

Abstract
Runx2 may play an important role in development of osteoarthritis (OA). However, the specific role of Runx2 in articular chondrocyte function and in OA development in adult mice has not been fully defined. In this study, we performed the destabilization of the medial meniscus (DMM) surgery at 12-week-old mice to induce OA in adult Runx2 Agc1CreER mice, in which Runx2 was specifically deleted in Aggrecan-expressing chondrocytes by administering tamoxifen at 8-weeks of age. Knee joint samples were collected 8- and 12-weeks post-surgery and analyzed through histology, histomorphometry and micro-computed tomography (μCT). Our results showed that severe OA-like defects were observed after DMM surgery in Cre-negative control mice, including articular cartilage degradation and subchondral sclerosis, while the defects were significantly ameliorated in Runx2 Agc1CreER KO mice. Immunohistochemical (IHC) results showed significantly reduced expression of MMP13 in Runx2 Agc1CreER KO mice compared to that in Cre-negative control mice. Results of quantitative reverse-transcription PCR (qRT-PCR) demonstrated that expression of the genes encoding for matrix degradation enzymes was significantly decreased in Runx2 Agc1CreER KO mice. Thus, our findings suggest that inhibition of Runx2 in chondrocytes could at least partially rescue DMM-induced OA-like defects in adult mice.
Introduction
Osteoarthritis (OA) is the most common degenerative joint disease, affecting close to 27 million Americans. The major pathological features of OA include progressive loss of articular cartilage, osteophyte formation, the increases in subchondral bone mass and synovial tissue inflammation and hyperplasia1, 2. Multiple animal models have been established to mimic the development of OA. Among them, DMM-induced OA is the most widely used OA animal model3.
During OA progression, articular chondrocytes undergo hypertrophic differentiation. Runx2 plays a pivotal role in regulation of genes important for chondrocyte differentiation, matrix degradation and osteoblast differentiation4, 5. Several studies reported that Runx2 expression levels are high in human OA cartilage6–8. To explore the role of Runx2 in OA progression, genetic animal models have been used. Mice with Runx2 overexpression (Runx2-Tg) display an increased number of cartilage protease expression in chondrocytes9. Overexpression of Runx2 also activates matrix degradation enzymes (MMP13 and ADAMTS5) through mitogen-activated protein kinase (MAPK) pathways10 and through direct regulation of Mmp13 gene transcription11. Previous work in our laboratory showed that deletion of Tgfbr2 in chondrocytes up-regulates Runx2 and accelerates OA progression12. A recent study reported that Runx2 has been identified as a novel potential target of miR-105 and the FGF2-p65-miR-105-Runx2 axis might play an important role in OA pathogenesis13. A previous study reported that heterozygous Runx2 global KO mice exhibited decreased cartilage destruction and osteophyte formation after induction of knee joint instability14. Since mice used in this study is global Runx2 heterozygous KO mice (Runx2 +/−), the specific effect of Runx2 on articular cartilage in adult mice remains undefined.
Aggrecan is a major extracellular matrix protein in articular cartilage. Aggrecan gene is expressed more robustly than Col2a1 gene, another cartilage matrix component in adult mice15, 16. To determine the role of Runx2 in OA development in adult mice, we have generated Runx2 Agc1CreER conditional KO mice by crossing Runx2 flox/flox mice17 with Agc1-CreER transgenic mice16. In the present studies, we determined if Runx2 specific deletion in chondrocytes in adult mice has chondro-protective effect on DMM-induced OA development.
Matrix metalloproteinase 13 (MMP13) is a potent enzyme that targets cartilage for degradation. MMP13 expression was low in normal and early degenerative cartilage but was strongly up-regulated in late-stage OA specimens18. Moreover, transgenic mice with constitutively active MMP13 expression in the hyaline cartilages and joints developed pathological changes in articular cartilage of mouse joints similar to those observed in human OA19. Clinical investigation suggests that MMP13 may be associated with cartilage degradation during OA development20. This clinical observation was further confirmed by the study with Mmp13 KO mice. Pharmacological inhibition of MMP13 activity has been demonstrated to be an effective strategy to decelerate articular cartilage loss in a murine model of injury-induced knee OA21.
The relationship of Runx2 and Mmp13 has been studied in the developing skeleton during the process of endochondral ossification. The DNA sequence of Runx2 binding site was originally described as osteoblast-specific elemenet-2 (OSE2), which is essential for expression of osteoblast-specific gene osteocalscin22, 23. The Mmp13 proximal promoter contains an OSE2 site conserved among different species, such as human, rabbit, mouse and rats24–27, 11. Runx2 binds to the OSE2 site in the Mmp13 promoter and increases Mmp13 gene transcription in cooperation with c-Fos and c-Jun binding to a neighboring AP-1 site26, 28–30. Moreover, co-transfection of Runx2 with the Mmp13 promoter in osteosarcoma UMR 106–01 cells has been shown to enhance Mmp13 promoter activity29. It has been showed that 148 bp upstream of Mmp13 transcription start site is sufficient and necessary for Mmp13 gene expression in bone, teeth and skin in vivo and the AP-1 and Runx2 binding sites are likely to regulate this Mmp13 proximal promoter activity. Runx2 also regulates Mmp13 during chondrocytes differentiation. A recent study reported that the interaction of Runx2 and Osterix, a downstream molecule of Runx2, cooperatively induces Mmp13 expression during chondrocyte differentiation31. In recent studies, we demonstrated, through mutation analysis and ChIP assays, that Runx2 activates Mmp13 expression by binding to the OSE2 site located in the proximal region of the human Mmp13 promoter in chondrocytes11.
Results
Agc1-CreER mice drive Cre recombination in articular chondrocytes
To efficiently target articular chondrocytes in adult mice, we used Agc1-CreER transgenic mice16. We evaluated the targeting efficiency and specificity of these mice by breeding them with ROSA mT/mG reporter mice. ROSA mT/mG mice are double-fluorescent Cre reporter mice that express membrane-targeted tandem dimer Tomato (mT) prior to Cre-mediated excision and membrane-targeted green fluorescent protein (mG) after excision. Also, mG labeling is Cre-dependent, complementary to mT at single cell resolution, and distinguishable by fluorescence-activated cell sorting: red before and green after recombination32. Analysis of histologic frozen sections from 3-month-old mice (tamoxifen was given to the mice at 2-month-old) using fluorescence microscopy showed that Agc1-CreER targeting cells are located in growth plate, articular cartilage and meniscus in Agc1-CreER; ROSA mT/mG mice (Fig. 1a).
Figure 1.
