

Selection acted repeatedly on regions that may regulate the expression of genes underlying coloration differences in seedeaters.
Keywords: Capuchino Seedeaters, Genomic Scan, sexual selection, Melanogenesis, Neotropics
Abstract
Instances of recent and rapid speciation are suitable for associating phenotypes with their causal genotypes, especially if gene flow homogenizes areas of the genome that are not under divergent selection. We study a rapid radiation of nine sympatric bird species known as capuchino seedeaters, which are differentiated in sexually selected characters of male plumage and song. We sequenced the genomes of a phenotypically diverse set of species to search for differentiated genomic regions. Capuchinos show differences in a small proportion of their genomes, yet selection has acted independently on the same targets in different members of this radiation. Many divergent regions contain genes involved in the melanogenesis pathway, with the strongest signal originating from putative regulatory regions. Selection has acted on these same genomic regions in different lineages, likely shaping the evolution of cis-regulatory elements, which control how more conserved genes are expressed and thereby generate diversity in classically sexually selected traits.
INTRODUCTION
Understanding the processes that shape biological diversity at the molecular level is a central goal of evolutionary biology. Nonmodel organisms with divergent traits can be powerful systems in which to discover the genetic basis of distinct phenotypes. In some cases, the same genes independently generate similar phenotypes in different taxa [for example, wing color patterns in butterflies (1)]. Although some phenotypic differences are caused by mutations in coding regions of causal genes (for example, coloration in deer mice) (2–4), others arise through selection on areas that regulate the expression of these genes (1). In addition, macromutations such as chromosomal inversions can suppress local recombination, leading to the formation of supergenes, which allow genes to coevolve and produce complex traits (for example, plumage coloration and mating behavior in ruffs and white-throated sparrows (5–7)]. Despite this wealth of knowledge, connecting the evolution of phenotype to the genetic mechanisms that generate reproductive isolation and, ultimately, speciation remains challenging in most systems. One of the best understood cases of the genetics of speciation in animals comes from Darwin’s finches, where the morphological traits that are under selection have been identified (8), and both the molecular mechanisms that generate those traits (9–11) and the effect of trait variation on reproductive isolation are known (12, 13).
From a genomic perspective, Darwin’s finches have offered two key advantages for researchers searching for the molecular basis of the phenotypes that distinguish these birds. First, the finches have speciated recently, which translates into a relatively low level of background genomic differentiation. Those few areas of the genome that are highly divergent among species contain candidate loci that may have shaped the evolution of the adaptive radiation or are still under strong selection, even in the face of gene flow (10, 11). Second, there are many different species in the radiation with comparable divergence times, which leads to similarly low background genomic differentiation across multiple possible comparisons. This allows researchers to compare the genomes of more than one pair of forms with similar differences in phenotype and assess the degree to which molecular evolution has happened in parallel (11).
The study of additional biological systems that share these tractable attributes, but which have been driven by forces other than natural selection on foraging-related phenotypes, can provide further insights into the genomics of traits that may lead to speciation. Here, we focus on a group of finch-like birds from continental South America, known as southern capuchino seedeaters (14, 15), which have diversified as a biological radiation. Capuchinos share many characteristics with Darwin’s finches (16), yet differ in that they seem to have diversified primarily via sexual selection on plumage traits that are likely melanin-based rather than via natural selection on foraging-related traits (14, 15). Capuchino seedeaters belong to the genus Sporophila and are in the same family as Darwin’s finches (Thraupidae) (17), and both radiations show comparable speciation rates that are much greater than those of all other groups within that large family (17). The southern capuchinos are nine predominantly sympatric species that occur in Neotropical grasslands (Fig. 1A): Sporophila bouvreuil (bou), Sporophila pileata (pil), Sporophila cinnamomea (cin), Sporophila ruficollis (ruf), Sporophila melanogaster (mel), Sporophila nigrorufa (nig), Sporophila palustris (pal), Sporophila hypochroma (hypoch), and Sporophila hypoxantha (hypox). Capuchinos are sexually dimorphic, and males from different species differ in secondary sexual characters, such as the plumage coloration patterns and songs that they use to attract mates and to defend their territories (14). Males defend their territories during simulated intrusions of conspecifics but not from sympatric male capuchinos of other species (18). The species in the group are otherwise indistinguishable morphologically (19, 20) [and very similar ecologically (21, 22)] to the extent that females and juveniles lacking male secondary sexual characters cannot be identified to species even in the hand (14, 15). Despite their phenotypic diversity in male plumage, southern capuchinos show extremely low levels of genetic differentiation (14) and, except for S. bouvreuil, cannot be assigned reliably to species even using thousands of genome-wide single-nucleotide polymorphisms (SNP) (15). This genetic homogeneity is a result of the groups’ relatively recent origin, which split from its sister species during the Pleistocene, and likely the product of both incomplete lineage sorting and ongoing genetic admixture (14, 15). The apparent genetic homogeneity among southern capuchinos, despite their distinct phenotypes that are maintained in sympatry, led us to hypothesize that these species differences in male plumage may be the result of strong selection at a few key loci. We therefore sequenced and compared the genomes of the nine southern capuchinos with the objective of locating such loci and to test whether the same targets of selection have independently shaped phenotypic diversity across different species.
Fig. 1. Genomic landscapes in southern capuchino seedeaters.
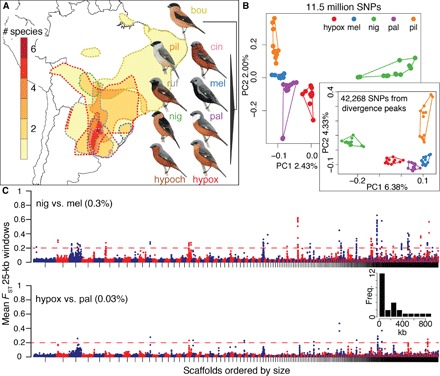
(A) Map indicating the extent of range overlap in the nine species; note that up to six species breed sympatrically in northeastern Argentina. The range of each species is outlined by dashed lines, with colors matching species names. The schematic phylogeny was obtained from Campagna et al. (see text for name abbreviations) (14, 15). (B) PCA including 56 individuals of five species genotyped at ~11.5 million SNPs and a second PCA (60 individuals) using SNPs from divergence peaks alone. Four outlier individuals were omitted from the first PCA (see fig. S1 and Materials and Methods for details). (C) Manhattan plots for nig versus mel (top) and hypox versus pal (bottom); 12 individuals per species. Each circle indicates the mean FST value for all the SNPs within a nonoverlapping 25-kb window. Scaffolds in the reference genome were sorted by decreasing size and are indicated by alternating colors. The threshold for calling divergent windows is indicated by the dashed red line, and the percentage of total elevated windows is noted next to each comparison. The inset is a histogram showing the width distribution (in kilobases) for the 25 divergence peaks we identified.
