

Abstract
Connexin (Cx) 36 is known to be a component of gap junctions, and has been suggested to play an important role in epilepsy. In order to determine dynamic changes of Cx36 protein expression in epilepsy and investigate the role of Cx36 in electroencephalographic activity and pathogenesis, we utilized kainic acid (KA) to induce epileptogenesis. We found that epileptic discharges began 71.8 ± 23.7 s after KA administration. Spike frequency and amplitude of epileptiform activity reached maximal levels at 30 ± 5.2 min. The maximum level of spike frequency and amplitude of epileptiform activity was 13.9 ± 0.3 Hz and 198 ± 14.3mV respectively. Employing Western blotting and immunohistochemistry, we demonstrated that hippocampal Cx36 protein expression was significantly increased 6 h after KA kindling compared to control or sham groups, but decreased in 3 d and 7d groups. Our results suggested that the dynamic change of Cx36 expression may play an important role inepilepsy, and the specific manipulation of Cx36 expression may be a potential target for the treatment of epilepsy.
Keywords: epilepsy, gap junction, connexin 36, in vivo
1. Introduction
Epilepsy is one of the most prevalent neurological disorders worldwide, with an increasing incidence annually [1], affecting 1% to 3% of the population worldwide [2]. It has been reported that gap junction channels play an important role in epileptogenesis and epilepsy propagation [3]. The gap junction channel consists of two hemichannels, composed of six connexin (Cx) proteins [4]. Cx36 was identified as the first Cx protein expressed predominantly in mammalian neurons, and is highly expressed in the inferior olivary complex and hippocampus, which are often areas exhibiting activity conducive of epileptic seizures [5-7].
Indeed, hippocampal Cx36 expression has been reported to be decreased two to three weeks after kainic acid (KA) treatment [8], while another study demonstrated that Cx36 expression is increased after partial kindling, but decreased after full kindling in epilepsy rat models [9]. However, using a 4-AP in vivo epilepsy model revealed that the expression of Cx36 protein was increased one hour after the onset of the first seizure [10, 11]. Taken together, these results imply that Cx36 may play an important role in epilepsy. However, the expression of Cx36 is controversial, and any potential role is yet to be fully elucidated.
The mechanisms of seizures in the rat model of epilepsy induced by kainic acid (KA) injection in the amygdala are well characterized [12-14]. The development of convulsive seizures is synchronous with the electrical activity of EEG monitoring [15]. This spontaneous convulsion can be repeated, which can simulate the occurrence, transmission and maintenance of epilepsy seen in humans [16]. Therefore, the KA-induced epilepsy animal model is considered a classic animal model of epilepsy, which has been widely used in study of the pathogenesis of epilepsy and antiepileptic drugs [13, 14].
To the best of our knowledge, our study is the first analysis to elucidate the dynamic expression of Cx36 protein in KA kindled epilepsy rats. We examined Cx36 expression, electroencephalographic activities and pathogenesis at different epileptic periods including 3 h, 6 h, 24 h, 3 d and 7 d after seizure onset to clarify the function of Cx36 in epilepsy.
2. Materials and methods
2.1. Materials
Adult male Wistar rats (200-250g, Experimental Animal Center of Bethune Medical Department) were used in this study, which had free access to food, water and housing at 25 ± 1 °C. Experiments were performed and approved by First Hospital of Jilin University and in accordance with the Chinese law for animal protection.
Rats were divided into three groups: control group (n=18), sham group (n=18) and KA group (n=90). KA group was subsequently divided into five subgroups: KA 3 h group (n=18), KA 6 h group (n=18), KA 24 h group (n=18), KA 3 d group (n=18), and KA 7 d group (n=18).
Kainic acid monohydrate was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in phosphate-buffered saline (PBS). 10% chloral hydrate was obtained from the First Hospital of Jilin University, and was injected intraperitoneally (i.p.) at a concentration of 3.5 mL/kg. Polyclonal anti-connexin 36 and polyclonal anti-MAP-2 antibodies were purchased from Santa Cruz (Dallas, TX, USA).
2.2. Surgery
The animals were anesthetized with 10% chloral hydrate (3.5 mL/kg) and fixed in a stereotaxic frame with the incisor bar positioned at -3.3 mm. The position of bilateral nuclear in right nucleus amygdala was 3.5 mm posterior to Bregma, 4.5 mm lateral to the midline and 8.0 mm below the surface of the skull. One µL KA (1 mg/mL) was injected into the nucleus amygdala with a 5 µL syringe (infusion rate 1 mL/min). For the sham group, 1 µL PBS was injected in the same way. According to the time course after KA injection, KA group was subsequently divided into KA 3 h group, KA 6 h group, KA 24 h group, KA 3 d group, and KA 7 d group.
