 15 min at 3000 rpm. An aliquot of 100 μL from the supernatant (plasma) was taken and precipitated by addition of 400 μL of cold ACN and frozen at −20 °C overnight. Samples were thawed and centrifuged at 3000 rpm, 4 °C for 20 min. Aliquots of the supernatants were submitted to LC/MS/MS analysis employing a validated method on an Agilent 1200 HPLC coupled to a Sciex API 5500 instrument. Standard pharmacokinetic parameters were calculated by noncompartmental analysis using dedicated software.
Physicochemistry
Reversed-Phase HPLC Method for Lipophilicity (logD)
logD (pH 7.5) was determined by reversed-phase HPLC at physiological pH, as described.73 A homologous series of n-alkan-2-ones (C3–C16, 0.02 mol in ACN) was used for calibration. Test compounds were applied as 0.67 mM DMSO stock solutions in ACN/H2O 1:1. The lipophilicity of compounds was then assessed by comparison to the calibration curve.
High-Throughput Determination of Aqueous Drug Solubility (100 mM in DMSO)
The high-throughput screening method to determine aqueous drug solubility was performed as described previously.74 Test compounds were applied as 1 mM solutions in DMSO. After the addition of a pH 6.5 buffer, the solution was shaken for 24 h at RT. Undissolved material was separated by filtration. The compound dissolved in the supernatant was diluted accordingly and quantified by HPLC-MS/MS.
Acknowledgments
We are indebted to M. Hitchcock, C. Scholten, U. Egner, S. Gründemann, U. Ganzer, G. Depke (Bayer AG), and P. J. Brown and J. Porter (SGC Oxford) for their support throughout the project. We thank J. Méndez and D. Daniels (Promega Corporation) for excellent advice with the NanoBRET assays, and DiscoverX Corp. for performing the BROMOscan screening. The expert technical assistance of J. Blaese, N. Dittmar, R. Karmauss, A. Mattstedt, S. Wolf, L. Schaelicke, V. Trepte, C. Bölitz, F. Knoth, H. Muckwar, I. Herms, and M. Wolfrum is gratefully acknowledged. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust [106169/ZZ14/Z].
Glossary
Abbreviations
- ACN
acetonitrile
- BD
bromodomain
- BEI
binding efficiency index
- BET
bromodomain and extra-terminal
- BRPF
bromodomain and PHD finger
- CREBBP
cyclic-AMP response element binding protein
- DAD
diode array detector
- DMSO
dimethyl sulfoxide
- EP300
adenoviral E1A binding protein
- equiv
equivalent, stoichiometric equivalents
- GP
general procedure
- HAT
histone acetyltransferase
- HPLC
high-performance liquid chromatography
- HTS
high-throughput screening
- ITC
isothermal titration calorimetry
- KAc
acetyl-lysine
- LCMS
liquid chromatography–mass spectrometry
- LLE
lipophilic ligand efficiency
- NMR
nuclear magnetic resonance spectroscopy
- tR
retention time
- RT
room temperature
- SAR
structure–activity relationship
- TAF1
TATA box binding protein-associated factor
- TR-FRET
time-resolved fluorescence energy transfer
- TSA
thermal shift assay
- UPLCMS
ultra-performance liquid chromatography–mass spectrometry
- vol
volume
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00306.
Binding mode of 5 in TAF1 BD2 (Figure S1), cellular activity of 7, 8, and 13 in NanoBRET assay (Table S1), TSA results for 13 in a panel of 48 BDs (Table S2), molecular properties and physicochemical profile of 5 (Table S3), BD selectivity profile of 5 (Table S4), antiproliferative activity of 5 (Table S5), and crystallographic data for 7 in BRPF2 BD and 5 in TAF1 BD2 (Table S6) (PDF)
Molecular formula strings (CSV)
Accession Codes
PDB code for the structure of the BRPF2 BD in complex with 7 is 5N49. PDB code for the structure of the TAF1 BD2 with 5 is 5MG2. Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
∥ (J.M.) Merck KGaA, Frankfurter Strasse 250, 64293 Darmstadt, Germany.