RESULTS
Individual capuchinos clustered by species in a principal components analysis (PCA) derived from 11.5 million SNPs, yet the percentage of variation explained by the first two principal components was low, suggesting that a small proportion of these SNPs could be driving the pattern (Fig. 1B and fig. S1). To search for divergent areas of the genome, we compared FST values for nonoverlapping 25-kb windows across the 10 possible pairwise comparisons of five species (those for which our sample sizes were larger). The mean differentiation across all windows/comparisons was low (mean FST = 0.008 and SD = 0.015 across ~43.5 thousand windows), yet we found a number of divergence peaks with highly elevated FST with respect to this low background. For example, Fig. 1C shows two Manhattan plots; the upper graph (nig versus mel) had the largest number of elevated windows (0.3%) among all comparisons and involved two allopatric species with small ranges. The bottom comparison (hypox versus pal) had an order of magnitude fewer elevated windows (0.03%) and compared two species with highly overlapping ranges. All other pairwise comparisons had a number of divergence peaks that ranged between the extremes shown in Fig. 1C (fig. S2). We identified a total of 25 divergence peaks with elevated FST (>0.2) that are candidate targets of selection driving species differences among capuchinos (Table 1); these peaks range in width from 25 to 840 kb (average, ~243 kb; inset in Fig. 1C). SNPs from these divergence peaks alone can be used to assign individuals to species in a PCA (inset in Fig. 1B). We found 99 SNPs that were fixed (FST = 1) in at least one comparison across all pairwise combinations of five capuchinos; these represented 65 different sites in the genome. Because our sample sizes were low for the remaining capuchino species (bou, cin, hypoch, and ruf), we did not use them to identify divergence peaks. A PCA using the SNPs from under the peaks showed some overlap between taxa when we included all nine species (mainly between ruf versus hypox and bou versus pil; fig. S1). It is therefore possible that there are some additional undetected areas of the genome that are involved in capuchino seedeater differentiation.
Table 1. Areas of the genome that are highly differentiated in capuchino seedeaters.
| Scaffold | Chromosome |
Peak size (kb) |
Highest FST for 5-kb window |
SNPs with FST > 0.85 (=1)* |
Melanogenesis gene |
Function of melanogenesis gene† |
Total no. of genes (known function)‡ |
Figure |
| 252 | 20 | 90 | 0.83 | 383 (57) | ASIP | Induces melanocytes to synthesize pheomelanin (yellow) instead of eumelanin (black/brown) |
6 (4) | Fig. 2 |
| 762 | 11 | 35 | 0.63 | 178 (22) | — | — | 9 (9) | Fig. 3 |
| 412 | 1A | 205 | 0.70 | 113 | KITL | Stimulates melanocyte proliferation |
1 (1) | Fig. 2 |
| 308 | Unknown | 140 | 0.65 | 112 (14) | — | — | 4 (4) | Fig. 3 |
| 430 | 1 | 85 | 0.48 | 105 | — | — | 4 (1) | Fig. 3 |
| 404 | Z | 765 | 0.60 | 92 (5) | SLC45A2 | Transports substances needed for melanin synthesis |
21 (20) | Fig. 2 |
| 257 (A) | Z | 500 | 0.53 | 70 | TYRP1 | Enzyme important for melanin biosynthesis |
7 (6) | Fig. 2 |
| 1717 | 4 | 30 | 0.78 | 70 | CAMK2D | Cell communication | 9 (7) | fig. S3 |
| 3622 | 1 | 385 | 0.48 | 37 | — | — | 44 (17) | — |
| 257 (B) | Z | 840 | 0.54 | 29 | MLANA | Plays a role in melanosome biogenesis |
28 (20) | Fig. 2 |
| 567 | 2 | 260 | 0.65 | 23 (1) | — | — | 8 (4) | — |
| 579 | 1 | 670 | 0.33 | 13 | — | — | 16 (13) | — |
| 1954 | 5 | 75 | 0.57 | 11 | — | — | 3 (2) | — |
| 59 | 15 | 85 | 0.35 | 8 | — | — | 6 (2) | — |
| 118 | 2 | 25 | 0.61 | 7 | — | — | 3 (2) | — |
| 257 | Z | 265 | 0.39 | 6 | — | — | 11 (6) | — |
| 404 | Z | 195 | 0.55 | 5 | — | — | 3 (3) | — |
| 257 | Z | 370 | 0.53 | 4 | — | — | 23 (10) | — |
| 766 | 4 | 65 | 0.53 | 4 | — | — | 6 (6) | — |
| 637 | Z | 40 | 0.45 | 4 | — | — | 0 | — |
| 1635 | 6 | 30 | 0.46 | 3 | — | — | 1 (1) | — |
| 257 | Z | 45 | 0.31 | 2 | — | — | 16 (15) | — |
| 263 | Z | 535 | 0.34 | 2 | MYO5A | Actin-based motor protein involved in melanosome transport |
24 (11) | fig. S3 |
| 791 | 1 | 50 | 0.40 | 1 | TYR or DCT§ |
TYR: Enzyme involved in converting tyrosine to melanin DCT: Regulates eumelanin and pheomelanin levels |
4 (3) | fig. S3 |
| 637 | Z | 300 | 0.30 | 1 | — | — | 0 | — |
*Scaffolds are ranked from top to bottom by the number of highly divergent SNPs (FST > 0.85).
†Information from DAVID (Database for Annotation, Visualization and Integrated Discovery) (https://david.ncifcrf.gov/) and GeneCards (www.genecards.org).
‡Total of 257 annotations, of which 246 were unique and 11 were predicted more than once. Approximately 63% (156) of the unique annotations matched a record in the UniProt database with a known name and function.
§Annotation predicted on the basis of protein similarity of both TYR and DCT, which are on chromosome 1 in the zebra finch.
Next, we asked whether the same divergence peaks were involved in differentiation across multiple combinations of capuchino species in ways that imply independent patterns of selection on the same loci. The left panels in Fig. 2 show examples of divergence peaks (5-kb windows) with the 10 different pairwise comparisons overlaid. Although no single area of the genome (that is, differentiation peak) was present in all comparisons, many are found in multiple comparisons. For example, the peak in Fig. 2A was present in 9 of the 10 comparisons, many of which involved pairs of species where the four taxa are different (for example, nig versus mel and pil versus hypox). Other divergence peaks were less ubiquitous yet are present across multiple pairs of species (left panels in Figs. 2 and 3 and fig. S3). To better understand the nature of the differences among species within the divergence peaks, we conducted PCAs with the SNPs from each of these areas separately. Figure 1B shows four clusters of haplotypes in the region under the most common peak in our data set. Other divergence peaks varied in the species where they were present and the extent to which they could be used to diagnose species in PCAs (center panels in Figs. 2 and 3 and fig. S3).