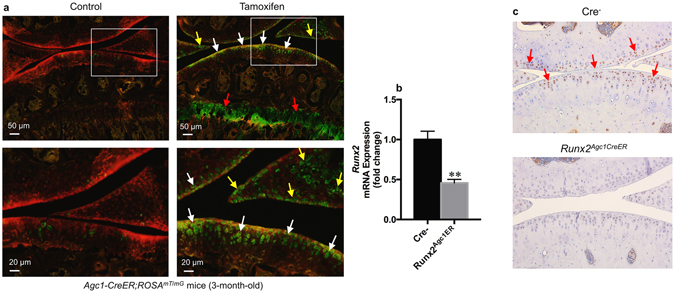
Directed Cre recombination in articular chondrocytes from Agc1-CreER mice. (a) Agc1-CreER mice target articular chondrocytes in adult mice. Agc1-CreER; ROSA mT/mG mice were generated by breeding Agc1-CreER transgenic mice with ROSA mT/mG reporter mice. Tamoxifen or vehicle control was administered into 2-month-old Agc1-CreER; ROSA mT/mG mice. Bone samples were harvested from 3-month-old mice after they were injected with tamoxifen at age of 2 months (1 mg/10 g body weight, i.p. injection, daily for 5 days). Histologic sections of Agc1-CreER; ROSA mT/mG mice with or without tamoxifen treatment were analyzed by fluorescence microscopy. The results showed that Agc1-CreER targeting GFP-positive cells (green color cells) are located in growth plate (red arrowheads), articular cartilage (white arrowheads) and meniscus (yellow arrowheads) in Agc1-CreER; ROSA mT/mG mice. (b) Significant decrease in Runx2 mRNA expression was observed in Runx2 Agc1CreER mice compared to their Cre-negative littermates. Total RNA was isolated from articular cartilage of 5-month-old Runx2 Agc1CreER mice and their Cre-negative littermates and real-time PCR assay was performed. All mice were administrated with tamoxifen at 2-months of age (**P < 0.01 versus Cre-negative mice, unpaired Student’s t-test; n = 3 mice per group). (c) Immunohistochemical (IHC) results showed that Runx2 protein levels were significantly decreased in articular cartilage of Runx2 conditional KO mice compared to Cre-negative mice (Red arrowheads show Runx2 positive cells, n = 3 mice per group).
The efficiency of Runx2 KO in the articular cartilage in Runx2Agc1CreER mice
To assess the efficiency of Runx2 KO in the articular cartilage in adult Runx2 Agc1CreER KO mice, we administrated tamoxifen to 2-month-old Runx2 Agc1CreER mice and their Cre-negative littermates. We then sacrificed them at the 5-months of age. Subsequently, the q RT-PCR and IHC staining were performed. The q RT-PCR analysis showed that Runx2 mRNA levels were significantly decreased by 60% in articular cartilage of Runx2 Agc1CreER KO mice compared to Cre-negative control mice (Fig. 1b). Consistent with this result, the IHC analysis showed that Runx2 protein levels were decreased in articular cartilage of Runx2 Agc1CreER KO mice (Fig. 1c).
OA progression was decelerated in Runx2Agc1CreER mice
To investigate if Runx2 deletion could prevent or decelerate DMM-induced OA-like defects, we crossed Runx2 flox/flox mice with Agc1-CreER transgenic mice16 to generate the Runx2 Agc1CreER conditional KO mice. Tamoxifen was administered into 8-week-old Runx2 Agc1CreER mice. Deletion of Runx2 had no significant effect on chondrocyte morphology (Figs 2a and 3a). To create an OA mouse model, DMM surgery was performed when the mice were 12-week-old. Knee joint samples were harvested 8- and 12-weeks post-surgery (n = 6–8 mice per group). Results of histological analysis showed that the OA-like phenotype, including fibrillation, clefting and cartilage degradation, was observed 8-weeks after DMM surgery and worsened at the 12-week time point in Cre-negative control mice. In contrast, much less articular cartilage excavation (but not statistical significant) was observed in 8-week-old Runx2 AgcCre1ER KO mice (Fig. 2a). In contrast, deletion of Runx2 significantly protected DMM-induced OA development at the time point 12-weeks after DMM surgery (Fig. 3a).
Figure 2.
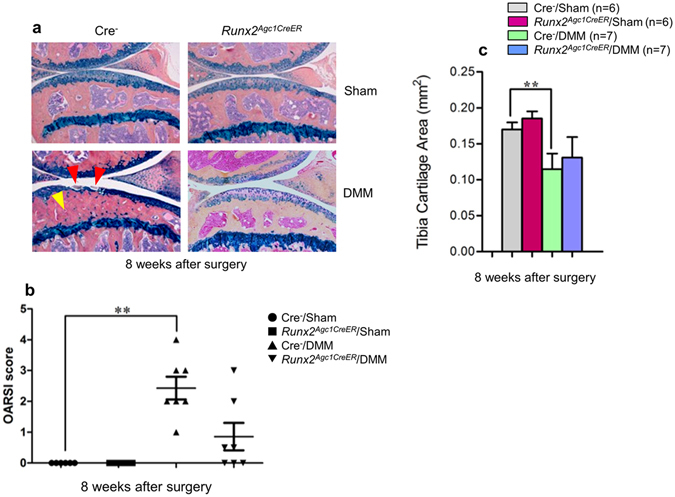
OA progression was decelerated in Runx2 Agc1CreER mice 8 weeks after DMM surgery. Tamoxifen was administered into 8-week-old male Cre-negative control and Runx2 Agc1CreER KO mice (1 mg/10 g body weight, i. p. injection, daily for 5 days). DMM surgery or Sham operation was performed when these mice were 12-week-old (right hind limbs). (a) Knee joints were harvested from Cre-negative and Runx2 Agc1CreER KO mice 8 weeks post-surgery and Alcian blue/Hematoxylin Orange G staining was performed. Histological results showed that articular cartilage degradation (red arrowheads) and subchondral sclerosis (yellow arrowhead) were observed in Cre-negative control mice after DMM surgery. In contrast, defects in articular cartilage degradation and subchondral sclerosis induced by DMM surgery was significantly protected in Runx2 Agc1CreER KO mice. (b,c) Histological sections were analyzed by OARSI scoring system and by histomorphometric method. The severity of OA-like phenotype was analyzed by grading histological sections using OARSI score system by two blinded observers. Articular cartilage area of tibia plateau was quantified by tracing the Alcian blue-positive staining areas using the OsteoMeasure system. These results demonstrated that DMM surgery caused significant OA-like defects in Cre-negative control mice. However, no significant difference, rather a decreased tendency of cartilage degeneration was observed in the mice 8-weeks after DMM surgery in Runx2 Agc1CreER KO mice compared to Cre-negative control mice. (**P < 0.01, one-way ANOVA followed by Tukey’s post-hoc test; n = 6–7 mice per group).