The authors declare the following competing financial interest(s): L.B., C.D.C., S.S., A.E.F.-M., S.J.H., A.t.L., T.S., D.S., J.M., V.P., J.W., I.V.H., and B.H. are or have been employees and/or are stockholders of Bayer AG.
Supplementary Material
References
- Shi J.; Wang E.; Milazzo J. P.; Wang Z.; Kinney J. B.; Vakoc C. R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Gelato K. A.; Shaikhibrahim Z.; Ocker M.; Haendler B. Targeting epigenetic regulators for cancer therapy: modulation of bromodomain proteins, methyltransferases, demethylases, and microRNAs. Expert Opin. Ther. Targets 2016, 20, 783–799. 10.1517/14728222.2016.1134490. [DOI] [PubMed] [Google Scholar]
- Ferri E.; Petosa C.; McKenna C. E. Bromodomains: structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Tomkinson N. C.; Prinjha R. K.; Humphreys P. G. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr. Opin. Chem. Biol. 2016, 33, 58–66. 10.1016/j.cbpa.2016.05.028. [DOI] [PubMed] [Google Scholar]
- Crawford T. D.; Romero F. A.; Lai K. W.; Tsui V.; Taylor A. M.; de Leon Boenig G.; Noland C. L.; Murray J.; Ly J.; Choo E. F.; Hunsaker T. L.; Chan E. W.; Merchant M.; Kharbanda S.; Gascoigne K. E.; Kaufman S.; Beresini M. H.; Liao J.; Liu W.; Chen K. X.; Chen Z.; Conery A. R.; Cote A.; Jayaram H.; Jiang Y.; Kiefer J. R.; Kleinheinz T.; Li Y.; Maher J.; Pardo E.; Poy F.; Spillane K. L.; Wang F.; Wang J.; Wei X.; Xu Z.; Xu Z.; Yen I.; Zawadzke L.; Zhu X.; Bellon S.; Cummings R.; Cochran A. G.; Albrecht B. K.; Magnuson S. Discovery of a potent and selective in vivo probe (GNE-272) for the bromodomains of CBP/EP300. J. Med. Chem. 2016, 59, 10549–10563. 10.1021/acs.jmedchem.6b01022. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Brown N.; Knapp S.; Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55, 7346–7359. 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M.; Philpott M.; Muller S.; Schulze J.; Badock V.; Eberspacher U.; Moosmayer D.; Bader B.; Schmees N.; Fernandez-Montalvan A.; Haendler B. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. 10.1074/jbc.M113.523019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Cote A.; Magnuson S. Disrupting acetyl-lysine recognition: progress in the development of bromodomain Inhibitors. J. Med. Chem. 2016, 59, 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- Popp T. A.; Tallant C.; Rogers C.; Fedorov O.; Brennan P. E.; Muller S.; Knapp S.; Bracher F. Development of selective CBP/P300 benzoxazepine bromodomain inhibitors. J. Med. Chem. 2016, 59, 8889–8912. 10.1021/acs.jmedchem.6b00774. [DOI] [PubMed] [Google Scholar]
- Myrianthopoulos V.; Gaboriaud-Kolar N.; Tallant C.; Hall M. L.; Grigoriou S.; Brownlee P. M.; Fedorov O.; Rogers C.; Heidenreich D.; Wanior M.; Drosos N.; Mexia N.; Savitsky P.; Bagratuni T.; Kastritis E.; Terpos E.; Filippakopoulos P.; Muller S.; Skaltsounis A. L.; Downs J. A.; Knapp S.; Mikros E. Discovery and optimization of a selective ligand for the switch/sucrose nonfermenting-related bromodomains of polybromo protein-1 by the use of virtual screening and hydration analysis. J. Med. Chem. 2016, 59, 8787–8803. 10.1021/acs.jmedchem.6b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frederiksen M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V.; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Muller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundstrom M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P. J.; Muller S. Open access chemical probes for epigenetic targets. Future Med. Chem. 2015, 7, 1901–1917. 