Fig. 2. Repeated selection on pigmentation genes in different capuchino species.
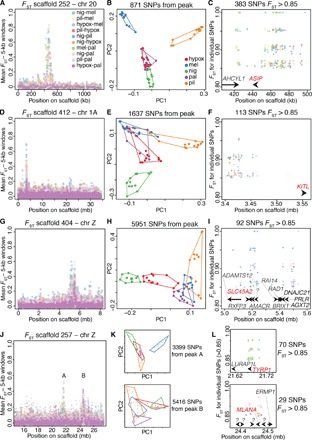
(A) Divergence peak on scaffold 252, which mapped to chromosome 20 in the zebra finch. The 10 possible pairwise comparisons across five capuchino species are overlaid (see color-coded legend to identify specific comparisons). Each circle is the mean FST value for all SNPs within a nonoverlapping 5-kb window. (B) PCA for 60 individuals of five species using the SNPs from under the peak in (A); see the legend to identify species. (C) FST and genomic location of individual SNPs with values of 0.85 and higher, color-coded by pairwise comparison as in (A). The positions of genes that are close to these highly divergent SNPs are indicated by arrows drawn to scale. Names in red note genes involved in the melanogenesis pathway. (D to F) As above, for the divergence peak on scaffold 412. (G to I) As above, for the divergence peak on scaffold 404. (J to L) As above, for the divergence peak on scaffold 257. (K and L) The top plot corresponds to the peak labeled “A” and the bottom one to the peak labeled “B” in (J). Annotations with question marks did not match known genes.
Fig. 3. Repeated selection on nonpigmentation genes in different capuchino species.
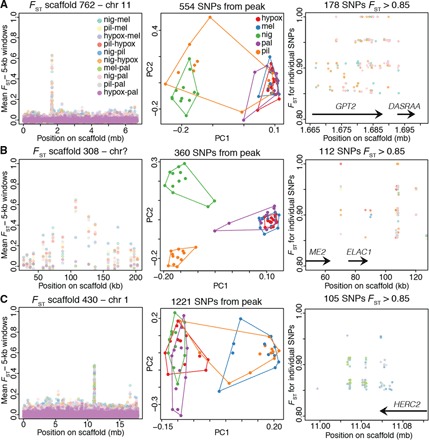
(A) Divergence peak for scaffold 762 (left), PCA obtained from SNPs under the peak (center), and FST values for highly divergent SNPs with gene annotations (right). The GPT2 gene is involved in alanine degradation and contains the only fixed difference (FST = 1) found in a coding region in our data set (fixed in nig versus hypox and nig versus mel). DASRA-A is a gene involved in the regulation of mitosis. (B) The divergence peak on scaffold 308 did not align reliably to a zebra finch chromosome. This peak contained the genes ME2 (involved in conversion of malate to pyruvate) and ELAC1 (involved in transfer RNA maturation). (C) The gene HERC2 was found in the peak on scaffold 430. This gene contains a regulator of the pigmentation gene OCA2. Other details as in Fig. 2.
We identified a total of 246 gene models within these divergent areas of the genome (an average of 10 per divergence peak), 156 of which matched genes of known functions in other species. We performed an enrichment analyses to understand if genes in this list were predominantly involved in certain pathways. The most prominent hit was the melanogenesis pathway (Kyoto Encyclopedia of Genes and Genomes pathway analysis, P = 2.0 × 10−3). We found nine melanogenesis genes in eight different divergence peaks (Table 1). The peak containing the gene coding for the Agouti-signaling protein (ASIP) had the largest number of highly divergent SNPs in our data set: 30% of all observed SNPs with FST > 0.85 and 58% of all SNPs with FST = 1 (Fig. 2C). Accordingly, this was the peak that showed the greatest increase in absolute sequence divergence (measured using the Dxy statistic) when comparing the region under the peak to the areas on the same scaffold outside of the peak (fig. S4). Other peaks contained a smaller number of these highly divergent SNPs (Figs. 2 and 3, Table 1, and fig. S3), and Dxy was, on average, indistinguishable inside and outside of the FST peaks (P > 0.05; fig. S4). The peaks containing melanogenesis genes accumulated 60% of SNPs with FST > 0.85 and 63% of fixed SNPs (across all pairwise comparisons). Figure 2 and fig. S3 show the eight divergence peaks containing melanogenesis genes, ranked from top to bottom by the number of highly divergent SNPs (FST > 0.85) present in these peaks (summarized in Table 1). The right panels in these figures indicate the position of these SNPs with respect to the closest gene models. We also identified three peaks that together accumulated 30% of the observed highly divergent SNPs and did not contain melanogenesis genes (Fig. 3); however, one of these peaks contained the gene HERC2. An intron within this gene functions as an enhancer that regulates the expression of the OCA2 pigmentation gene in humans, involved in controlling eye, hair, and skin color (23). The remaining peaks in Fig. 3 did not contain genes that could be easily associated to plumage or other phenotypes. The remaining 14 peaks (Table 1) accounted for only 10% of the observed highly divergent SNPs. Finally, because the melanocortin 1 receptor (MC1R) is known to affect plumage coloration in many bird species and interact directly with ASIP (24), we asked whether this gene showed divergence among capuchino seedeaters and had been overlooked in our analyses. MC1R was present in our reference genome assembly yet did not show differences in any of the pairwise comparisons across our five capuchino species (fig. S5).
Nearly all the fixed sites we observed (99%) were located in noncoding areas of the genome. The most common peak in our data set concentrated 58% of these fixed differences within several thousand kilobases up and downstream of the ASIP gene (Fig. 2C). We found that these areas contained positions that were highly conserved across the genomes of distantly related birds (turkey, chicken, budgerigar, and zebra finch), comparable in their levels of conservation to certain coding positions on the exons of ASIP (Fig. 4). It is therefore likely that these regions contain cis-regulatory elements that are necessary to control the expression of ASIP, and a similar situation may be true for the other differentiated regions found in close proximity to genes.
Fig. 4. Conserved genomic regions across multiple bird species.
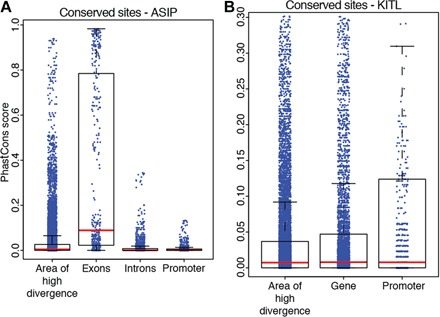
(A) PhastCons scores in regions close to ASIP. The y axis represents the probability of a nucleotide being conserved in a multigenome alignment of the budgerigar, zebra finch, chicken, and turkey genomes. For the promoter, we used a 1-kb region upstream of the gene. The area of high divergence is shown in Fig. 2C. (B) Same as in (A) for KIT ligand (KITL). In this case, the gene model did not contain intron and exon boundaries.
