

Abstract
Objective
To compare outcomes of infants and children who underwent lung transplantation for genetic disorders of surfactant metabolism (SFTPB, SFTPC, ABCA3, NKX2-1) over 2 epochs (1993–2003 and 2004–2015) at St. Louis Children’s Hospital.
Study design
We retrospectively reviewed clinical characteristics, mortality, and short- and long-term morbidities of infants (transplanted at <1 year, n=28) and children (transplanted >1 year, n=16) and compared outcomes by age at transplantation (infants vs. children) and by epoch of transplantation.
Results
Infants were transplanted more frequently for surfactant protein-B or ABCA3 deficiency, while children were transplanted more frequently for SFTPC mutations or ABCA3 deficiency. Infants experienced shorter times from listing to transplant (p=0.014), were more likely to be mechanically ventilated at time of transplant (p<0.0001), were less likely to develop bronchiolitis obliterans post-transplant (p=0.021), and were more likely to have speech and motor delays (p = <0.0001) than children. Despite advances in genetic diagnosis, immunosuppressive therapies, and supportive respiratory and nutritional therapies, mortality did not differ between infants and children (p = 0.076) or between epochs. Kaplan-Meier analyses demonstrated that children transplanted in epoch 1 (1993–2003) were more likely to develop systemic hypertension (p=0.049) and less likely to develop post-transplant lymphoproliferative disorder than children transplanted in epoch 2 (2004–2015) (p=0.051).
Conclusion
Post-lung transplant mortality and morbidities remain substantial for infants and children with genetic disorders of surfactant metabolism.
Keywords: surfactant protein B, surfactant protein C, ABCA3, SFTPB, SFTPC, NKX2.1, childhood interstitial lung disease, neonatal respiratory distress syndrome, chILD, RDS, pediatric lung transplantation
Pulmonary surfactant is a phospholipid-protein complex that lowers surface tension and prevents alveolar collapse at end-expiration. Surfactant proteins B and C contribute to the surface tension-lowering properties of surfactant.(1) ATP-binding cassette member A3 (ABCA3) transports phospholipids into lamellar bodies where surfactant is assembled and processed.(2) Thyroid transcription factor-1 (TTF1), encoded by NKX2-1, regulates transcription of surfactant-associated genes including SFTPB, SFTPC, and ABCA3.(3) Genetic disruption of SFTPB (NM 198843.2, Gene ID 6439), SFTPC (NM 003018.3, Gene ID 6440), ABCA3 (NM 001089.2, Gene ID 21), or NKX2-1 (NM 001079668, Gene ID 7080) expression or protein function can result in severe, progressive neonatal respiratory distress syndrome (RDS) among term or late preterm infants and childhood interstitial lung disease (chILD) (Table 1; available at www.jpeds.com).(4–16)
Table 1.
Genetic Disorders of Surfactant Metabolism
| Surfactant Protein B Deficiency | ABCA3 Deficiency Neonate | ABCA3 Deficiency Infant | ABCA3 Deficiency Child | Surfactant Protein C Associated Lung Disease | NKX2-1 Deficiency Neonate | NKX2-1 Deficiency Child | |
|---|---|---|---|---|---|---|---|
| Presentation | Acute severe neonatal respiratory distress (RDS) shortly after birth5 | Acute severe neonatal RDS shortly after birth35 | Respiratory distress or interstitial lung disease (chILD) between 1month and 1 year35 | Interstitial lung disease (chILD) after 1 year of life35 | Interstitial lung disease (chILD) and/or failure to thrive during infancy and childhood; less commonly as neonatal RDS10 | Acute severe neonatal RDS shortly after birth, hypothyroidism, hypotonia16 | Interstitial lung disease (chILD), recurrent respiratory infections, chorea, ataxia, developmental delay, hypothyroidism16 |
| Frequency | 1 per million European-descent (ED)4 | As frequent as 1/3100 ED; as frequent as 1/18,000 African-descent (AD)13 | As frequent as 1/3100 ED; as frequent as 1/18,000 AD13 | As frequent as 1/3100 ED; as frequent as 1/18,000 AD13 | Unknown4 | Unknown16 | Unknown16 |
| Disease Course | Lethal in newborn period5 | Lethal for infants with 2 loss of function (frameshift, nonsense) mutations, variable for other genotypes35 | Variable35 | Variable35 | Variable22 | Variable16 | Variable16 |
| Inheritance | Recessive/compound heterozygous | Recessive/compound heterozygous | Recessive/compound heterozygous | Recessive/compound heterozygous | Dominant, sporadic | Dominant, sporadic | Dominant, Sporadic |
Although modest pulmonary responses to empiric medical therapies including corticosteroids, azithromycin, hydroxychloroquine, and prolonged mechanical ventilation have been observed in a subset of patients with genetic disorders of surfactant metabolism, (17–22) many affected infants and children progress to lung transplantation. Infant and pediatric lung transplantation has been performed in a few United States centers since the early 1990s and has permitted survival for infants and children with genetically mediated end-stage lung disease.(23) However, lung transplant patients are at higher risk of death and transplant-related morbidities (24, 25) than other organ transplant recipients.
Over the past two decades, advances in DNA sequencing have permitted earlier definitive diagnosis,(26) targeted and less toxic immunosuppressive therapies have been developed, additional non-invasive ventilation strategies have emerged, and the importance of nutritional status has been increasingly recognized. The effects of these advances on outcomes for infants and children transplanted for genetic disorders of surfactant metabolism have not been recently described. (27) Here, we compare pre-transplant characteristics, mortality, and transplant-related morbidities for infants and children who underwent lung transplantation for genetic disorders of surfactant metabolism over the past 2 decades at St. Louis Children’s Hospital.
Methods
For all infants (<1 year of age, n=35) and children (>1 year of age, n=16) who underwent bilateral lung transplantation for genetic disorders of surfactant metabolism at St. Louis Children’s Hospital between 1993 and 2015, we collected age at listing, respiratory support at time of transplant, wait time to transplant, survival at 1 and 5 years post-transplant, and common transplant-related morbidities including cytomegalovirus (CMV) infection, seizures, hypertension, renal insufficiency, bronchiolitis obliterans, and post-transplant lymphoproliferative disorder (PTLD) (Table II).
Table 2.