Figure 3.
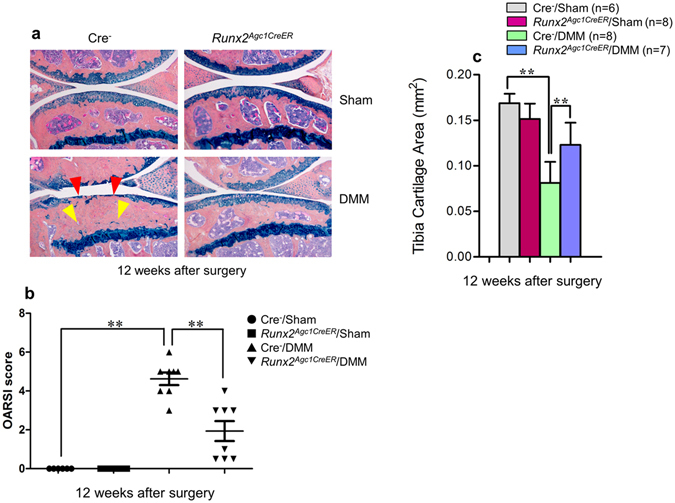
OA progression was protected in Runx2 Agc1CreER mice 12 weeks after DMM surgery. (a) Knee joints were harvested from Cre-negative and Runx2 Agc1CreER KO mice 12 weeks post-surgery and Alcian blue/Hematoxylin Orange G staining was performed. Histological results showed that articular cartilage degradation (red arrowheads) and subchondral sclerosis (yellow arrowheads) were observed in Cre-negative control mice after DMM surgery. In contrast, defects in articular cartilage degradation and subchondral sclerosis induced by DMM surgery was significantly protected in Runx2 Agc1CreER mice. (b) Histological sections were analyzed by OARSI scoring system. The severity of OA-like phenotype was graded by OARSI scoring system by two blinded observers. The results demonstrated that DMM surgery caused significant OA-like defects in Cre-negative control mice. In contrast, DMM-induced OA-like defects (12-weeks post-surgery) were significantly protected in Runx2 Agc1CreER KO mice (**P < 0.01, one-way ANOVA followed by Tukey’s post-hoc test; n = 6–8 mice per group). (c) Histological sections were analyzed by histomorphometric method. Articular cartilage area of tibia plateau was quantified by tracing the Alcian blue-positive staining areas using the OsteoMeasure system. DMM surgery led to significant loss of articular cartilage in Cre-negative control mice. In contrast, this effect was significantly protected by deletion of Runx2 observed in Runx2 Agc1CreER KO mice (12-weeks after DMM surgery) (**P < 0.01, one-way ANOVA followed by Tukey’s post-hoc test; n = 6–8 mice per group).
The evaluation using OARSI scoring system revealed that there was no significant difference, rather a decreased tendency of cartilage degeneration in the mice 8-weeks after DMM surgery in Runx2 Agc1CreER KO mice (Fig. 2b). With time progression, significantly reduced cartilage degeneration was observed in Runx2 Agc1CreER KO mice 12-weeks after DMM surgery (Fig. 3b). We then quantify the OA progression by performing histomorphometry using the OsteoMeasure system. The results showed that at the time point 8-weeks after DMM surgery, although there was no significant difference between the Runx2 Agc1CreER KO mice and Cre-negative controls, the tendency of increased articular cartilage area was observed in Runx2 Agc1CreER KO mice (Fig. 2c). At the time point of 12-weeks after DMM surgery, deletion of Runx2 significantly protected DMM-induced cartilage degradation (Fig. 3c). The subchondral sclerosis was observed 8- and 12-weeks after DMM surgery in Cre-negative control mice and this phenotype was also rescued in the Runx2 Agc1CreER KO mice (Figs 2a and 3a). Consistent with this result, the data of μCT analysis showed that subchondral bone mass was increased in Cre-negative control mice 12-weeks after DMM surgery and this effect was rescued in the Runx2 Agc1CreER KO mice (Fig. 4).
Figure 4.
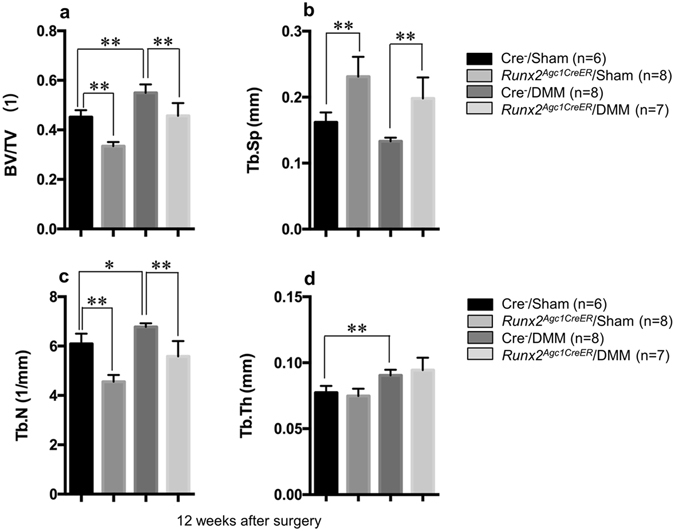
Micro-CT data display evidence of significantly increased bone mass of subchondral bone of knee joint in Cre negative mice with DMM surgery compared with Cre negative mice with sham surgery, and the increased bone mass was significantly reduced in Runx2 Agc1CreER KO mice. All the mice were administrated tamoxifen at 2-months of age, followed by the DMM surgery performed at 3-months of age. Mice were sacrificed 3 months after surgery. (*P < 0.05, **P < 0.01, one-way ANOVA followed by Tukey’s post-hoc test; n = 6–8 mice per group).
The increased MMP13 protein levels by DMM surgery were rescued by Runx2 deletion
IHC results showed that, MMP13 expression was weak in articular cartilage and was restricted mainly in deep zone (below the tidemark) and adjacent to subchondral bone in Sham operated mice (Fig. 5). However, MMP13 expression was increased in articular cartilage in Cre-negative control mice 8- and 12-weeks after DMM surgery (Fig. 5). MMP13 was expressed not only in deep zone but also in middle and superficial zones of articular cartilage in the mice after DMM surgery (Fig. 5). Since superficial and mid zones of cartilage of Cre-negative mice were no longer present in the mice 12 weeks after DMM surgery, the MMP13 expression at this time point were only observed in deep zone of articular cartilage. The increased MMP13 expression was reduced in both time points 8- and 12-weeks after DMM surgery in Runx2 Agc1CreER KO mice (Fig. 5).