The SNPs showing fixed differences in at least one pairwise comparison among capuchinos (total of 99) were found on five different divergence peaks (Table 1), which mapped to at least four different zebra finch chromosomes (one could not be confidently assigned to a zebra finch chromosome). Despite being on different divergence peaks, some of these SNPs were in high linkage disequilibrium (LD; Fig. 5). The strength of the LD and the divergence peaks that were involved depended on the species (Fig. 5). In some cases, most positions within most peaks showed moderate to high LD (for example, nig), whereas in others (for example, hypox), high LD was restricted to a few positions between certain peaks. Few positions and peaks tend to show moderate to high LD across all taxa, suggesting that the processes (for example, low gene flow and/or selection) that could generate LD are species-specific.
Fig. 5. LD among divergence peaks.
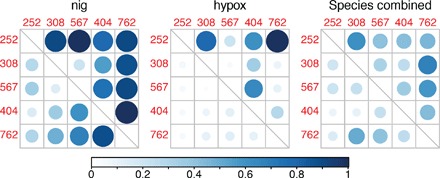
The r2 statistic was calculated among all possible combinations of positions that were fixed (FST = 1) in at least one pairwise comparison between species. Fixed SNPs were found in a total of five divergence peaks (the peaks on scaffolds 252, 308, 567, 404, and 762). Lower triangular matrices display the r2 value averaged across all the positions in that comparison. The upper triangular matrices display the highest r2 value that was observed. The calculation was carried out within species (nig and hypox shown) and when all species were pooled together. The size of the circle and the shade of blue (see scale) indicate the magnitude of the r2 value.
Because most divergence peaks identified among capuchino species contained more than one gene, we asked whether some of these areas could coincide with chromosomal inversions (or structural variants in general). Recombination is largely suppressed within inversions and can lead to the formation of clusters of coadapted genes (25). We compared long sequence reads (average of ~8 kb) obtained from pil to the hypox reference genome and identified a total of 500 putative structural variants between these two species (352 inversions, 133 deletions, and 15 insertions; fig. S6). These structural variants were much smaller than the average divergence peak (~1.8 kb versus 243 kb), and only four small inversions [average size, ~450 base pairs (bp)] were located within areas of high differentiation. Most structural rearrangements (~71%) did not involve areas of the genome that contained annotated genes, and the only inversion that harbored more than one gene comprised two annotations (fig. S6). It is thus unlikely that, at least between hypox and pil, the high divergence of certain areas of the genome is driven by structural rearrangements.
DISCUSSION
Despite their marked differences in male plumage, southern capuchino seedeaters are differentiated only in a small proportion of their genomes. The identity of these rare differentiated genomic regions differs somewhat among capuchinos, yet in many cases, the same divergent regions are present in comparisons across many pairs of species. This convergence of differentiation across multiple independent pairs of species implies that selection has acted repeatedly in different lineages on the same genomic targets to shape phenotypes.
Many of the most highly differentiated areas of the capuchino genome contain genes that are part of the melanogenesis pathway. The area upstream of ASIP is the most ubiquitous peak, showing the strongest signal of differentiation. More generally, we observe narrow divergence peaks involving different genes from the melanogenesis pathway that are generally on different chromosomes. Plumage coloration is generally important for reproductive isolation in birds (26), and differences in genes that control melanin-based variation in plumage have been found in different pairs of incipient avian taxa [for example, carrion and hooded crows (27, 28), flycatchers of the Solomon Islands (29), blue-winged and golden-winged warblers (30)]. The differences we observe in capuchinos are mostly in noncoding areas of the genome; therefore, our findings are consistent with the evolution of cis-regulatory elements. These candidate regulatory elements may vary by species yet control the expression of the same set of genes, generating the markedly different phenotypes we observe in the capuchinos. In particular, the regulation of the expression of melanogenesis genes may also lead to pigmentation differences across the plumage patches within each species (for example, throat versus back) (Fig. 1A).
Three factors could have contributed to the rapid evolution of phenotypic diversity in the capuchinos. First, a previous study used coalescent modeling to infer a very large effective population size for the ancestor of the radiation (15). The amount of genetic variation a population can sustain is proportional to its size, with large populations providing more possible substrates for rapid evolution from standing genetic variation to take place (31). In addition, differences could have accumulated among species in allopatry and eventually been exchanged via hybridization, leading to novel phenotypes as has been described for Heliconius butterflies (32). In particular, the differentiation and exchange via hybridization of regulatory elements that control the expression of more conserved genes have been found to drive phenotypic diversity in Heliconius (33). A similar situation could have contributed to phenotypic diversity in the capuchinos, which also show modular variation in their plumage, with the same patches having different colors depending on the species (for example, throat, back, or belly can be black, white, cinnamon, or rufous in different species; Fig. 1A). Finally, 10 of the 25 divergence peaks that we identified are located on the Z chromosome. Sex chromosomes play an important role in speciation, as evidenced by Haldane’s rule [the predominant inviability or sterility of hybrids of the heterogametic sex (34)] and the large X (or Z) effect [the disproportionate effect of loci located on sex chromosomes on hybrid fitness (35, 36)]. A possible explanation for these empirical observations is that selection is able to act on favorable recessive mutations with fitness consequences on hybrids when the loci are on sex chromosomes (35). This is because these recessive mutations are exposed to selection when the sex chromosome is in the heterogametic sex. For these reasons, divergence in genes located on sex chromosomes could have facilitated rapid evolution in capuchinos.
We have identified genomic substrates that lead to phenotypic diversity in capuchinos, differences that are likely relevant to mate recognition and, eventually, reproductive isolation in these strongly sexually dimorphic species. Because the most divergent areas across the genomes of capuchinos contain pigmentation genes, this leads to the question of whether we have found the genes responsible for maintaining these lineages as separate species. The pigmentation genes are linked to other loci for which the connection between genotype and phenotype is harder to make, and because we do not understand the contribution of these additional genes to species differences, we cannot conclude that pigmentation genes alone are driving speciation in capuchinos. Differences in coloration could promote prezygotic isolation if mate choice is strong enough to maintain the phenotypic integrity of capuchino species, even though many species breed in local-scale sympatry. However, we also cannot, at this point, discard the possibility that postzygotic incompatibilities exist between species, perhaps associated with these same divergent regions of the genome. As natural selection has shaped the beaks of finches in the Galapagos, leading to the generation of biological diversity, our study suggests that sexual selection may have shaped the plumage and songs of male capuchinos, generating yet another extraordinary rapid radiation of finch-like birds.