Clinical Characteristics and Post-Transplant Morbidities for Infants and Children Transplanted for Genetics Disorders of Surfactant Metabolism during Epoch 1 (1993–2003) and Epoch 2 (2004–2015)
| Infants | Children | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Epoch 1 (1993–2003) |
Epoch 2 (2004–2015) |
p-value | Epoch 1 (1993–2003) |
Epoch 2 (2004–2015) |
p-value | |
|
| ||||||
| Number listed | 19 | 16 | 9 | 8 | ||
|
| ||||||
| Number transplanted | 16 (0.84) | 12 (0.75) | 9 (1.0) | 7 (0.88) | ||
|
| ||||||
| Genetic etiology | ||||||
| SFTPB | 10 (0.53) | 4 (0.25) | 0.26 | 0 (0) | 0 (0) | |
| SFTPC | 0 (0) | 1 (0.06) | 6 (0.67) | 5 (0.63) | 1.0 | |
| ABCA3 | 7 (0.37) | 10 (0.63) | 3 (0.33) | 2 (0.25) | ||
| NKX2-1 | 2 (0.10) | 1 (0.06) | 0 (0) | 1 (0.12) | ||
|
| ||||||
| Sex Female | 11 (0.58) | 8 (0.50) | 0.64 | 6 (0.67) | 5 (0.63) | 1.0 |
| Male | 8 (0.42) | 8 (0.50) | 3 (0.33) | 3 (0.37) | ||
|
| ||||||
| Race/Ethnicity | ||||||
| European descent | 18 (0.95) | 9 (0.56) | 0.0017 | 8 (0.89) | 6 (0.75) | 0.72 |
| African descent | 1 (0.05) | 0 (0) | 1 (0.11) | 1 (0.125) | ||
| Hispanic | 0 (0) | 2 (0.125) | 0 (0) | 0 (0) | ||
| Middle Eastern | 0 (0) | 3 (0.19) | 0 (0) | 0 (0) | ||
| Other | 0 (0) | 2 (0.125) | 0 (0) | 1 (0.125) | ||
|
| ||||||
| Age at listing (months ± SD) | 2.3 ± 2.2 | 2.9 ± 1.9 | 0.45 | 41 ± 45 | 99±47 | 0.020 |
|
| ||||||
| Death prior to transplantation | 3/19 (0.16) | 4/16 (0.25) | 0.68 | 0/9 (0) | 1/8 (0.13) | 0.47 |
|
| ||||||
| Age at transplant (months ± SD) | 4.1 ± 2.1 | 5.6 ± 2.3 | 0.81 | 45 ± 49 | 118 ± 54 | 0.015 |
|
| ||||||
| Wait time to transplant (months ± SD) | 1.6 ± 1.3 | 2.2 ± 1.7 | 0.34 | 4.2 ± 4.3 | 16 ± 13 | 0.055 |
|
| ||||||
| Mechanically ventilated at transplant | 13/16 (0.81) | 12/12 (1.0) | 0.24 | 1/9 (0.11) | 1/7 (0.14) | 1.0 |
|
| ||||||
| ECMO prior to transplant (% listed) | 7/19 (0.37) | 0/16 (0) | 0.0092 | 0/9 (0) | 1/8 (0.13) | 0.47 |
|
| ||||||
| Survival at 1 year | 13/16 (0.81) | 10/12 (0.83) | 1.0 | 9/9 (1.0) | 7/7 (1.0) | NA |
|
| ||||||
| Survival at 5 years | 9/16 (0.56) | 5/9 (0.56)* | 1.0 | 7/9 (0.78) | 4/5 (0.80) * | 1.0 |
|
| ||||||
| CMV infection | 1/16 (0.06) | 1/12 (0.08) | 1.0 | 2/9 (0.22) | 0/7 (0) | 0.48 |
|
| ||||||
| Seizures | 3/16 (0.19) | 0/12 (0) | 0.24 | 0/9 (0) | 0/7 (0) | NA |
|
| ||||||
| Hypertension by 5 years post-transplant | 2/9 (0.22) | 2/6 (0.33)† | 1.0 | 4/9 (0.44)† | 0/4 (0) | 0.23 |
|
| ||||||
| Renal insufficiency by 5 years post-transplant | 2/10 (0.20)‡ | 0/5 (0) | 0.52 | 1/8 (0.13)‡ | 0/4 (0) | 1.0 |
|
| ||||||
| Bronchiolitis obliterans by 5 years post-transplant | 4/11 (0.36)§ | 2/6 (0.33)§ | 1.0 | 5/9 (0.56)§ | 3/5 (0.60)§ | 1.0 |
|
| ||||||
| PTLD by 5 years post-transplant | 2/9 (0.22) | 0/5 (0) | 0.51 | 0/7 (0) | 1/4 (0.25) | 0.36 |
|
| ||||||
| Gastrostomy tube | 11/16 (0.69) | 11/12 (0.92) | 0.20 | 7/9 (0.78) | 7/7 (1.0) | 0.48 |
|
| ||||||
| Growth impairment | 5/14(0.36)ǁ | 9/12 (0.75) | 0.062 | 6/9 (0.67) | 4/7 (0.57) | 1.0 |
|
| ||||||
| Speech delay | 10/12(0.83)ǁ | 9/10 (.90)ǁ | 1.0 | 1/9 (0.11) | 1/7 (0.14) | 1.0 |
|
| ||||||
| Motor delay | 9/12(0.75)ǁ | 10/12 (0.83) | 1.0 | 2/9 (0.22) | 0/7 (0) | 0.48 |
|
| ||||||
| Hearing loss | 5/11(0.45)ǁ | 2/10 (0.2)ǁ | 0.36 | 0/9 (0) | 0/7 (0) | NA |
Data presented as number (percent) of individuals
Does not include children alive, but not yet 5 years post-transplant
Numerator and denominator include patients with hypertension who died prior to 5 years post-transplant
Numerator and denominator include patients with renal insufficiency who died prior to 5 years post-transplant
Numerator and denominator include patients with BO who died prior to 5 years post-transplant
Information not available for all infants
We assigned hypertension based on need for antihypertensive medication and/or documentation by nephrologist or pulmonologist in the medical record. We assigned renal insufficiency based on available laboratory values or clinical characteristics including elevated creatinine for age, glomerular filtration rate, need for dialysis or renal transplant, and/or documentation of diagnosis of chronic kidney disease by nephrologist or pulmonologist. Bronchiolitis obliterans was assigned based on the International Society of Heart and Lung Transplant histologic grading of transbronchial biopsy or autopsy (28) and/or documentation of diagnosis by pulmonologist. PTLD was assigned based on positron emission tomography (PET) scan or lymph node biopsy results. We assessed growth impairment based on standardized growth charts.(29) We captured other outcomes including need for gastrostomy tube placement, motor and speech delays described in formal testing (Bayley II, Peabody motor scale) and/or provider notes, and hearing loss based on audiograms when available and/or need for hearing aids.