Figure 5.
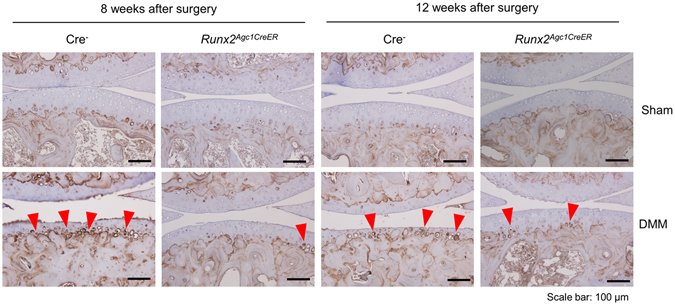
Loss of Runx2 reversed MMP13 up-regulation induced by DMM surgery. DMM surgery and Sham operation was performed in 12-week-old male Cre-negative and Runx2 Agc1CreER KO mice. MMP13 expression was analyzed by IHC assay. Increased MMP13 expression was detected and more MMP13-positive cells were located toward articular surface in Cre-negative control mice 8 and 12-weeks after DMM surgery (MMP13-positive cells: red arrowheads). Deletion of Runx2 significantly down-regulated MMP13 up-regulation induced by DMM (scale bar = 100 μm).
The expression of chondrocyte marker genes was reduced in articular chondrocytes derived from Runx2Agc1CreER KO mice
Primary articular chondrocytes were isolated from 4-day-old Runx2 Agc1CreER mice and control littermates and were treated with 4-hydroxy tamoxifen (1 μM) for 24 hours, followed by real-time PCR assay. The results showed that there was 56% reduction in Runx2 mRNA expression in articular chondrocytes derived from Runx2 Agc1CreER mice (Fig. 6a). Expression of Mmp9 and Mmp13 was decreased by 56 and 59% in Runx2 deficient cells (Fig. 6b and c). Similar to Mmp, expression of Adamts family members was also regulated by Runx212, 13. Expression of Adamts4 and Adamts5 was decreased by 62 and 19%, respectively (Fig. 6e and f), and expression of Adamts7 and Adamts12 was decreased by 23 and 67%, respectively (Fig. 6g and h) in Runx2 deficient chondrocytes. To further determine Runx2 regulation of gene expression in articular cartilage in vivo, we administrated tamoxifen to 2-month-old Runx2 Agc1CreER mice and their Cre-negative littermates and isolated mRNA from articular cartilage of Cre-negative and Runx2 Agc1CreER KO mice. The qRT-PCR data showed the similar gene expression patterns as those observed in the primary articular chondrocytes isolated from 4-day-old Runx2 Agc1CreER mice treated with 4-OH tamoxifen (Fig. 7a–h).
Figure 6.
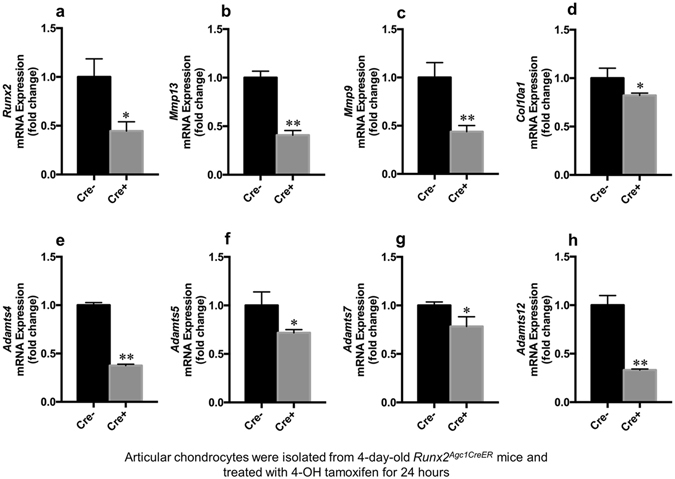
Loss of Runx2 in chondrocytes isolated from early postnatal mice causes decreased expression of genes encoding to matrix degradation enzymes. (a–h) Primary articular chondrocytes were isolated from 4-day-old Runx2 Agc1CreER mice (Cre+) and Cre-negative control mice (Cre−) and treated with 4-hydroxy tamoxifen (1 μM) for 24 hours, followed by real-time PCR assay. Expression of Mmp9, Mmp13, Adamts4, Adamts5, Adamts7 and Adamts12 was significantly reduced in Runx2 deficient chondrocytes (*P < 0.05 and **P < 0.01, Unpaired Student’s t-test; n = 3 mice per group).
Figure 7.
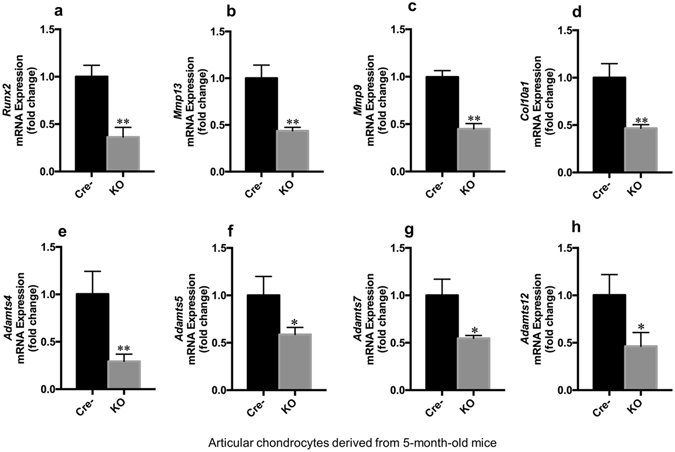
Loss of Runx2 in adult articular chondrocytes leads to decreased expression of genes encoding to matrix degradation enzymes. (a–h) Total RNA was extracted from articular chondrocytes isolated from 5-month-old Runx2 Agc1CreER KO mice (KO) or Cre-negative control mice (Cre−) followed by real-time PCR assay. All these mice were administrated with tamoxifen at the two-months of age and received DMM surgery at 12-week-old. Expression of Mmp9, Mmp13, Adamts4, Adamts5, Adamts7 and Adamts12 was significantly reduced in articular chondrocytes derived from Runx2 Agc1CreER KO mice (*P < 0.05 and **P < 0.01, unpaired Student’s t-test; n = 3 mice per group).