MATERIALS AND METHODS
Reference genome assembly and annotation
We assembled the genome of a S. hypoxantha male (sample MACN 5231) by combining data from short-read Illumina sequencing with those from long-read Pacific Biosciences sequencing. Because southern capuchino seedeaters constitute a phylogenetic hard polytomy, S. hypoxantha shows similar genetic distances to all other members of the clade (14, 15). We therefore do not expect our choice of species for the reference genome to have a strong effect on the mapping quality of sequences from different capuchino species. One 180-bp fragment library and two mate-pair libraries (insert sizes of 3 and 8 kb, respectively) were prepared by the Weill Cornell Medical College genomics core facility. Each library was sequenced on an Illumina HiSeq 2500 lane, obtaining paired-end, 101-bp reads. One large insert library (15 to 20 kb) was prepared by the Duke Center for Genomic and Computational Biology core facility and sequenced on 14 cells of a Pacific Biosciences RS II instrument. The three Illumina lanes generated ~1343 million raw paired-end reads (estimated depth of coverage of 112×). The 14 Pacific Biosciences cells produced a total of ~1 million reads with an average subread length of 10,430 bp (estimated depth of coverage of 9.1×).
We performed the genome assembly with ALLPATHS-LG version 49148 (37), which uses the data from the Illumina fragment and mate-pair libraries. The assembly was completed in ~256.3 hours (10.7 days) on a 64-core computer (512-gigabyte RAM, 13-terabyte hard drive) from the Cornell Computational Biology Service Unit BioHPC Lab. We obtained assembly statistics with QUAST version 2.3 (38). This first assembly had a total length of 1.14 Gb distributed across 5636 scaffolds, an N50 of 5.9 Mb, and 9.5% Ns.
We improved our first assembly using PBJelly version 12.9.14 (39), which is a program designed to use long Pacific Biosciences reads to fill or reduce gaps. We ran three iterations of PBJelly using the output of one run as the input of the following one. After the final PBJelly run, the total length of the assembly was 1.17 Gb and consisted of 5120 scaffolds, an N50 of 8.7 Mb, and 4.8% Ns. We assessed the completeness of our reference assembly by searching for a vertebrate set of 3023 single-copy orthologs using BUSCO version 1.2 (40). Our S. hypoxantha reference genome contained a single and complete copy of 82.3% of the genes in the vertebrate set. A fragment of an additional 7.7% of this set of genes was present in our assembly. Finally, 0.9% of the BUSCO vertebrate set was found more than once, and 9.1% was missing from the S. hypoxantha reference genome.
We annotated the S. hypoxantha genome by first generating a library of the repetitive sequences present in our assembly with RepeatModeler version 1.08 (www.repeatmasker.org/). We subsequently produced gene models by running two iterations of the MAKER version 2.31.8 pipeline (41). The first iteration produces ab initio gene predictions by training the algorithms with data from expressed sequence tags (ESTs) and protein databases and subsequently refines these gene models. We used EST and protein data from the zebra finch assembly Taeniopygia_guttata-3.2.4 downloaded from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/). The second iteration uses the gene models predicted by the first MAKER run as input, which improves the performance of the ab initio gene predictors. This pipeline produced a total of 14,667 gene models, representing 75.5% of the 19,437 genes annotated in the zebra finch genome. A total of 95.9% of the proteins predicted by MAKER for the S. hypoxantha reference genome matched zebra finch proteins in a BLAST search (42).
We obtained the putative chromosomal location of the different scaffolds with divergence peaks (see below) by aligning them to the zebra finch assembly (Taeniopygia_guttata-3.2.4) using the Satsuma synteny module from the Satsuma version 3.1 pipeline (43). We assigned the scaffold to the chromosome with the top hit and inspected the results in MizBee (44).
Population-level sequencing and variant discovery
We sequenced the genomes of a total of 72 individuals (71 males and 1 S. melanogaster female) from nine capuchino species: 12 S. nigrorufa (nig), 12 S. pileata (pil), 12 S. melanogaster (mel), 12 S. hypoxantha (hypox), 12 S. palustris (pal), 3 S. bouvreuil (bou), 3 S. cinnamomea (cin), 3 S. hypochroma (hypoch), and 3 S. ruficollis (ruf). See table S1 for further details on sampling. DNA was extracted using the DNeasy blood and tissue kit (Qiagen), and 200 ng of each sample were used to prepare individually barcoded libraries following the TruSeq Nano DNA library preparation kit protocol, with an inset size of 550 bp. The 72 libraries were pooled using concentrations of adapter-ligated DNA determined through digital polymerase chain reaction (PCR) into three groups of 24 samples. Each pool was sequenced on an Illumina NextSeq 500 lane at the Cornell Institute for Biotechnology core facility. We obtained a total of 2664 million paired-end reads, with a length of 151 bp. On the basis of the number of reads obtained for individual libraries, we expected the depth of coverage to range between 1.7× and 10.2×.
We assessed the quality of individual libraries using fastqc version 0.11.5 (www.bioinformatics.babraham.ac.uk/projects/fastqc) and performed sequence trimming, adapter removal, and quality filtering with AdapterRemoval version 2.1.1 (45). We allowed a minimum Phred quality score of 10 and merged overlapping paired-end reads. We subsequently aligned the filtered reads to the reference S. hypoxantha genome with the very sensitive, local option implemented in Bowtie 2 version 2.2.8 (46). We obtained alignment statistics using Qualimap version 2.1.1 (47). The average alignment rate across all samples was 98.4 ± 0.8% and was comparably high between the species to which the reference genome belonged to and the remaining species (98.4 ± 0.6% for S. hypoxantha versus 98.5 ± 0.9% for all other species combined). After filtering and aligning sequences to the reference genome, the depth of coverage ranged between 1.4× and 9.8×.
We converted .sam files into .bam format and subsequently sorted and indexed these files with SAMtools version 1.3 (48). We marked PCR duplicates with Picard Tools version 2.1.1 (https://broadinstitute.github.io/picard/), realigned around indels, and fixed mate pairs after realigning with GATK version 3.5 (49). We simultaneously performed SNP variant discovery and genotyping for the 72 samples with the unified genotyper module in GATK. We removed variants based on the following hard filtering parameters: QD < 2, FS > 40.0, MQ < 20.0, and HaplotypeScore > 12.0. We subsequently filtered out variants that were not biallelic, had a minor allele frequency smaller than 0.1, with mean depth of coverage smaller than 2 or greater than 50, and/or with more than 15 missing individuals (20% missing data across the data set). This pipeline produced 11,530,110 SNPs genotyped for 72 individuals of the nine southern capuchino species (mean depth of coverage across all sites per species: nig, 3.8×; pil, 4.1×; mel, 4.8×; hypox, 4.3×; pal, 5.7×; bou, 7.0×; cin, 4.1×; hypoch, 5.9×; ruf, 5.2×).