We used Fisher exact tests, Chi square, and Student t-tests to compare clinical characteristics for infants and children from the two decades of experience: epoch 1 (1993–2003) and epoch 2 (2004–2015). We used Kaplan-Meier analyses to compare survival and transplant-related morbidities between infants and children and between the 2 epochs of transplant experience at St. Louis Children’s Hospital. Statistical analyses were performed using SAS® 9.2, Cary, NC. This study was approved by the Human Research Protection Office at Washington University School of Medicine.
Results
Infants
We compared clinical characteristics for infants listed (n=35) and transplanted (n=28) for genetic disorders of surfactant metabolism by epoch (1993–2003 (n=19 listed, 16 transplanted) and 2004–2015 (n=16 listed, 12 transplanted)) (Table II). Seven infants were listed but died awaiting transplant. Five children who were listed but later inactivated were not included in the analyses (Table III). Most infants underwent transplantation for surfactant protein-B or ABCA3 deficiency. Median time to follow up was 67 months for infants in epoch 1 and 59 months for infants in epoch 2. There was no difference in sex distribution between infants from the two epochs. More infants from epoch 2 were of Hispanic or Middle Eastern descent (p = 0.0017). The ages of the infants at listing and transplant, time spent awaiting transplant, and need for mechanical ventilation at time of transplant were similar for infants between the 2 epochs. Significantly more infants received extracorporeal membrane oxygenation (ECMO) prior to transplant during the first epoch (p = 0.0092). The majority (11/16) of infants transplanted in epoch 1 received a combination of cyclosporine and azathioprine for maintenance immunosuppression, while most (10/12) of the infants transplanted in epoch 2 were treated with tacrolimus and mycophenolate mofetil. The immunosuppressive regimens of 4 infants in epoch 1 were changed from cyclosporine and azathioprine to tacrolimus and mycophenolate mofetil for declining lung function. All infants from both epochs received corticosteroids as a part of their immunosuppressive regimen.
Table 3. Clinical Characteristics of Children Inactivated from Transplant List.
Five children were listed, but later inactivated due to improved or stable lung function. Clinical data for 4 of the 5 children are presented here. LPM: liters per minute of oxygen
| Genetic Etiology | Age of symptom onset (months) | Age at listing (months) | Maximum respiratory support | Respiratory support at time of listing | Respiratory support at time of inactivation | Reason for inactivation | Medications | Respiratory support and age at last follow up |
|---|---|---|---|---|---|---|---|---|
|
SFTPC c.218T>C p.I73T |
14 | 24 | Invasive endotracheal mechanical ventilation | 4 LPM | 3 LPM | Stable lung function | Pulse solumedrol, hydroxychloroquine, furosemide, albuterol | 3 LPM; 7.5 years |
|
SFTPC c.325-1 G>A |
4 | 72 | 2 LPM | 2 LPM | 1–2 LPM | Stable lung function | Prednisone, furosemide, albuterol | 1 LPM; 11.5 years |
|
SFTPC c.565T>G p.C189G |
0.5 | 24 | Invasive endotracheal mechanical ventilation | 0.5 LPM | Room air | Improved lung function | Pulse solumedrol, prednisone, cyclosporine, IVIG | Room air; 13 years |
|
ABCA3 c.3609_3611delCTT (p.F1203del)/c.4751delT (p.L1584Rfs*50) |
16 | 18 | 15 LPM | 3 LPM | 3 LPM | Stable lung function | Prednisone, hydroxychloroquine, azithromycin | 3 LPM; 9 years |
The 1-year (81% vs. 83%) and 5-year (56% vs. 56%) survival rates for transplanted infants were similar for both epochs (Table II) as was mortality evaluated by Kaplan-Meier analysis (Figure 3; available at www.jpeds.com). Deaths within the first year among infants were due to non-CMV infection (n=3) and graft-related complications including pulmonary vein stenosis (n=1) and bronchial stenosis (n=1). Deaths after the first year were due to bronchiolitis obliterans and infection. Despite a change in primary immunosuppressive regimens between the 2 epochs, there were no differences in short-term (CMV infection, seizures) or long-term (hypertension, renal insufficiency, bronchiolitis obliterans, or PTLD) morbidities between infants transplanted from the two epochs (Table II). Most infants received a gastrostomy tube for nutritional supplementation, and many infants had significant growth impairment, speech and motor delays, and hearing loss (Table II).
Figure 3.
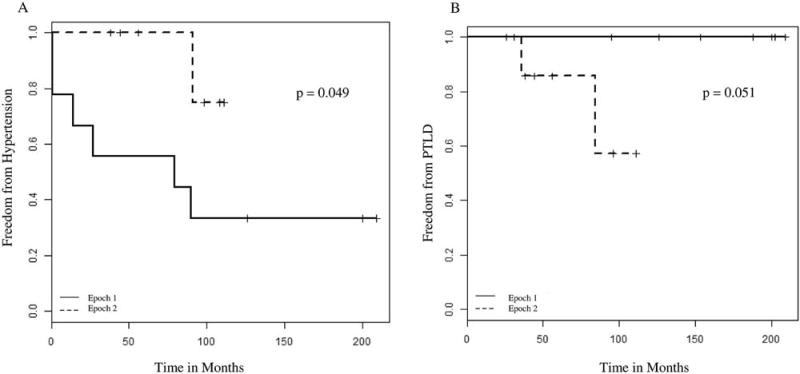
A, Kaplan-Meier analysis of freedom from hypertension: children, epoch 1 (1993–2003) compared with epoch 2 (2004–2015). B, Kaplan-Meier analysis of freedom from PTLD: children, epoch 1 (1993–2003) compared with epoch 2 (2004–2015)
Children
Most of the 16 children (age >1 year at transplant) listed and transplanted for genetic disorders of surfactant metabolism had SFTPC associated lung disease or ABCA3 deficiency (Table II). There were no differences in sex or race/ethnicity between children of the 2 epochs. One child died on ECMO while awaiting transplant. Children transplanted during epoch 2 were older at listing (99 months vs. 41 months, p=0.020) and transplant (118 months vs. 45 months, p=0.015). Children in epoch 2 waited longer for transplant (16 months vs. 4.2 months), but this difference was not statistically significant, p=0.055). Few children were mechanically ventilated at the time of transplant. Median time to follow up was 94 months for children in epoch 1 and 54 months for children in epoch 2.