10.4155/fmc.15.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein B. J.; Lalonde M. E.; Cote J.; Yang X. J.; Kutateladze T. G. Crosstalk between epigenetic readers regulates the MOZ/MORF HAT complexes. Epigenetics 2014, 9, 186–193. 10.4161/epi.26792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L.; Yan K.; Zou J.; Zhao H.; Bertos N. R.; Park M.; Wang E.; Yang X. J. The chromatin regulator Brpf1 regulates embryo development and cell proliferation. J. Biol. Chem. 2015, 290, 11349–11364. 10.1074/jbc.M115.643189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima Y.; Miyagi S.; Saraya A.; Negishi M.; Endoh M.; Endo T. A.; Toyoda T.; Shinga J.; Katsumoto T.; Chiba T.; Yamaguchi N.; Kitabayashi I.; Koseki H.; Iwama A. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 2011, 118, 2443–2453. 10.1182/blood-2011-01-331892. [DOI] [PubMed] [Google Scholar]
- Yan K.; You L.; Degerny C.; Ghorbani M.; Liu X.; Chen L.; Li L.; Miao D.; Yang X. J. The chromatin regulator BRPF3 preferentially activates the HBO1 acetyltransferase but is dispensable for mouse development and survival. J. Biol. Chem. 2016, 291, 2647–2663. 10.1074/jbc.M115.703041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. I.; Kim M. S.; Jang Y. K. The BRPF2/BRD1-MOZ complex is involved in retinoic acid-induced differentiation of embryonic stem cells. Exp. Cell Res. 2016, 346, 30–39. 10.1016/j.yexcr.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Shima H.; Yamagata K.; Aikawa Y.; Shino M.; Koseki H.; Shimada H.; Kitabayashi I. Bromodomain-PHD finger protein 1 is critical for leukemogenesis associated with MOZ-TIF2 fusion. Int. J. Hematol. 2014, 99, 21–31. 10.1007/s12185-013-1466-x. [DOI] [PubMed] [Google Scholar]
- Bennett J.; Fedorov O.; Tallant C.; Monteiro O.; Meier J.; Gamble V.; Savitsky P.; Nunez-Alonso G. A.; Haendler B.; Rogers C.; Brennan P. E.; Muller S.; Knapp S. Discovery of a chemical tool inhibitor targeting the bromodomains of TRIM24 and BRPF. J. Med. Chem. 2016, 59, 1642–1647. 10.1021/acs.jmedchem.5b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demont E. H.; Bamborough P.; Chung C. W.; Craggs P. D.; Fallon D.; Gordon L. J.; Grandi P.; Hobbs C. I.; Hussain J.; Jones E. J.; Le Gall A.; Michon A. M.; Mitchell D. J.; Prinjha R. K.; Roberts A. D.; Sheppard R. J.; Watson R. J. 1,3-dimethyl benzimidazolones are potent, selective inhibitors of the BRPF1 bromodomain. ACS Med. Chem. Lett. 2014, 5, 1190–1195. 10.1021/ml5002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer W. S.; Poncet-Montange G.; Liu G.; Petrocchi A.; Reyna N.; Subramanian G.; Theroff J.; Yau A.; Kost-Alimova M.; Bardenhagen J. P.; Leo E.; Shepard H. E.; Tieu T. N.; Shi X.; Zhan Y.; Zhao S.; Barton M. C.; Draetta G.; Toniatti C.; Jones P.; Geck Do M.; Andersen J. N. Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. J. Med. Chem. 2016, 59, 1440–1454. 10.1021/acs.jmedchem.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Barnett H. A.; Becher I.; Bird M. J.; Chung C. W.; Craggs P. D.; Demont E. H.; Diallo H.; Fallon D. J.; Gordon L. J.; Grandi P.; Hobbs C. I.; Hooper-Greenhill E.; Jones E. J.; Law R. P.; Le Gall A.; Lugo D.; Michon A. M.; Mitchell D. J.; Prinjha R. K.; Sheppard R. J.; Watson A. J.; Watson R. J. GSK6853, a chemical probe for inhibition of the BRPF1 bromodomain. ACS Med. Chem. Lett. 2016, 7, 552–557. 