Population genomic analyses
We searched for divergent areas of the genome by calculating FST values using VCFtools version 0.1.14 (50) and the five southern capuchino species with sample sizes of 12 individuals (hypox, mel, nig, pal, and pil). We calculated FST in three different ways across the 10 possible pairwise comparisons involving five species (for example, nig versus mel, hypox versus pal). We used three strategies: (i) calculated average FST values for nonoverlapping 25-kb windows, (ii) zoomed in to scaffolds of interest and calculated average FST values for nonoverlapping 5-kb windows, and (iii) calculated FST values for individual SNPs. We built Manhattan plots and conducted PCA in R version 3.3.0 (51) with the packages “qqman” and SNPRelate version 3.3 (52), respectively. The PCA derived from 11.5 million SNPs was run both with and without four outlier individuals (two S. melanogaster and two S. pileata; see details in fig. S1). Downstream analyses were conducted with and without these four individuals and produced similar results.
We identified divergence peaks in the 10 pairwise comparisons using the average FST value calculated for the nonoverlapping 25-kb windows, discarding regions with less than two windows and windows with less than 10 SNPs. We took a conservative approach and only selected regions that showed an FST value elevated above 0.2. Because the average FST across all comparisons was 0.008, these criteria only selected regions that fall between 12 and 13 SDs above the FST mean. We subsequently narrowed our selection of candidate regions by retaining only those that had at least one individual SNP with an FST of 0.85 or higher. We thus filtered out regions with an elevated average FST that did not contain individual outlier sites that could be putative targets of selection. We identified a total of 25 divergent regions across the 10 possible pairwise FST comparisons.
We estimated absolute sequence divergence by calculating the summary statistic Dxy for each site and obtaining an average for nonoverlapping 5-kb windows with a custom perl script. Dxy was calculated as the minor allele frequency in species A times the major allele frequency in species B plus the product of the major allele frequency in species A and the minor allele frequency in species B. The per-site minor allele frequency was obtained using AGSD version 0.911 (53).
We estimated LD using VCFtools to calculate the r2 statistic. The calculations were carried out with the 99 SNPs that showed fixed differences (FST = 1) in at least one pairwise comparison between species. We recorded the average and the highest r2 value when comparing more than one pair of sites between two peaks. Calculations were conducted for each species separately and for all taxa pooled together. For the former, we included one outgroup from each of the remaining species because, in many cases, the position was not variable within species and otherwise could not be used to calculate LD.
Identification of genes in divergent regions
We inspected the subset of regions identified as divergence peaks in Geneious version 9.1.5 (54) and compiled a list of gene models within 50 kb of each region. We obtained information on these annotations of interest from the UniProt database (www.uniprot.org/) and searched for enriched pathways using a Homo sapiens database with DAVID v6.7 (55). Some divergent regions did not overlap with annotated genes and thus could putatively contain elements that regulate gene expression and are targets of selection. We searched for conserved elements within these regions by aligning them to the medium ground finch (Geospiza fortis) reference genome (geoFor1) using BLAT (56). We subsequently downloaded the PhastCons bird conservation track (57) from the UCSC (University of California Santa Cruz) genome browser (58). PhastCons scores range from 0 to 1 and represent the probability of a nucleotide belonging to a conserved element in a multigenome alignment. The bird PhastCons track is derived from an alignment of the budgerigar (Melopsittacus undulatus), zebra finch (Taeniopygia guttata), chicken (Gallus gallus), and turkey (Meleagris gallopavo) genomes.
Structural variant discovery
We searched for structural variants between our S. hypoxantha reference genome and S. pileata samples. We first pooled three S. pileata DNA samples in equal proportions and used this pool to prepare a large insert library (15 to 20 kb), as described above. Because we were interested in finding potential structural variants between these two species, we were able to follow this pooled strategy that allowed us to obtain the required amount of DNA for this type of library (>15 μg). The library was sequenced on six cells of a Pacific Biosciences RS II instrument, producing 398,217 reads with an average subread length of 7884 bp (estimated depth of coverage of 2.6×). The S. pileata Pacific Biosciences long reads were then aligned to the S. hypoxantha reference genome using BLASR version 2014-09-24 (59). The .sam file produced by BLASR was converted to .bam, sorted, and indexed using SAMtools. Finally, we identified putative structural variants using the PBHoney pipeline from PBSuite version 15.8.24 (60) using a buffer size of 10 kb (B parameter).
This list of candidate structural variants could include spurious rearrangements derived from problematic areas in our reference genome, as well as rearrangements that segregate at low frequency in S. hypoxantha but happen to be carried by the individual used to build the reference genome. To filter our list of candidate structural variants between S. hypoxantha and S. pileata, we searched for structural variants between the reference genome and the 12 S. hypoxantha individuals used for population-level resequencing. We pooled the short Illumina reads for the 12 S. hypoxantha individuals (estimated depth of coverage of ~50×) and aligned them to the reference genome using BWA-MEM version 0.7.13 (61), alignment files were sorted with SAMtools, PCR duplicates were marked with Picard Tools, and information about discordantly matching paired-end reads and split reads was extracted with SAMtools. We subsequently ran the lumpyexpress pipeline from the Lumpy version 0.2.13 package (62). Lumpy integrates information from reads that align discontinuously (split reads) and mate pairs that align discordantly with respect to each other (paired-end) to identify the potential breakpoints of structural rearrangements. Each split and paired-end read is considered a piece of evidence supporting the variant. This strategy identified a list of putative rearrangements that we subsequently filtered by retaining only those that were supported by at least 24 reads (average of 2× depth of coverage in each individual).
Finally, we discarded any potential structural variants identified between the S. hypoxantha reference genome and S. pileata that were also present between the S. hypoxantha reference genome and the pool of S. hypoxantha individuals. These variants are likely misassembled regions in the reference genome or variants that are present in the reference genome but not fixed within S. hypoxantha. We retained a total of 352 inversions, 133 deletions, and 15 insertions and searched for annotated genes within the limits of the breakpoints in Geneious.