Most (6/9) children transplanted in epoch 1 were initiated on cyclosporine and azathioprine for immunosuppression; however, the majority (5/6) were subsequently transitioned to tacrolimus and mycophenolate mofetil due to declining lung function. Most (6/7) children transplanted during epoch 2 were treated with tacrolimus and mycophenolate mofetil, and only one child received cyclosporine and azathioprine. All children from both epochs received steroids as a part of their immunosuppressive regimen.
All children transplanted for genetic disorders of surfactant metabolism survived to 1 year after transplant, and 5-year survival rates were similar between the 2 epochs (78% vs. 80%, p=1.0, Table II) with no differences detected by Kaplan-Meier survival curve analysis (Figure 4; available at www.jpeds.com). Deaths among children were due to bronchiolitis obliterans and non-PTLD malignancy. There were no differences in univariate analyses for short-(CMV infection, seizures) or long-term morbidities (hypertension, renal insufficiency, bronchiolitis obliterans, PTLD) at 5 years post-transplant for children from the 2 epochs (Table II). Using Kaplan-Meier analyses, we found that more children transplanted during epoch 1 developed hypertension (p=0.049, Figure 1, A; available at www.jpeds.com) while more children from epoch 2 developed PTLD (p = 0.051, Figure 1, B). The incidence of bronchiolitis obliterans was similar between the 2 epochs (56% vs. 60%, p=1.0, Table II), with no differences detected by Kaplan-Meier analysis. Most transplanted children had growth impairment and received gastrostomy tubes (Table II). Few children in either epoch had significant developmental delays, and none had hearing loss at last recorded follow up (Table II).
Figure 4.
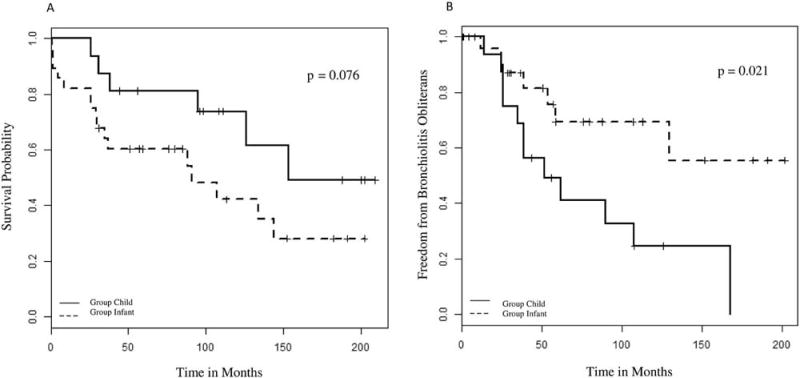
A, Kaplan-Meier analysis of mortality: infants compared with children, 1993–2015. B, Kaplan-Meier analysis of bronchiolitis obliterans: infants compared with children, 1993–2015.
Figure 1.
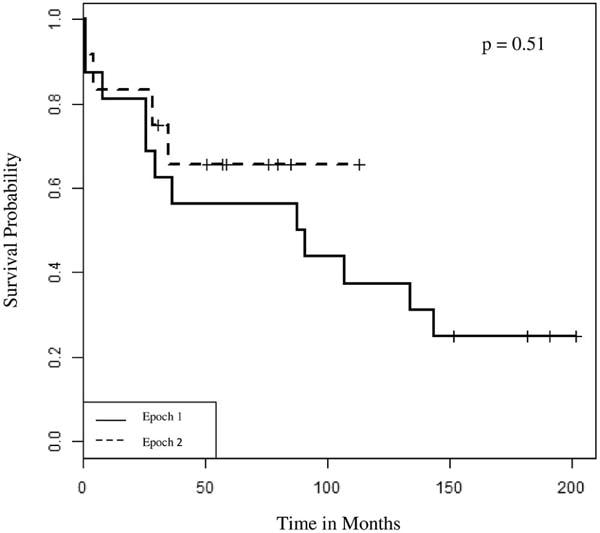
Kaplan-Meier Analysis of Mortality: Infants, Epoch 1 (1993–2003) compared to Epoch 2 (2004–2015).
Infants vs. Children
To compare outcomes for infants and children transplanted for genetic disorders of surfactant metabolism, we combined data from the 2 epochs (Table IV). There were no differences in sex and race/ethnicity between the infants and children. Infants were more likely to be transplanted for surfactant protein-B deficiency while children were more likely to be transplanted for SFTPC-associated lung disease (p<0.0001); both infants and children underwent transplant for ABCA3 deficiency (Table IV). Children waited longer for donor lungs (9.3 months vs. 1.9 months, p = 0.014, Table IV) and were less likely to be mechanically ventilated at the time of transplant (13% vs. 89%, p = <0.0001, Table IV) than infants. One infant from epoch 1 received a second transplant 4 months after first transplant due to chronic graft dysfunction. There were no differences in survival at 1 or 5 years between the infants and children (Table IV, Figure 2, A).
Table 4.