Discussion
OA is the most common degenerative joint disorder and a major cause of disability. To investigate the mechanism of the development of OA, several mouse models mimicking human OA were reported in recent years. Among them the well-accepted OA mouse model is DMM-induced OA model. Compared to anterior cruciate ligament transaction (ACLT) model, the OA severity and location of the lesion in DMM model are similar to the lesions observed in aging-related spontaneous mouse model of OA. In addition, DMM model has sufficient sensitivity to show disease modification3. The major symptom of OA is the progressive cartilage breakdown and eventually completely loss of articular cartilage1, 2, 21. Chondrocytes are the sole cell type in articular cartilage. To better understand the function of specific gene, such as Runx2, in articular cartilage in vivo, it is desirable to delete this gene specifically in joint tissues in adult mice16.
Aggrecan is a major extracellular matrix (ECM) protein in both growth plate and articular cartilage. A previous study15 showed that there was no difference in the localization of Col2a1 and Aggrecan expression in young mice (4–9 weeks old). They express throughout the entire articular cartilage of both medial and lateral tibiae. In contrast to young mice (4–9 weeks old), Col2a1 expression was not detected in older mice (36–50 weeks old). However, there was strong signal for Aggrecan mRNA expression throughout the entire articular cartilage of the medial and lateral tibiae in STR/ort mouse strain. In this study we have used the Agc1-CreER mice to delete Runx2 in articular chondrocytes. The results of frozen sections of Agc1-CreER; ROSA mT/mG mice showed that articular chondrocytes can be efficiently targeted by Agc1-CreER mice. Furthermore, the results of qRT-PCR and IHC staining showed significantly decreased mRNA and protein levels of Runx2 in Runx2 Agc1CreER KO mice compared to Cre-negative mice. These findings suggest that Runx2 could be efficiently deleted in articular cartilage in Runx2 Agc1CreER KO mice.
A recent study showed that Mmp13 and Adamts5 up-regulation may be mediated by Runx2 10. Studies of mutation analysis in the Mmp13 promoter and the ChIP assays further demonstrated that Runx2 directly binds to its blinding site at the Mmp13 promoter in chondrocytes11. Recent study also showed that expression of Adamts7 and Adamts12 could also be regulated by Runx2 in human chondrocytes13.
Runx2 is a critical transcription factor for chondrocyte maturation5 and its role in OA development has not been fully defined. Runx2 expression levels increase in human OA cartilage6–8. Additionally, Runx2 overexpression activated ECM-degrading enzymes (MMP13 and ADAMTS5) through direct interaction with the Runx2 binding sites at the promoters of these genes and through mitogen-activated protein kinase (MAPK) pathway in chondrocytes10. A previous study showed that after induction of knee joint instability, Runx2 global heterozygous KO mice exhibited decreased cartilage destruction and osteophyte formation14. The study using Runx2 heterozygous KO mice did provide important information about the role of Runx2 in OA pathogenesis. However, the Runx2 deficiency in the heterozygous Runx2 global KO mice is not chondrocyte-specific and Runx2 deletion in those mice is not specific at adult stage so the possibilities of embryonic effect (carrying-over to the adult stage) and indirect effect of Runx2 on articular cartilage could not be ruled out in those studies. In the present studies, we have deleted Runx2 in Aggrecan-expressing chondrocytes and investigated the specific effect of Runx2 in articular chondrocytes at the adult stage. Our studies provided additional and critical information about the role of Runx2 in OA development. Since many genetic OA mouse models showed Runx2 up-regulation, suggesting that Runx2 may be a central molecule mediating downstream target gene expression during OA development, it would be very important to clearly define if Runx2 is indeed required in OA development and progression.
A recent study in our laboratory showed that Mmp13 and Adamts5 up-regulation may be mediated by Runx2 11. Studies of mutation analysis in Mmp13 promoter and the ChIP assays further demonstrated that Runx2 directly binds to the OSE2 site at the Mmp13 promoter11, 12. Recent study also showed that expression of Adamts7 and Adamts12 could also be regulated by Runx2 in human chondrocytes13.
To better understand the role of Runx2 in adult mice during the initiation and progression of OA, in this study we generated Runx2 Agc1CreER mice by crossing Runx2 flox/flox mice with Agc1-CreER transgenic mice. Tamoxifen was administered to the mice at 8-week-old when growth plate development is basically completed. DMM surgery was performed in both Runx2 Agc1CreER KO mice and Cre-negative controls at 12-weeks-old. Our results demonstrated that a progressive OA-like phenotype was observed 8- and 12-weeks after DMM surgery in Cre-negative control mice. The evaluation of histology results using the OARSI scoring system33 showed that Runx2 deletion has a protective effect on the DMM-induce OA. Consistently, the histomorphometric analysis of changes in cartilage area at the proximal tibiae showed the similar results. These findings provide evidence showing that Runx2 deletion has a protective role in the DMM-induced OA.
The critical event in OA development is the progressive loss of articular cartilage. Two major structural components of articular cartilage are collagen and aggrecan. Accordingly, enzymes mainly targeting collagen and aggrecan degradation are MMPs and ADAMTS2, 34, 35. A previous study showed that increased expression of Mmp9 and Mmp13 was found in the cartilage during OA development35. In the present studies, the inhibition of Mmp9 and Mmp13 expression was found in Runx2 deficient chondrocytes, suggesting that Runx2 is the upstream regulator of Mmp9 and Mmp13 expression.
MMP13 is the most potent enzyme among collagenases for degradation of type II collagen36, 37. In addition to cleaves type II collagen, it also targets the degradation of aggrecan, collagen types IV and type IX, gelatin, osteonectin, and perlecan in cartilage38. The recent studies demonstrated that Mmp13 may be a downstream target gene of Runx2 11, 12. In the present studies, we observed the significant reduction of Mmp13 expression in primary articular chondrocytes of Runx2 Agc1CreER KO mice. IHC result showed that MMP13 was mainly expressed in the deep zone of articular cartilage, adjacent to subchondral bone and below the tidemark in the Sham operated mice. In mice with DMM surgery, an obvious increase in MMP13 expression was observed in deep zone, as well as in middle and superficial zones of the articular cartilage 8 weeks after DMM surgery. However, superficial and middle zones of the cartilage of Cre-negative mice were no longer present 12 weeks after DMM surgery, so MMP13 expression at this time points were only observed in the deep zone of the articular cartilage. In contrast, the increased MMP13 expression was rescued in Runx2 Agc1CreER KO mice after DMM surgery.