Supplementary Material
Acknowledgments
We thank the members of the Fuller Evolutionary Biology laboratory group (particularly B. Butcher, D. Toews, S. Taylor, N. Mason, S. Aguillon, J. Walsh-Emond, P. Dean-Coe, N. Hofmeister, J. Berv, and B. Van Doren), C. Dardia, J. Lewis, D. Lijtmaer, R. Razavi, and two anonymous reviewers for the feedback on this project. We thank the government agencies of Brazil (CNPq, CAPES-PROEX, FAPESP, ICMBio, SISBIO 36881-1, and CEMAVE 361788) and Argentina for the grants and permits. For tissue loans, we are indebted to the Museo Argentino de Ciencias Naturales “Bernardino Rivadavia” (MACN, CONICET, Argentina), University of Kansas Natural History Museum (United States), and Museu de Ciências e Tecnologia of the Pontifícia Universidade Católica do Rio Grande do Sul (Brazil). Funding: This project was funded by NSF grant DEB 1555754 to I.J.L., by PICT 2014-2154 (Agencia Nacional de Promoción Científica y Tecnológica) to P.L.T., and by a Genomics Scholar fellowship from the Center for Vertebrate Genomics (Cornell University) to L.C. Author contributions: L.C., M.R., L.F.S., C.S.F., P.L.T., and I.J.L. designed the study. L.C., L.F.S., and M.R. collected the data. L.C. analyzed the data and wrote the manuscript with help from all coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Genomic data have been archived in GenBank (BioProject ID PRJNA382416). Illustrations in Fig. 1A were obtained with permission from Hoyo et al. (63).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602404/DC1
fig. S1. Clustering of individuals by species.
fig. S2. Genomic landscapes of differentiation in 10 pairwise comparisons of five species.
fig. S3. Repeated selection on pigmentation genes in different capuchino species II.
fig. S4. Absolute sequence divergence inside and outside of peak areas.
fig. S5. MC1R is not differentiated in capuchinos.
fig. S6. Structural variants found comparing two capuchino species (hypox and pil).
table S1. Details on the samples used in this study.
REFERENCES AND NOTES
- 1.Martin A., Papa R., Nadeau N. J., Hill R. I., Counterman B. A., Halder G., Jiggins C. D., Kronforst M. R., Long A. D., McMillan W. O., Reed R. D., Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc. Natl. Acad. Sci. U.S.A. 109, 12632–12637 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linnen C. R., Poh Y.-P., Peterson B. K., Barrett R. D. H., Larson J. G., Jensen J. D., Hoekstra H. E., Adaptive evolution of multiple traits through multiple mutations at a single gene. Science 339, 1312–1316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theron E., Hawkins K., Bermingham E., Ricklefs R. E., Mundy N. I., The molecular basis of an avian plumage polymorphism in the wild: A melanocortin-1-receptor point mutation is perfectly associated with the melanic plumage morph of the bananaquit, Coereba flaveola. Curr. Biol. 11, 550–557 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Brawand D., Wagner C. E., Li Y. I., Malinsky M., Keller I., Fan S., Simakov O., Ng A. Y., Lim Z. W., Bezault E., Turner-Maier J., Johnson J., Alcazar R., Noh H. J., Russell P., Aken B., Alföldi J., Amemiya C., Azzouzi N., Baroiller J.-F., Barloy-Hubler F., Berlin A., Bloomquist R., Carleton K. L., Conte M. A., D’Cotta H., Eshel O., Gaffney L., Galibert F., Gante H. F., Gnerre S., Greuter L., Guyon R., Haddad N. S., Haerty W., Harris R. M., Hofmann H. A., Hourlier T., Hulata G., Jaffe D. B., Lara M., Lee A. P., MacCallum I., Mwaiko S., Nikaido M., Nishihara H., Ozouf-Costaz C., Penman D. J., Przybylski D., Rakotomanga M., Renn S. C. P., Ribeiro F. J., Ron M., Salzburger W., Sanchez-Pulido L., Santos M. E., Searle S., Sharpe T., Swofford R., Tan F. J., Williams L., Young S., Yin S., Okada N., Kocher T. D., Miska E. A., Lander E. S., Venkatesh B., Fernald R. D., Meyer A., Ponting C. P., Streelman J. T., Lindblad-Toh K., Seehausen O., Di Palma F., The genomic substrate for adaptive radiation in African cichlid fish. Nature 513, 375–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Küpper C., Stocks M., Risse J. E., dos Remedios N., Farrell L. L., McRae S. B., Morgan T. C., Karlionova N., Pinchuk P., Verkuil Y. I., Kitaysky A. S., Wingfield J. C., Piersma T., Zeng K., Slate J., Blaxter M., Lank D. B., Burke T., A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 48, 79–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamichhaney S., Fan G., Widemo F., Gunnarsson U., Thalmann D. S., Hoeppner M. P., Kerje S., Gustafson U., Shi C., Zhang H., Chen W., Liang X., Huang L., Wang J., Liang E., Wu Q., Lee S. M.-Y., Xu X., Höglund J., Liu X., Andersson L., Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nat. Genet. 48, 84–88 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Tuttle E. M., Bergland A. O., Korody M. L., Brewer M. S., Newhouse D. J., Minx P., Stager M., Betuel A., Cheviron Z. A., Warren W. C., Gonser R. A., Balakrishnan C. N., Divergence and functional degradation of a sex chromosome-like supergene. Curr. Biol. 26, 344–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boag P. T., Grant P. R., Intense natural selection in a population of Darwin’s finches (Geospizinae) in the Galapagos. Science 214, 82–85 (1981). [DOI] [PubMed] [Google Scholar]
- 9.Abzhanov A., Kuo W. P., Hartmann C., Grant B. R., Grant P. R., Tabin C. J., The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches. Nature 442, 563–567 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Lamichhaney S., Berglund J., Almén M. S., Maqbool K., Grabherr M., Martinez-Barrio A., Promerová M., Rubin C.-J., Wang C., Zamani N., Grant B. R., Grant P. R., Webster M. T., Andersson L., Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Lamichhaney S., Han F., Berglund J., Wang C., Almén M. S., Webster M. T., Grant B. R., Grant P. R., Andersson L., A beak size locus in Darwin’s finches facilitated character displacement during a drought. Science 352, 470–474 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Podos J., Correlated evolution of morphology and vocal signal structure in Darwin’s finches. Nature 409, 185–188 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Ratcliffe L. M., Grant P. R., Species recognition in Darwin’s finches (Geospiza, Gould). III. Male responses to playback of different song types, dialects and heterospecific songs. Anim. Behav. 33, 290–307 (1985). [Google Scholar]
- 14.Campagna L., Benites P., Lougheed S. C., Lijtmaer D. A., Di Giacomo A. S., Eaton M. D., Tubaro P. L., Rapid phenotypic evolution during incipient speciation in a continental avian radiation. Proc. R. Soc. B 279, 1847–1856 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campagna L., Gronau I., Silveira L. F., Siepel A., Lovette I. J., Distinguishing noise from signal in patterns of genomic divergence in a highly polymorphic avian radiation. Mol. Ecol. 24, 4238–4251 (2015). [DOI] [PubMed] [Google Scholar]
- 16.P. R. Grant, B. R. Grant, 40 Years of Evolution: Darwin’s Finches on Daphne Major Island (Princeton Univ. Press, 2014). [Google Scholar]