Clinical Characteristics and Post-Transplant Morbidities of Infants and Children Transplanted for Genetics Disorders of Surfactant Metabolism (1993–2015)
| Infants | Children | ||
|---|---|---|---|
|
| |||
| Number listed | 35 | 17 | |
|
| |||
| Number transplanted (% listed) | 28 (0.80) | 16 (0.94) | |
|
| |||
| Genetic etiology | |||
| SFTPB | 14 (0.40) | 0 (0) | <0.000 |
| SFTPC | 1 (0.03) | 11 (0.65) | 1 |
| ABCA3 | 17 (0.49) | 5 (0.29) | |
| NKX2-1 | 3 (0.08) | 1 (0.06) | |
|
| |||
| Sex Female | 19 (0.54) | 11 (0.65) | 0.48 |
| Male | 16 (0.46) | 6 (0.35) | |
|
| |||
| Race/Ethnicity | 0.57 | ||
| European descent | 27 (0.77) | 14 (0.82) | |
| African descent | 1 (0.03) | 2 (0.12) | |
| Hispanic | 2 (0.06) | 0 (0) | |
| Middle Eastern | 3 (0.08) | 0 (0) | |
| Other | 2 (0.06) | 1 (0.06) | |
|
| |||
| Death prior to transplantation | 7/35 (0.20) | 1/17 (0.06) | 0.25 |
|
| |||
| Wait time to transplant (months ± SD) | 1.9 ±1.5 | 9.3 ±11 | 0.014 |
|
| |||
| Mechanically ventilated at transplant | 25/28 (0.89) | 2/16 (0.13) | <0.0001 |
|
| |||
| ECMO prior to transplant (% listed) | 7/35 (0.20) | 1/17 (0.06) | 0.25 |
|
| |||
| Survival at 1 year post transplant | 23/28 (0.82) | 16/16 (1.0) | 0.14 |
|
| |||
| Survival at 5 years post transplant | 14/25 (0.56) * | 11/14 (0.79) * | 0.19 |
|
| |||
| CMV infection | 2/28 (0.07) | 2/16 (0.13) | 0.61 |
|
| |||
| Seizures | 3/28 (0.11) | 0/16 (0) | 0.29 |
|
| |||
| Hypertension by 5 years post transplant | 4/15 (0.27)† | 4/13 (0.31)† | 1.0 |
|
| |||
| Renal insufficiency by 5 years post transplant | 2/15 (0.13)‡ | 1/12 (0.08)‡ | 1.0 |
|
| |||
| Bronchiolitis obliterans by 5 years post transplant | 6/17 (0.35)§ | 8/14 (0.57)§ | 0.22 |
|
| |||
| PTLD by 5 years post transplant | 2/14 (0.14) | 1/11 (0.09) | 1.0 |
|
| |||
| Gastrostomy tube | 22/28 (0.79) | 14/16 (0.88) | 0.69 |
|
| |||
| Growth impairment | 14/26 (0.54)ǁ | 10/16 (0.63) | 0.58 |
|
| |||
| Speech delay | 19/22 (0.86)ǁ | 2/16 (0.13) | <0.0001 |
|
| |||
| Motor delay | 19/24 (0.79)ǁ | 2/16 (0.13) | <0.0001 |
|
| |||
| Hearing loss | 7/21 (0.33)ǁ | 0/16 (0) | 0.012 |
Data presented as number (percent) of individuals
Does not include infants or children alive, but not yet 5 years post-transplant
Numerator and denominator include patients with hypertension who died prior to 5 years post-transplant
Numerator and denominator include patients with renal insufficiency who died prior to 5 years post-transplant
Numerator and denominator include patients with BO who died prior to 5 years post-transplant
Information not available for all infants
Figure 2.
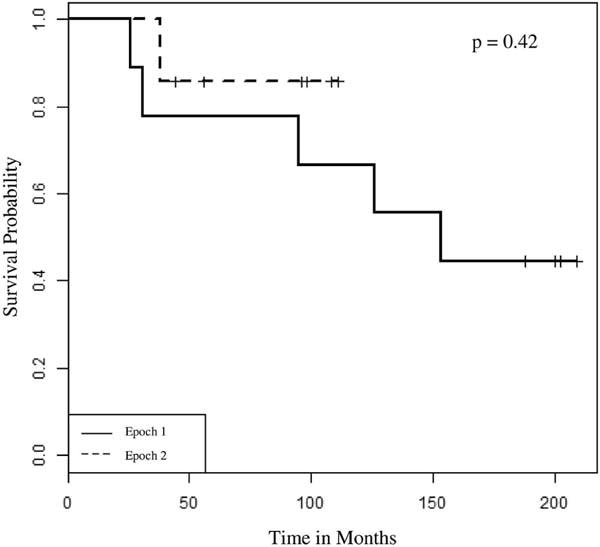
Kaplan-Meier analysis of mortality: children, epoch 1 (1993–2003) compared with epoch 2 (2004–2015).
Using Kaplan-Meier analysis, infants were less likely to develop bronchiolitis obliterans (p=0.021, Figure 2, B) and more likely to have motor and speech delays (79–86% vs. 13%, p <0.0001, Table IV) and hearing loss (33% vs. 0%, p =0.012, Table IV) than children.
Discussion
Mortality and long-term morbidities remain considerable for infants and children who undergo lung transplantation for genetic disorders of surfactant metabolism. Despite advances in genetic diagnosis, immunosuppressive therapies, and supportive respiratory and nutritional care, 5-year mortality was similar for both infants and children for the 2 epochs. The overall five year survival rates of infants (56%) and children (79%) transplanted for genetic disorders of surfactant metabolism at our single center are similar to those reported for infants and children transplanted for cystic fibrosis and pulmonary vascular disease.(30–32)
As observed in prior studies of infants with surfactant protein-B and ABCA3 deficiency, most infants from both epochs were critically ill at the time of transplant, requiring mechanical ventilation (including high frequency oscillatory ventilation) or ECMO.(33, 34) However, the timing of presentation of 6 infants (range 2 weeks-5 months) and 5 children with ABCA3 deficiency emphasizes the variability in disease course associated with biallelic missense, splice site, and in-frame insertion/deletions in ABCA3.(35)
We did not find any differences in age of infants at listing for transplant or the rate of death prior to transplant between epochs 1 and 2, suggesting that increased availability of definitive diagnostic sequencing and advances in neonatal intensive care including additional non-invasive ventilation strategies, increased availability of nitric oxide, nutritional support, and empiric medical therapies have not significantly impacted survival to transplant for infants with genetic disorders of surfactant metabolism. Transplanted infants were more likely to have motor and speech delays than children, likely reflecting greater pre-transplant severity of illness and the variable prognostic accuracy of neurologic evaluation in infancy. In addition, prolonged exposure to sedative medications, intermittent hypoxic episodes, suboptimal nutrition, and decreased developmental stimulation while awaiting transplant may contribute to poor developmental outcomes among transplanted infants. Our study was limited in that not all infants and children underwent formal developmental testing, and we may not have accounted for “catch-up” development or school readiness/performance.