10.1021/acsmedchemlett.6b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- Kandiah E.; Trowitzsch S.; Gupta K.; Haffke M.; Berger I. More pieces to the puzzle: recent structural insights into class II transcription initiation. Curr. Opin. Struct. Biol. 2014, 24, 91–97. 10.1016/j.sbi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Louder R. K.; He Y.; Lopez-Blanco J. R.; Fang J.; Chacon P.; Nogales E. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature 2016, 531, 604–609. 10.1038/nature17394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. J.; Page D. C. Functional substitution for TAF(II)250 by a retroposed homolog that is expressed in human spermatogenesis. Hum. Mol. Genet. 2002, 11, 2341–2346. 10.1093/hmg/11.19.2341. [DOI] [PubMed] [Google Scholar]
- Domingo A.; Amar D.; Grutz K.; Lee L. V.; Rosales R.; Bruggemann N.; Jamora R. D.; Cutiongco-Dela Paz E.; Rolfs A.; Dressler D.; Walter U.; Krainc D.; Lohmann K.; Shamir R.; Klein C.; Westenberger A. Evidence of TAF1 dysfunction in peripheral models of X-linked dystonia-parkinsonism. Cell. Mol. Life Sci. 2016, 73, 3205–3215. 10.1007/s00018-016-2159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rawe J. A.; Wu Y.; Dorfel M. J.; Rope A. F.; Au P. Y.; Parboosingh J. S.; Moon S.; Kousi M.; Kosma K.; Smith C. S.; Tzetis M.; Schuette J. L.; Hufnagel R. B.; Prada C. E.; Martinez F.; Orellana C.; Crain J.; Caro-Llopis A.; Oltra S.; Monfort S.; Jimenez-Barron L. T.; Swensen J.; Ellingwood S.; Smith R.; Fang H.; Ospina S.; Stegmann S.; Den Hollander N.; Mittelman D.; Highnam G.; Robison R.; Yang E.; Faivre L.; Roubertie A.; Riviere J. B.; Monaghan K. G.; Wang K.; Davis E. E.; Katsanis N.; Kalscheuer V. M.; Wang E. H.; Metcalfe K.; Kleefstra T.; Innes A. M.; Kitsiou-Tzeli S.; Rosello M.; Keegan C. E.; Lyon G. J. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am. J. Hum. Genet. 2015, 97, 922–932. 10.1016/j.ajhg.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown M. R.; Shaw D. L.; Fu H.; Liu S.; Xu X.; Marineau J. J.; Huang Y.; Zhang X.; Buckley D. L.; Kadam A.; Zhang Z.; Blacklow S. C.; Qi J.; Zhang W.; Bradner J. E. Biased multicomponent reactions to develop novel bromodomain inhibitors. J. Med. Chem. 2014, 57, 9019–9027. 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish P. V.; Filippakopoulos P.; Bish G.; Brennan P. E.; Bunnage M. E.; Cook A. S.; Federov O.; Gerstenberger B. S.; Jones H.; Knapp S.; Marsden B.; Nocka K.; Owen D. R.; Philpott M.; Picaud S.; Primiano M. J.; Ralph M. J.; Sciammetta N.; Trzupek J. D. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J. Med. Chem. 2012, 55, 9831–9837. 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C. W. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587–596. 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Fedorov O.; Filippakopoulos P.; Martin S.; Picaud S.; Tumber A.; Wells C.; Olcina M. M.; Freeman K.; Gill A.; Ritchie A. J.; Sheppard D. W.; Russell A. J.; Hammond E. M.; Knapp S.; Brennan P. E.; Conway S. J. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J. Med. Chem. 2013, 56, 3217–3227. 10.1021/jm301588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Cote A.; Nasveschuk C. G.; Taylor A. M.; Harmange J. C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S. Bangalore Srinivasamurthy, N.; Birudukota, S.; Albrecht, B. K. Discovery, design, and optimization of isoxazole azepine BET inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Vakoc C. R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M.; Gelato K. A.; Fernandez-Montalvan A.; Siegel S.; Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. 10.2217/epi.14.91. [DOI] [PubMed] [Google Scholar]