- 17.Burns K. J., Shultz A. J., Title P. O., Mason N. A., Barker F. K., Klicka J., Lanyon S. M., Lovette I. J., Phylogenetics and diversification of tanagers (Passeriformes: Thraupidae), the largest radiation of Neotropical songbirds. Mol. Phylogenet. Evol. 75, 41–77 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Benites P., Campagna L., Tubaro P. L., Song based species discrimination in a rapid Neotropical radiation of grassland seedeaters. J. Avian Biol. 46, 55–62 (2015). [Google Scholar]
- 19.Meyer de Schauensee R., A review of the genus Sporophila. Proc. Acad. Natl. Sci. Phila. 104, 153–196 (1952). [Google Scholar]
- 20.Machado É., Silveira L. F., Plumage variability and taxonomy of the Capped Seedeater Sporophila bouvreuil (Aves: Passeriformes: Emberizidae). Zootaxa 2781, 49–62 (2011). [Google Scholar]
- 21.Areta J. I., Repenning M., Systematics of the Tawny-bellied Seedeater (Sporophila hypoxantha). I. Geographic variation, ecology, and evolution of vocalizations. Condor 113, 664–677 (2011). [Google Scholar]
- 22.Di Giacomo A. S., Vickery P. D., Casañas H., Spitznagel O. A., Ostrosky C., Krapovickas S., Bosso A. J., Landscape associations of globally threatened grassland birds in the Aguapey river Important Bird Area, Corrientes, Argentina. Bird Conserv. Int. 20, 62–73 (2010). [Google Scholar]
- 23.Visser M., Kayser M., Palstra R.-J., HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 22, 446–455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mundy N. I., A window on the genetics of evolution: MC1R and plumage colouration in birds. Proc. R. Soc. B 272, 1633–1640 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rieseberg L. H., Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358 (2001). [DOI] [PubMed] [Google Scholar]
- 26.T. Price, Speciation in Birds (Roberts and Company, 2007). [Google Scholar]
- 27.Poelstra J. W., Vijay N., Bossu C. M., Lantz H., Ryll B., Müller I., Baglione V., Unneberg P., Wikelski M., Grabherr M. G., Wolf J. B. W., The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 344, 1410–1414 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Vijay N., Bossu C. M., Poelstra J. W., Weissensteiner M. H., Suh A., Kryukov A. P., Wolf J. B. W., Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat. Commun. 7, 13195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uy J. A. C., Cooper E. A., Cutie S., Concannon M. R., Poelstra J. W., Moyle R. G., Filardi C. E., Mutations in different pigmentation genes are associated with parallel melanism in island flycatchers. Proc. R. Soc. B 283, 20160731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toews D. P. L., Taylor S. A., Vallender R., Brelsford A., Butcher B. G., Messer P. W., Lovette I. J., Plumage genes and little else distinguish the genomes of hybridizing warblers. Curr. Biol. 26, 2313–2318 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Barrett R. D. H., Schluter D., Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Heliconius Genome Consortium , Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallbank R. W. R., Baxter S. W., Pardo-Diaz C., Hanly J. J., Martin S. H., Mallet J., Dasmahapatra K. K., Salazar C., Joron M., Nadeau N., McMillan W. O., Jiggins C. D., Evolutionary novelty in a butterfly wing pattern through enhancer shuffling. PLOS Biol. 14, e1002353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haldane J. B. S., Sex ratio and unisexual sterility in hybrid animals. J. Genet. 12, 101–109 (1922). [Google Scholar]
- 35.Charlesworth B., Coyne J. A., Barton N. H., The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130, 113–146 (1987). [Google Scholar]
- 36.Ellegren H., Genomic evidence for a large-Z effect. Proc. R. Soc. B 276, 361–366 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gnerre S., MacCallum I., Przybylski D., Ribeiro F. J., Burton J. N., Walker B. J., Sharpe T., Hall G., Shea T. P., Sykes S., Berlin A. M., Aird D., Costello M., Daza R., Williams L., Nicol R., Gnirke A., Nusbaum C., Lander E. S., Jaffe D. B., High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gurevich A., Saveliev V., Vyahhi N., Tesler G., QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.English A. C., Richards S., Han Y., Wang M., Vee V., Qu J., Qin X., Muzny D. M., Reid J. G., Worley K. C., Gibbs R. A., Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLOS ONE 7, e47768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M., BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Cantarel B. L., Korf I., Robb S. M. C., Parra G., Ross E., Moore B., Holt C., Alvarado A. S., Yandell M., MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 43.Grabherr M. G., Russell P., Meyer M., Mauceli E., Alföldi J., Di Palma F., Lindblad-Toh K., Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics 26, 1145–1151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer M., Munzner T., Pfister H., MizBee: A multiscale synteny browser. IEEE Trans. Vis. Comput. Graph. 15, 897–904 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Lindgreen S., AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.García-Alcalde F., Okonechnikov K., Carbonell J., Cruz L. M., Götz S., Tarazona S., Dopazo J., Meyer T. F., Conesa A., Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2016); https://www.R-project.org/.
- 52.Zheng X., Levine D., Shen J., Gogarten S. M., Laurie C., Weir B. S., A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A., Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Kent W. J., BLAT—The BLAST-like alignment tool. Genome Res. 12, 656–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siepel A., Bejerano G., Pedersen J. S., Hinrichs A. S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L. W., Richards S., Weinstock G. M., Wilson R. K., Gibbs R. A., Kent W. J., Miller W., Haussler D., Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaisson M. J., Tesler G., Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinformatics 13, 238 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.English A. C., Salerno W. J., Reid J. G., PBHoney: Identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics 15, 180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H., Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 (2013). [Google Scholar]
- 62.Layer R. M., Chiang C., Quinlan A. R., Hall I. M., LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.J. del Hoyo, A. Elliott, J. Sargatal, D. A. Christie, E. de Juana, Handbook of the Birds of the World Alive (Lynx Edicions, 2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602404/DC1
fig. S1. Clustering of individuals by species.
fig. S2. Genomic landscapes of differentiation in 10 pairwise comparisons of five species.
fig. S3. Repeated selection on pigmentation genes in different capuchino species II.
fig. S4. Absolute sequence divergence inside and outside of peak areas.
fig. S5. MC1R is not differentiated in capuchinos.
fig. S6. Structural variants found comparing two capuchino species (hypox and pil).
table S1. Details on the samples used in this study.