Bronchiolitis obliterans remains a leading cause of death after the first year of transplant for pediatric lung transplant recipients.(30) Similar to a previous report from our institution,(36) we also found a decreased incidence of bronchiolitis obliterans among infant lung transplant recipients compared with children (Figure 2, B, p=0.021); however, post-transplant mortality among infants and children were similar (Figure 2, A) and consistent with other studies.(30, 31) In addition, rates of bronchiolitis obliterans were similar between the 2 epochs for infants and children. A lower incidence of bronchiolitis obliterans among infant recipients may reflect immaturity of the infant immune system at time of transplant.(36) Although more children in epoch 2 developed PTLD compared with epoch 1 (Figure 1, B), the overall incidence of PTLD among infants and children in our study was similar to previous reports.(36) Records of pre-transplantation Epstein-Barr virus status were incomplete and therefore not included in our analyses.
Prior studies from eras corresponding to epoch 1 and epoch 2 found similar rates of renal dysfunction (30–40%)(30, 37) and suggest that renal insufficiency and hypertension (up to 70% of patients) (30) may develop due to side-effects of chronic immunosuppressive medications. The lower rates of renal insufficiency and hypertension at 5 years post-transplant observed in our study compared with others may be due to incomplete recording of diagnoses in the medical chart. Fewer children developed hypertension in epoch 2 (Figure 1, A), possibly related to modifications to immunosuppressive regimens.
Growth impairment continues to be a major challenge for both infant and child lung transplant recipients.(37, 38) Poor nutritional status prior to transplantation independently increases post-transplant mortality among adult recipients.(39) The majority of infants and children in our study underwent gastrostomy tube placement for nutritional supplementation and exhibited growth impairment post-transplant. A previous study from our institution reported an average post-transplant linear growth of 64% of predicted in the first year of follow-up for infant and child lung transplant recipients.(38)
Although responses to empiric medical therapies including corticosteroids, azithromycin, and hydroxychloroquine have been observed in some infants and children with ABCA3 deficiency or SFTPC mutations,(17, 18, 20, 32, 40) others progress to transplantation or death despite these therapies.(35, 41) Even though infants with biallelic loss of function (nonsense, frameshift) mutations in SFTPB and ABCA3 present with severe respiratory failure at birth and die without lung transplantation by 1 year of age (35, 42, 43), genotype-phenotype correlations for infants and children with SFTPC, NKX2-1, and ‘other’ (missense, splice site, in-frame insertion/deletions) ABCA3 mutations are more variable and more difficult to predict.(44–46) This variability in disease course is highlighted by the observation that five children listed for transplant were listed but later inactivated due to improved or stable lung function (Table III). Four of these children have SFTPC mutations and 1 child is compound heterozygous for a frameshift and an in-frame deletion in ABCA3. Less predictable genotype-phenotype correlations and variability in disease coursecomplicates discussions with families regarding lung transplantation.(20) Tracheostomy and chronic ventilation in combination with empiric anti-inflammatory agents (corticosteroids, azithromycin, hydroxychloroquine, and/or azathioprine) have been suggested as alternatives to lung transplantation for infants and children with SFTPC mutations and severe, persistent respiratory failure, with some reported children decannulated and weaned from all respiratory support over 2–6 years. (19, 22) In addition to further characterization of the natural histories of infants and children with variable genotype-phenotype correlations (SFTPC, NKX2-1, and ‘other’ ABCA3 mutations), studies of factors (genetic and environmental) that contribute to prolonged survival without transplantation, functional studies of disease mechanisms, and randomized clinical trials of pharmacotherapeutics are needed to determine whether anecdotal responses to empiric therapies are related to disease amelioration or the natural waxing/waning disease course of chILD.(47)
Although transplantation may permit short-term survival for infants and children with otherwise lethal genetic lung disease, it is associated with chronic medical challenges and long-term morbidities. Counseling which reflects these realities likely contributes to parental decisions not to pursue lung transplantation for approximately 50% of families.(27, 34) In addition to medical complications of transplantation, families encounter considerable social challenges as frequent follow up at transplant centers is required in the months and years after transplant for surveillance bronchoscopies, pulmonary function tests, and laboratory evaluations. As genetic disorders of surfactant metabolism are rare, a multi-disciplinary care delivery model by a team of physicians (lung transplant pulmonologists, neonatologists, cardiothoracic surgeons, geneticists, nephrologists, neurologists), social workers, nutritionists, physical and occupational therapists, audiologists, and families of prior transplant recipients is needed to optimize care for affected patients. Interdisciplinary coordination can inform families of the risks and benefits of lung transplantation, optimize pre- and post-transplant care, support families with their decisions, and communicate genetic recurrence risk. Prospective studies such as those currently in development through the Children’s Interstitial Lung Disease Research Network (chILDRN) to characterize the natural history, strategies to optimize pre-transplant health status, and approaches to reduce the incidence of bronchiolitis obliterans are needed to improve outcomes for infants and children with genetic disorders of surfactant metabolism.
Acknowledgments
Support by the National Institutes of Health (K08 HL105891 [to J.W.], K12 HL120002 [to F.C.], R01 HL065174 [to F.C.], R01 HL082747 [to F.C.]), American Lung Association (to J.W.), American Thoracic Society (to J.W.), and Children’s Discovery Institute and the Saigh Foundation (to F.C.).
List of Abbreviations
- ABCA3
ATP-binding cassette member A3
- SFTPB
Surfactant Protein B Gene
- SFTPC
Surfactant Protein C Gene
- RDS
respiratory distress syndrome
- chILD
childhood interstitial lung disease
- PTLD
post-transplant lymphoproliferative disorder
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Edited by RS and WFB
The authors declare no conflicts of interest.
We thank Craig Cole for assisting with data acquisition from the St. Louis Children’s Pediatric Lung Transplant Database.