MMP13 is also a maker of the chondrocyte hypertrophy39 and Runx2 could regulate the Mmp13 expression in hypertrophic chondrocytes11, 40. During OA development, articular chondrocytes undergo hypertrophy leading to extracellular matrix degradation and articular cartilage breakdown1, 41. Consistent with these findings, in the present studies, we found that the expanded expression of MMP13 was observed in middle and superficial zones of the cartilage in Cre-negative control mice after DMM surgery, suggesting that chondrocyte hypertrophy is increased in this OA mouse model. Deletion of Runx2 could significantly inhibit MMP13 expression in Runx2 Agc1CreER KO mice, even after DMM surgery. The ADADMTS family consists of large members of aggrecanases and they share several distinct modules. Both Adamts4 and Adamts5 are responsible for aggrecan degradation in a human model of OA42. The previous study showed that Adamts5 up-regulation may be mediated by Runx2 in Tgfbr2 Col2CreER KO mice12. The most recent study demonstrated that Adamts7 and Adamts12 are also regulated by Runx2 13. In the present studies, we observed down-regulation of Adamts 4, 5, 7, 12 mRNA expression in Runx2 deficient chondrocytes derived from new born Runx2 Agc1CreER mice treated with 4-OH TM and in articular cartilage of adult Runx2 Agc1CreER KO mice. Col10a1 is the most specific hypertrophic chondrocyte maker. The previous studies showed that Runx2 directly targets Col10a1 transcription through interaction with the cis-enhancing elements43. Consistent with this finding, we found a significant down-regulation of Col10a1 expression in Runx2 deficient articular chondrocytes.
Runx2 plays a critical role in osteoblast and chondrocyte differentiation in mice and in humans44, 45 and is a key transcription factor for chondrocyte hypertrophy and osteoblast differentiation17. In chondrocyte-specific Runx2 KO mice, DMM-induced cartilage degradation was inhibited. A close relationship and a cross-talk may exist between the articular cartilage and subchondral bone. Moreover, cartilage and subchondral bone are in close proximity and soluble proteins produced in the cartilage are likely to be able to move from one compartment to the other. Perturbing cartilage is expected to preferentially affect subchondral bone46. This may explain why chondrocyte-specific Runx2 KO contributes to the inhibition of subchondral bone sclerosis.
In addition to articular cartilage degeneration, subchondral bone sclerosis is also a characteristic of OA12, 47, 48. Runx2 plays a critical role in osteoblast and chondrocyte differentiation in mice and in humans44, 45 and is a key transcription factor for chondrocyte and osteoblast differentiation17. In chondrocyte-specific Runx2 KO mice, DMM-induced cartilage degradation was inhibited. A close relationship and a cross-talk may exist between the articular cartilage and subchondral bone. Moreover, cartilage and subchondral bone are in close proximity and soluble proteins produced in the cartilage are likely to be able to move from one compartment to the other. Perturbing cartilage is expected to preferentially affect subchondral bone. This may explain why chondrocyte-specific Runx2 KO contributes to the inhibition of subchondral bone sclerosis.
In summary, we performed chondrocyte-specific Runx2 deletion in adult Runx2 KO mice and demonstrated that Runx2 deletion has chondro-protective effect on DMM-induced OA development and progression. Since we have targeted chondrocytes at adult stage, the potential effects of Runx2 deletion during embryonic development and indirect effect of Runx2 on growth plate cartilage development have been ruled out in the current studies. Our studies indicate that deletion of Runx2 in Aggrecan-expressing mature articular chondrocytes prevents DMM-induced OA development. The chondro-protective effect of Runx2 deletion could be due to the inhibition of genes encoding for multiple matrix degradation enzymes. Our studies suggest that Runx2 could serve as a molecular target for drug development for OA treatment.
Methods
DMM-induced OA model
Tamoxifen (Sigma, St. Louis, MO, USA) was administered into 2-month-old Runx2 Agc1CreER mice and Cre-negative littermates by intraperitoneal (i. p.) injection (1 mg/10 g body weight) for 5 consecutive days49. DMM surgery was performed on the right knee of mice to induce knee OA in 12-week-old mice50. Sham operation was performed by opening and exposing the structures of the right knee and then closing the skin incision without manipulating the joint tissue on 3-month-old Runx2 Agc1CreER mice and Cre-negative control mice. Pre- and post-surgery, mice were provided with analgesia (2.5 mg/kg banamine, i. p. injection) every 24 hours for 72 hours and the sutures were removed 10 days after surgery. The right legs were harvested 8 and 12 weeks post-surgery (n = 6–8 in each group), processed, sectioned and stained. The animal protocol of this study has been approved by the IACUC of the Rush University Medical Center and all experimental methods and procedures were carried out in accordance with the approved guidelines.
The generation of Runx2Agc1CreER conditional KO mice
Runx2 flox-neo mice were provided by Dr. Takeshi Takarada17 (Okayama University, Japan). Runx2 floxed mice were generated mice carrying a Conditional Runx2 allele with exon 4, which encodes the Runt domain, flanked by loxP sites. Runx2 flox-neo mice were crossed with FLPe transgenic mice to eliminate the neomycin cassette. To generate Runx2 Agc1CreER conditional KO mice, Runx2 flox/flox mice were crossed with Agc1-CreER transgenic mice. Agc1-CreER transgenic mice were obtained from Jackson laboratories. The resulting Agc1-CreER; Runx2 flox/flox (Runx2 Agc1CreER) mice were administered with tamoxifen (1 mg/10 g body weight/day, i. p. injection, for 5 days) at age two-month-old and were sacrificed at ages 5 or 6 months (8 and 12 weeks post DMM surgery) for histologic analysis. Cre-negative littermates were used as controls.
Cre recombination efficiency
To determine whether Agc1-CreER mice could target articular chondrocytes efficiently in adult mice, Agc1-CreER transgenic mice were bred with ROSA mT/mG reporter mice32 (obtained from Jackson Laboratories). Tamoxifen was administered into 2-month-old mice. Mice were sacrificed at age of 3 months. Histologic sections were analyzed using a fluorescence microscope.
Micro-computed tomography (μCT)
Prior to histologic processing, we evaluated formalin-fixed mouse legs by μCT using a μCT-35 cone-beam scanner (Scanco Medical) with a 55 kVp source and a 145 μAmp current. We scanned the mouse legs at a resolution of 12 μm. Morphometric analysis was performed on 50 slices extending proximally, beginning with the first slice in which the tibia condyles had fully merged. The subchondral bone was segmented from the cortical shell manually on key slices using a contouring tool, and the contours were morphed automatically to segment the trabecular bone on all slices. The morphometry was reconstructed and analyzed.