- Sdelci S.; Lardeau C. H.; Tallant C.; Klepsch F.; Klaiber B.; Bennett J.; Rathert P.; Schuster M.; Penz T.; Fedorov O.; Superti-Furga G.; Bock C.; Zuber J.; Huber K. V.; Knapp S.; Muller S.; Kubicek S. Mapping the chemical chromatin reactivation landscape identifies BRD4-TAF1 cross-talk. Nat. Chem. Biol. 2016, 12, 504–510. 10.1038/nchembio.2080. [DOI] [PubMed] [Google Scholar]
- Crawford T. D.; Tsui V.; Flynn E. M.; Wang S.; Taylor A. M.; Cote A.; Audia J. E.; Beresini M. H.; Burdick D. J.; Cummings R.; Dakin L. A.; Duplessis M.; Good A. C.; Hewitt M. C.; Huang H. R.; Jayaram H.; Kiefer J. R.; Jiang Y.; Murray J.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S.; Cochran A. G. Diving into the water: inducible binding conformations for BRD4, TAF1(2), BRD9, and CECR2 bromodomains. J. Med. Chem. 2016, 59, 5391–5402. 10.1021/acs.jmedchem.6b00264. [DOI] [PubMed] [Google Scholar]
- BAY-299 A probe for BRD1 and TAF1; Structural Genomics Consortium: Oxford; http://www.thesgc.org/chemical-probes/BAY-299 (accessed Apr 5, 2017).
- Machleidt T.; Woodroofe C. C.; Schwinn M. K.; Mendez J.; Robers M. B.; Zimmerman K.; Otto P.; Daniels D. L.; Kirkland T. A.; Wood K. V. NanoBRET-A novel BRET platform for the analysis of protein-protein interactions. ACS Chem. Biol. 2015, 10, 1797–1804. 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- O̅ki M.Recent advances in atropisomerism. In Topics in Stereochemistry; Elliel E. L., Allinger N., Willen S. H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2007; pp 1–81. [Google Scholar]
- Bringmann G.; Gulder T.; Gulder T. A.; Breuning M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 2011, 111, 563–639. 10.1021/cr100155e. [DOI] [PubMed] [Google Scholar]
- Clayden J.; Moran W. J.; Edwards P. J.; LaPlante S. R. The challenge of atropisomerism in drug discovery. Angew. Chem., Int. Ed. 2009, 48, 6398–6401. 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]
- Laplante S. R.; L D. F.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P.; Edwards P. J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54, 7005–7022. 10.1021/jm200584g. [DOI] [PubMed] [Google Scholar]
- LaPlante S. R.; Edwards P. J.; Fader L. D.; Jakalian A.; Hucke O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 6, 505–513. 10.1002/cmdc.201000485. [DOI] [PubMed] [Google Scholar]
- Igoe N.; Bayle E. D.; Fedorov O.; Tallant C.; Savitsky P.; Rogers C.; Owen D. R.; Deb G.; Somervaille T. C.; Andrews D. M.; Jones N.; Cheasty A.; Ryder H.; Brennan P. E.; Muller S.; Knapp S.; Fish P. V. Design of a biased potent small molecule inhibitor of the bromodomain and PHD finger-containing (BRPF) proteins suitable for cellular and in vivo studies. J. Med. Chem. 2017, 60, 668–680. 10.1021/acs.jmedchem.6b01583. [DOI] [PubMed] [Google Scholar]
- Eggert E.; Hillig R. C.; Koehr S.; Stockigt D.; Weiske J.; Barak N.; Mowat J.; Brumby T.; Christ C. D.; Ter Laak A.; Lang T.; Fernandez-Montalvan A. E.; Badock V.; Weinmann H.; Hartung I. V.; Barsyte-Lovejoy D.; Szewczyk M.; Kennedy S.; Li F.; Vedadi M.; Brown P. J.; Santhakumar V.; Arrowsmith C. H.; Stellfeld T.; Stresemann C. Discovery and characterization of a highly potent and selective aminopyrazoline-based in vivo probe (BAY-598) for the protein lysine methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. 