References
- 1.Weaver TE, Conkright JJ. Function of surfactant proteins B and C. Annu Rev Physiol. 2001;63:555–78. doi: 10.1146/annurev.physiol.63.1.555. [DOI] [PubMed] [Google Scholar]
- 2.Ban N, Matsumura Y, Sakai H, Takanezawa Y, Sasaki M, Arai H, et al. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. 2007 Mar 30;282:9628–34. doi: 10.1074/jbc.M611767200. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda K, Clark JC, Shaw-White JR, Stahlman MT, Boutell CJ, Whitsett JA. Gene structure and expression of human thyroid transcription factor-1 in respiratory epithelial cells. J Biol Chem. 1995 Apr 7;270:8108–14. doi: 10.1074/jbc.270.14.8108. [DOI] [PubMed] [Google Scholar]
- 4.Garmany TH, Wambach JA, Heins HB, Watkins-Torry JM, Wegner DJ, Bennet K, et al. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res. 2008 Jun;63:645–9. doi: 10.1203/PDR.0b013e31816fdbeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nogee LM, de Mello DE, Dehner LP, Colten HR. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med. 1993 Feb 11;328:406–10. doi: 10.1056/NEJM199302113280606. [DOI] [PubMed] [Google Scholar]
- 6.Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med. 2005 Oct 15;172:1026–31. doi: 10.1164/rccm.200503-504OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole FS, Hamvas A, Rubinstein P, King E, Trusgnich M, Nogee LM, et al. Population-based estimates of surfactant protein B deficiency. Pediatrics. 2000 Mar;105:538–41. doi: 10.1542/peds.105.3.538. [DOI] [PubMed] [Google Scholar]
- 8.Hamvas A, Nogee LM, deMello DE, Cole FS. Pathophysiology and treatment of surfactant protein-B deficiency. Biol Neonate. 1995;67(Suppl 1):18–31. doi: 10.1159/000244204. [DOI] [PubMed] [Google Scholar]
- 9.Hamvas A, Nogee LM, White FV, Schuler P, Hackett BP, Huddleston CB, et al. Progressive lung disease and surfactant dysfunction with a deletion in surfactant protein C gene. Am J Respir Cell Mol Biol. 2004 Jun;30:771–6. doi: 10.1165/rcmb.2003-0323OC. [DOI] [PubMed] [Google Scholar]
- 10.Nogee LM, Dunbar AE, 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001 Feb 22;344:573–9. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 11.Poterjoy BS, Vibert Y, Sola-Visner M, McGowan J, Visner G, Nogee LM. Neonatal respiratory failure due to a novel mutation in the surfactant protein C gene. J Perinatol : official journal of the California Perinatal Association. 2010 Feb;30:151–3. doi: 10.1038/jp.2009.97. [DOI] [PubMed] [Google Scholar]
- 12.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004 Mar 25;350:1296–303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 13.Wambach JA, Wegner DJ, Depass K, Heins H, Druley TE, Mitra RD, et al. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics. 2012 Dec;130:e1575–82. doi: 10.1542/peds.2012-0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devriendt K, Vanhole C, Matthijs G, de Zegher F. Deletion of thyroid transcription factor-1 gene in an infant with neonatal thyroid dysfunction and respiratory failure. N Engl J Med. 1998 Apr 30;338:1317–8. doi: 10.1056/NEJM199804303381817. [DOI] [PubMed] [Google Scholar]
- 15.Krude H, Schutz B, Biebermann H, von Moers A, Schnabel D, Neitzel H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002 Feb;109:475–80. doi: 10.1172/JCI14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamvas A, Deterding RR, Wert SE, White FV, Dishop MK, Alfano DN, et al. Heterogeneous pulmonary phenotypes associated with mutations in the thyroid transcription factor gene NKX2-1. Chest. 2013 Sep;144:794–804. doi: 10.1378/chest.12-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayes D, Jr, Lloyd EA, Fitch JA, Bush A. ABCA3 transporter deficiency. Am J Respir Crit Care Med. 2012 Oct 15;186:807. doi: 10.1164/ajrccm.186.8.807a. [DOI] [PubMed] [Google Scholar]
- 18.Rosen DM, Waltz DA. Hydroxychloroquine and surfactant protein C deficiency. N Engl J Med. 2005 Jan 13;352:207–8. doi: 10.1056/NEJM200501133520223. [DOI] [PubMed] [Google Scholar]
- 19.van Hoorn J, Brouwers A, Griese M, Kramer B. Successful weaning from mechanical ventilation in a patient with surfactant protein C deficiency presenting with severe neonatal respiratory distress. BMJ Case Rep. 2014 doi: 10.1136/bcr-2013-203053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroner C, Reu S, Teusch V, Schams A, Grimmelt AC, Barker M, et al. Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur Respir J. 2015 Jul;46:197–206. doi: 10.1183/09031936.00129414. [DOI] [PubMed] [Google Scholar]
- 21.Rabach I, Poli F, Zennaro F, Germani C, Ventura A, Barbi E. Is treatment with hydroxychloroquine effective in surfactant protein C deficiency? Arch Bronconeumol. 2013 May;49:213–5. doi: 10.1016/j.arbres.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Liptzin DR, Patel T, Deterding RR. Chronic ventilation in infants with surfactant protein C mutations: an alternative to lung transplantation. Am J Respir Crit Care Med. 2015 Jun 1;191:1338–40. doi: 10.1164/rccm.201411-1955LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huddleston CB, Sweet SC, Mallory GB, Hamvas A, Mendeloff EN. Lung transplantation in very young infants. J Thorac Cardiovasc Surg. 1999 Nov;118:796–804. doi: 10.1016/s0022-5223(99)70048-6. [DOI] [PubMed] [Google Scholar]
- 24.Dharnidharka VR, Lamb KE, Zheng J, Schechtman KB, Meier-Kriesche HU. Lack of significant improvements in long-term allograft survival in pediatric solid organ transplantation: A US national registry analysis. Pediatr Transplant. 2015 Aug;19:477–83. doi: 10.1111/petr.12465. [DOI] [PubMed] [Google Scholar]