Histology and immunohistochemistry
Knee joint tissues were fixed in 4% paraformaldehyde for 48 hours, decalcified with 10% formic acid (commercially-available decalcification solution) for ten days, dehydrated with graded ethanol, and embedded in paraffin. Serial mid-sagittal sections (3-μm thick) were cut and stained with Alcian blue/hematoxylin and eosin (AB/H&E) for morphologic analysis51. Histomorphometric measurements were performed with OsteoMeasure software (OsteoMetrics, Inc., Atlanta, GA, USA). AB/H&E-stained areas were outlined on projected images of each histologic section to determine articular cartilage area12. IHC was performed on the 3-μm thick tissue sections and sections were baked at 60 °C overnight. Slides were then deparaffinized, rehydrated, and washed twice in dH2O for 5 minutes each. The antigen retrieval was performed with Antigen Unmasking solution (Vector Laboratories, H-3300) in 95 °C for 10 minutes. Slides were then quenched in 3% hydrogen peroxide for 10 minutes at room temperature. Slides were incubated with 0.5% Triton X-100 (Sigma-Aldrich, 9002-93-1) for 1 hour, and washed with PBS for 3 times and then blocked with Avidin/Biotin Blocking Kit (Invitrogen, 004303). Slides were then washed again with PBS for 3 times and then blocked with the blocking serum at 10% normal goat serum (Vector Laboratories, S-1000) in 1% BSA for 30 minutes at room temperature. Slides were then incubated with primary antibodies against MMP13 (Mouse anti-Human, MAB 13424, 1:100 dilution) or Runx2 (Mouse IgG, MBL, D130-3, 1:200 dilution) at 4 °C overnight. On the second day, secondary biotinylated goat anti-mouse antibody (Vector Laboratories, BA-9200) was added for 30 minutes, followed by incubation with VECTASTAIN Elite ABC HRP Kit (Vector Laboratories, PK-6100) for 30 minutes. Positive staining was detected by ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories, PK-6100). Slides were then counterstained with CAT Hematoxylin (Biocare Medical, CATHE-GL), dehydrated with graded ethanol and cleared with 3 changes of Xylene and then coversliped.
Grading of cartilage structure
Histology sections of knee joint (tibia, sagittal view) were stained with Alcian blue/Orange G and graded by two blinded observers based on the scoring system developed by Glasson et al.33. In brief, each section was assigned a grade 0–6: 0, normal cartilage; 0.5, loss of Safranin O staining without structural changes; 1, small fibrillations without loss of cartilage; 2, vertical clefts down to the layer below the superficial layer; 3–6, vertical clefts or erosion to the calcified cartilage (<25% (grade 3), 25–50% (grade 4), 50–75% (grade 5) and >75% (grade 6) of the articular surface is affected)33. The maximal score was used to represent severity of the OA progression of each mouse.
Cell culture and real-time polymerase chain reaction (PCR) analysis
Primary articular chondrocytes were isolated from articular cartilage of 4-day-old neonatal mice, as described previously52. The isolated cells were treated with 4-OH tamoxifen (1 μM) for 24 hours. Total mRNA was extract with Trizol (Invitrogen Life Technologies, CA, USA). 1 μg total RNA was used to synthesize complementary DNA (cDNA) using an iScripts cDNA Synthesis kit (Quanta Biosciences, MD, USA). Real-time PCR amplification was performed using specific primers of genes encoding for matrix degradation enzymes and a SYBR Green real-time PCR kit (Quanta Biosciences, MD, USA). The primer names and sequences were listed in Table 1. Data were collected from cells of 3 independent mice (n = 3).
Table 1.
The names of sequences of primers used in this project.
| Genes | Primer sequence (forward primers) | Primer sequence (reverse primers) |
|---|---|---|
| Runx2 | GACTGTGGTTACCGTCATGGC | ACTTGGTTTTTCATAACAGCGGA |
| Mmp9 | GCAGAGGCATACTTGTACCG | TGATGTTATGATGGTCCCACTTG |
| Mmp13 | CTTCTTCTTGTTGAGCTGGACTC | CTGTGGAGGTCACTGTAGACT |
| Adamts4 | ATGGCCTCAATCCATCCCAG | GCAAGCAGGGTTGGAATCTTTG |
| Adamts5 | GGAGCGAGGCCATTTACAAC | CGTAGACAAGGTAGCCCACTTT |
| Adamts7 | GCAGGCTTCGTCTGCTTTCTA | GCCATCAGATAAGGGTTGGTGG |
| Adamts12 | GACCCGAGGCAAGAACATTTT | CCCAGTTGACCGTCAGATTGA |
| Col10a1 | TTCTGCTGCTAATGTTCTTGACC | GGGATGAAGTATTGTGTCTTGGG |
| Actin | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
Statistical analysis
Data are presented as the mean ± SD. For experiments comparing two groups of data, unpaired Student t-test was performed. For data that multiple groups are involved, one-way analysis of variance (ANOVA) was performed followed by Turkey’s post-hoc test. P values less than 0.05 were considered significant.
Acknowledgements
We would like to express our gratitude to Ms. Lily Yu for her help on processing and staining histological samples. This work was supported by National Institutes of Health Grants R01 AR054465 and R01 AR070222 to D.C. This work was also partially supported by the Natural Science Foundation of China (grant 81371999) to D.C. This study was also partially sponsored by grants from Shenzhen Science and Technology Innovation Committee (JCYJ20160331114205502 and JCYJ20150626090344603), China to D.C. and J.L.
Author Contributions
Di Chen contributed to the experimental design and data interpretation. Lifan Liao, Shanxing Zhang, Jianhong Gu, Jian Huang, Lan Zhao, Chundo Oh, Jun Li and Baoli Wang carried out all experiments. Takeshi Takarada and Yukio Yoneda contributed to this work by providing Runx2 flox/flox mice and advising on how to breed and genotype the mice. Lifan Liao, Meiqing Wang and Di Chen contributed to the manuscript preparation and revision.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Meiqing Wang, Email: mqwang@fmmu.edu.cn.
Di Chen, Email: di_chen@rush.edu.