10.1021/acs.jmedchem.5b01890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram M. S.; Shen Y.; Szewczyk M. M.; Wu H.; Senisterra G.; Li F.; Butler K. V.; Kaniskan H. U.; Speed B. A.; dela Sena C.; Dong A.; Zeng H.; Schapira M.; Brown P. J.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Liu J.; Vedadi M.; Jin J. A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Szewczyk M. M.; Eram M. S.; Smil D.; Kaniskan H. U.; Ferreira de Freitas R.; Senisterra G.; Li F.; Schapira M.; Brown P. J.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Liu J.; Vedadi M.; Jin J. Discovery of a potent, selective, and cell-active dual inhibitor of protein arginine methyltransferase 4 and protein arginine methyltransferase 6. J. Med. Chem. 2016, 59, 9124–9139. 10.1021/acs.jmedchem.6b01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Bamborough P.; Bannister A. J.; Becher I.; Bit R. A.; Che K. H.; Chung C. W.; Dittmann A.; Drewes G.; Drewry D. H.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Michon A. M.; Molnar J.; Robson S. C.; Tomkinson N. C.; Kouzarides T.; Prinjha R. K.; Humphreys P. G. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J. Med. Chem. 2016, 59, 1425–1439. 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L. J.; Koegl M.; Bader G.; Cockcroft X. L.; Fedorov O.; Fiegen D.; Gerstberger T.; Hofmann M. H.; Hohmann A. F.; Kessler D.; Knapp S.; Knesl P.; Kornigg S.; Muller S.; Nar H.; Rogers C.; Rumpel K.; Schaaf O.; Steurer S.; Tallant C.; Vakoc C. R.; Zeeb M.; Zoephel A.; Pearson M.; Boehmelt G.; McConnell D. Structure-based design of an in vivo active selective BRD9 inhibitor. J. Med. Chem. 2016, 59, 4462–4475. 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R.; Lobell M.; Goller A. H.; Krenz U.; Schoenneis R.; Clark R. D.; Hillisch A. Best of both worlds: combining pharma data and state of the art modeling technology to improve in silico pKa prediction. J. Chem. Inf. Model. 2015, 55, 389–397. 10.1021/ci500585w. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Erlanson D. A.; Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H.; Richmond N. J. Validity of ligand efficiency metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. 10.1021/ml500146d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz M. D.; Dusuel S.; Schmidt K. P.; Vidal J. Topological phase transitions in the golden string-net model. Phys. Rev. Lett. 2013, 110, 147203. 10.1103/PhysRevLett.110.147203. [DOI] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G.; Cohen S. X.; Lamzin V. S.; Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Smart O. S.; Womack T. O.; Sharff A.; Flensburg C.; Keller P.; Paciorek W.; Vonrhein C.; Bricogne G.. Grade v1.102. http://www.globalphasing.com (accessed Sep 17, 2016).
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. D. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7, 2899–2908. 10.1039/c1mb05099k. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Niesen F. H.; Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. 10.1007/978-1-61779-337-0_7. [DOI] [PubMed] [Google Scholar]
- Minick D. J.; Frenz J. H.; Patrick M. A.; Brent D. A. A comprehensive method for determining hydrophobicity constants by reversed-phase high-performance liquid chromatography. J. Med. Chem. 1988, 31, 1923–1933. 10.1021/jm00118a010. [DOI] [PubMed] [Google Scholar]
- Onofrey T.; Kazan G. Performance and correlation of a 96-well high-throughput screening method to determine aqueous drug solubility. Lit. No. AN1731EN00 (2003). Millipore Corporation Application Note.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.