- 25.Kim JJ, Marks SD. Long-term outcomes of children after solid organ transplantation. Clinics. 2014;69(Suppl 1):28–38. doi: 10.6061/clinics/2014(Sup01)06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013 Aug 1;188:376–94. doi: 10.1164/rccm.201305-0923ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faro AHA. Lung Transplantation for Inherited Disorders of Surfactant Metabolism. Neoreviews. 2008;9:468–76. [Google Scholar]
- 28.Stewart S, Fishbein MC, Snell GI, Berry GJ, Boehler A, Burke MM, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007 Dec;26:1229–42. doi: 10.1016/j.healun.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 29.Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, Flegal KM, Guo SS, Wei R, et al. CDC growth charts: United States. Adv Data. 2000 Jun;8:1–27. [PubMed] [Google Scholar]
- 30.Benden C, Edwards LB, Kucheryavaya AY, Christie JD, Dipchand AI, Dobbels F, et al. The Registry of the International Society for Heart and Lung Transplantation: Sixteenth Official Pediatric Lung and Heart-Lung Transplantation Report–2013; focus theme: age. J Heart Lung Transplant: the official publication of the International Society for Heart Transplantation. 2013 Oct;32:989–97. doi: 10.1016/j.healun.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Khan MS, Heinle JS, Samayoa AX, Adachi I, Schecter MG, Mallory GB, et al. Is lung transplantation survival better in infants? Analysis of over 80 infants. J Heart Lung Transplant. 2013 Jan;32:44–9. doi: 10.1016/j.healun.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 32.Rama JA, Fan LL, Faro A, Elidemir O, Morales DL, Heinle JS, et al. Lung transplantation for childhood diffuse lung disease. Pediatr Pulmonol. 2013 May;48:490–6. doi: 10.1002/ppul.22634. [DOI] [PubMed] [Google Scholar]
- 33.King EL, Shackelford GD, Hamvas A. High-frequency oscillation and paralysis stabilize surfactant protein-B–deficient infants. J Perinatol. 2001 Oct-Nov;21:421–5. doi: 10.1038/sj.jp.7210555. Epub 2002/03/16. eng. [DOI] [PubMed] [Google Scholar]
- 34.Palomar LM, Nogee LM, Sweet SC, Huddleston CB, Cole FS, Hamvas A. Long-term outcomes after infant lung transplantation for surfactant protein B deficiency related to other causes of respiratory failure. J Pediatr. 2006 Oct;149:548–53. doi: 10.1016/j.jpeds.2006.06.004. Epub 2006/10/03. eng. [DOI] [PubMed] [Google Scholar]
- 35.Wambach JA, Casey AM, Fishman MP, Wegner DJ, Wert SE, Cole FS, et al. Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. 2014 Jun 15;189:1538–43. doi: 10.1164/rccm.201402-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elizur A, Faro A, Huddleston CB, Gandhi SK, White D, Kuklinski CA, et al. Lung transplantation in infants and toddlers from 1990 to 2004 at St. Louis Children’s Hospital. Am J Transplant. 2009 Apr;9:719–26. doi: 10.1111/j.1600-6143.2009.02552.x. [DOI] [PubMed] [Google Scholar]
- 37.Hmiel SP, Beck AM, de la Morena MT, Sweet S. Progressive chronic kidney disease after pediatric lung transplantation. Am J Transplant. 2005 Jul;5:1739–47. doi: 10.1111/j.1600-6143.2005.00930.x. [DOI] [PubMed] [Google Scholar]
- 38.Sweet SC, Spray TL, Huddleston CB, Mendeloff E, Canter CE, Balzer DT, et al. Pediatric lung transplantation at St. Louis Children’s Hospital, 1990–1995. Am J Respir Crit Care Med. 1997 Mar;155:1027–35. doi: 10.1164/ajrccm.155.3.9116982. [DOI] [PubMed] [Google Scholar]
- 39.Lederer DJ, Wilt JS, D’Ovidio F, Bacchetta MD, Shah L, Ravichandran S, et al. Obesity and underweight are associated with an increased risk of death after lung transplantation. Am J Respir Crit Care Med. 2009 Nov 1;180:887–95. doi: 10.1164/rccm.200903-0425OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thouvenin G, Nathan N, Epaud R, Clement A. Diffuse parenchymal lung disease caused by surfactant deficiency: dramatic improvement by azithromycin. BMJ Case Rep. 2013 doi: 10.1136/bcr-2013-009988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winter J, Essmann S, Kidszun A, Aslanidis C, Griese M, Poplawska K, et al. Neonatal respiratory insufficiency caused by an (homozygous) ABCA3-stop mutation: a systematic evaluation of therapeutic options. Klin Padiatr. 2014 Apr;226:53–8. doi: 10.1055/s-0033-1363687. [DOI] [PubMed] [Google Scholar]
- 42.Turcu S, Ashton E, Jenkins L, Gupta A, Mok Q. Genetic testing in children with surfactant dysfunction. Arch Dis Child. 2013;98:490–5. doi: 10.1136/archdischild-2012-303166. [DOI] [PubMed] [Google Scholar]
- 43.Tredano M, Griese M, de Blic J, Lorant T, Houdayer C, Schumacher S, et al. Analysis of 40 sporadic or familial neonatal and pediatric cases with severe unexplained respiratory distress: relationship to SFTPB. American journal of medical genetics Part A. 2003;119A:324–39. doi: 10.1002/ajmg.a.20058. [DOI] [PubMed] [Google Scholar]
- 44.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, Markin C, Speer M, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002 May 1;165:1322–8. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 45.Percopo S, Cameron HS, Nogee LM, Pettinato G, Montella S, Santamaria F. Variable phenotype associated with SP-C gene mutations: fatal case with the I73T mutation. Eur Respir J. 2004 Dec;24:1072–3. doi: 10.1183/09031936.04.00092304. [DOI] [PubMed] [Google Scholar]
- 46.Abou Taam R, Jaubert F, Emond S, Le Bourgeois M, Epaud R, Karila C, et al. Familial interstitial disease with I73T mutation: A mid- and long-term study. Pediatr Pulmonol. 2009 Feb;44:167–75. doi: 10.1002/ppul.20970. [DOI] [PubMed] [Google Scholar]
- 47.Fan LL, Dishop MK, Galambos C, Askin FB, White FV, Langston C, et al. Diffuse Lung Disease in Biopsied Children 2–18 Years of Age: Application of the chILD Classification Scheme. Ann Am Thorac Soc. 2015 Aug 20; doi: 10.1513/AnnalsATS.201501-064OC. [DOI] [PMC free article] [PubMed] [Google Scholar]