References
- 1.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–34. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 2.Wang M, et al. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann N Y Acad Sci. 2011;1240:61–9. doi: 10.1111/j.1749-6632.2011.06258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061–9. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Chen CG, Thuillier D, Chin EN, Alliston T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum. 2012;64:3278–89. doi: 10.1002/art.34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida CA, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18:952–63. doi: 10.1101/gad.1174704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, et al. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. 2004;12:963–73. doi: 10.1016/j.joca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Zhong, L., Huang, X., Karperien, M. & Post, J.N. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int J Mol Sci17 (2016). [DOI] [PMC free article] [PubMed]
- 8.Hasegawa A, et al. Cellular and extracellular matrix changes in anterior cruciate ligaments during human knee aging and osteoarthritis. Arthritis Res Ther. 2013;15:R29. doi: 10.1186/ar4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadri A, et al. Inhibition of bone resorption blunts osteoarthritis in mice with high bone remodelling. Ann Rheum Dis. 2010;69:1533–8. doi: 10.1136/ard.2009.124586. [DOI] [PubMed] [Google Scholar]
- 10.Tetsunaga T, et al. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthritis Cartilage. 2011;19:222–32. doi: 10.1016/j.joca.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Wang M, et al. Conditional activation of β-catenin signaling in mice leads to severe defects in intervertebral disc tissue. Arthritis Rheum. 2012;64:2611–23. doi: 10.1002/art.34469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen J, et al. Deletion of the transforming growth factor beta receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013;65:3107–19. doi: 10.1002/art.38122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji Q, et al. miR-105/Runx2 axis mediates FGF2-induced ADAMTS expression in osteoarthritis cartilage. J Mol Med (Berl) 2016;94:681–94. doi: 10.1007/s00109-016-1380-9. [DOI] [PubMed] [Google Scholar]
- 14.Kamekura S, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54:2462–70. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- 15.Chambers MG, Kuffner T, Cowan SK, Cheah KS, Mason RM. Expression of collagen and aggrecan genes in normal and osteoarthritic murine knee joints. Osteoarthritis Cartilage. 2002;10:51–61. doi: 10.1053/joca.2001.0481. [DOI] [PubMed] [Google Scholar]
- 16.Henry SP, et al. Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis. 2009;47:805–14. doi: 10.1002/dvg.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takarada T, et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J Bone Miner Res. 2013;28:2064–9. doi: 10.1002/jbmr.1945. [DOI] [PubMed] [Google Scholar]
- 18.Bau B, et al. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002;46:2648–57. doi: 10.1002/art.10531. [DOI] [PubMed] [Google Scholar]
- 19.Neuhold LA, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roach HI, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52:3110–24. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15:R5. doi: 10.1186/ar4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–69. doi: 10.1128/MCB.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–54. doi: 10.1016/S0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 24.Jimenez MJ, et al. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol. 1999;19:4431–42. doi: 10.1128/MCB.19.6.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winchester SK, Selvamurugan N, D’Alonzo RC, Partridge NC. Developmental regulation of collagenase-3 mRNA in normal, differentiating osteoblasts through the activator protein-1 and the runt domain binding sites. J Biol Chem. 2000;275:23310–8. doi: 10.1074/jbc.M003004200. [DOI] [PubMed] [Google Scholar]
- 26.Hess J, Porte D, Munz C, Angel P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J Biol Chem. 2001;276:20029–38. doi: 10.1074/jbc.M010601200. [DOI] [PubMed] [Google Scholar]
- 27.Mengshol JA, Vincenti MP, Brinckerhoff CE. IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 2001;29:4361–72. doi: 10.1093/nar/29.21.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porte D, et al. Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene. 1999;18:667–78. doi: 10.1038/sj.onc.1202333. [DOI] [PubMed] [Google Scholar]
- 29.D’Alonzo RC, Selvamurugan N, Karsenty G, Partridge NC. Physical interaction of the activator protein-1 factors c-Fos and c-Jun with Cbfa1 for collagenase-3 promoter activation. J Biol Chem. 2002;277:816–22. doi: 10.1074/jbc.M107082200. [DOI] [PubMed] [Google Scholar]
- 30.Jimenez MJ, et al. A regulatory cascade involving retinoic acid, Cbfa1, and matrix metalloproteinases is coupled to the development of a process of perichondrial invasion and osteogenic differentiation during bone formation. J Cell Biol. 2001;155:1333–44. doi: 10.1083/jcb.200106147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimura R, et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J Biol Chem. 2012;287:33179–90. doi: 10.1074/jbc.M111.337063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 33.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18(Suppl 3):S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 34.Davidson RK, et al. Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res Ther. 2006;8:R124. doi: 10.1186/ar2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kevorkian L, et al. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004;50:131–41. doi: 10.1002/art.11433. [DOI] [PubMed] [Google Scholar]
- 36.Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271:1544–50. doi: 10.1074/jbc.271.3.1544. [DOI] [PubMed] [Google Scholar]
- 37.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J Clin Invest. 1996;97:2011–9. doi: 10.1172/JCI118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiomi T, Lemaitre V, D’Armiento J, Okada Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol Int. 2010;60:477–96. doi: 10.1111/j.1440-1827.2010.02547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci USA. 2014;111:12097–102. doi: 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirata M, et al. C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum Mol Genet. 2012;21:1111–23. doi: 10.1093/hmg/ddr540. [DOI] [PubMed] [Google Scholar]
- 41.Felson DT. Clinical practice. Osteoarthritis of the knee. N Engl J Med. 2006;354:841–8. doi: 10.1056/NEJMcp051726. [DOI] [PubMed] [Google Scholar]
- 42.Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. J Cell Biochem. 2011;112:3507–14. doi: 10.1002/jcb.23298. [DOI] [PubMed] [Google Scholar]
- 43.Li F, et al. Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J Bone Miner Res. 2011;26:2899–910. doi: 10.1002/jbmr.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee B, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–10. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 45.Mundlos S, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–9. doi: 10.1016/S0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 46.Findlay DM, Kuliwaba JS. Bone-cartilage crosstalk: a conversation for understanding osteoarthritis. Bone Res. 2016;4:16028. doi: 10.1038/boneres.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suri S, Walsh DA. Osteochondral alterations in osteoarthritis. Bone. 2012;51:204–11. doi: 10.1016/j.bone.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 48.Zhen G, et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19:704–12. doi: 10.1038/nm.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M, Li S, Xie W, Wang B, Chen D. Col2CreER(T2), a mouse model for a chondrocyte-specific and inducible gene deletion. Eur Cell Mater. 2014;28:236–45. doi: 10.22203/eCM.v028a16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sampson ER, et al. Establishment of an index with increased sensitivity for assessing murine arthritis. J Orthop Res. 2011;29:1145–51. doi: 10.1002/jor.21368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shu B, et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci. 2011;124:3428–40. doi: 10.1242/jcs.083659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3:1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]