2.3. EEG recording
One surface Ag-AgCl non-polarizable electrode was fixed on the parietal cortex (parietal cortex: AP = -4.0 mm, L = 3.0 mm and V = 1.5mm from the Bregma) by dental cement. In addition, ground electrodes were fixed on the tip of nose and the neck muscle respectively with the same characteristics as the electrodes mentioned above. The electrode wires were attached to a socket connector. Animals were connected to PowerLab (AD Instruments, New South Wales, Australia), a computerized EEG recording system. The EEG was recorded for 1 h using a power filter with a low-frequency filter at 30 Hz and a high frequency filter at 1 s.
2.4. Tissue preparation
Rats were decapitated under deep anaesthesia, and then the brains were immediately removed. The hippocampus was collected and frozen in liquid nitrogen stored at -80 °C. Other brains were collected in 10% formalin prior to paraffin embedding and cut into 10 pm slices on a microtome and mounted on glass slides prepared for histopathological and immunohistochemical analysis. The slices were analyzed by microscopy (Olympus, Osaka, Japan).
2.5. Western blotting
Hippocampus tissue lysates were prepared in protein extraction buffer (150 mM NaCl, 0.25% sodium deoxycholate, 0.25% sodium dodecylsulfate, 1 mM ethylenedia-minetetraacetic acid, 50 mM Tris-HCl, pH 7.4). Lysates were then centrifuged for 2 min at 12,000 rpm at 4 °C. Protein concentration in each sample was determined by the Bradford method using Bio-Rad DC protein assay reagents. Equal amounts of proteins (5 µg per lane for β-actin, 10 µg per lane for Cx36) were resolved by 12% SDS-polyacrylamide gels and transferred onto a polyvinylidene difluoride membrane. The membrane was blocked with 3% BSA in PBS for 1 hour at room temperature, and then incubated overnight at 4 °C with primary antibodies (rabbit polyclonal anti-cx 36 1:200, and rabbit monoclonal anti-β-actin 1:2000, Santa Cruz Biotechnology). After washing with PBS-0.05% Tween-20 three times for 10 min, the membrane was incubated with anti-rabbit secondary antibody conjugated with peroxidase (1:2000 for both Cx36 and β-actin; Santa Cruz Biotechnology) for 1 h at room temperature, followed with washing three times with PBS-0.05% Tween-20. The protein level was detected with enhanced chemoluminescence. The antibody binding was exposed to an X-ray film (Fujifilm, Tokyo, Japan). The band density was quantified using Image-Pro Plus (v. 6.0, Media Cybernetics, Silver Spring, MD, USA).
2.6. Immunohistochemistry
Paraffin-embedded sections were placed at 60 °C for 15 min, incubated in xylene at room temperature for 15 min, and then transferred sequentially into 100% EtOH, 95% EtOH, 70% EtOH, and 50% EtOH for 4 min at room temperature. Sections were rinsed in deionized water and stored in PBS. Co-localization of Cx36 with MAP-2 in hippocampus was examined by double-labelled immunofluorescence with the rabbit polyclonal antibody to rat Cx36 (1:100, Santa Cruz, USA) and the mouse polyclonal antibody against rat MAP-2 (1:100, Santa Cruz, USA). The blocking solution used was 10% normal goat serum in PBS and antibodies were diluted in 1% goat serum. The sections were incubated for 60 min with the Cx36 antibody, and the affixed antibody was detected with FITC goat anti-rabbit IgG (1:400, Santa Cruz, USA). After washing, MAP-2 antibody was incubated and detected with TRITC labelled goat anti-mouse IgG (1:400, Santa Cruz, USA). Coverslips were mounted with mounting medium containing 4,6-diamidino-2-phenylindole (DAPI) (VWR International Aps, Denmark) to identify nuclei. Co-expression was visualized using confocal microscopy (Olympus, Osaka, Japan, FV-1000).
2.7. Statistical analysis
All statistical procedures were performed using SPSS (14.0) statistical software package (Armonk, NY, USA). The data were analyzed by one-way ANOVA and were expressed as mean ± SD. The level of statistical significance was set at p<0.05.
3. Results
3.1. Electroencephalographic activities showed an epileptiform activity in kainic acid induction
Kainic acid (KA) induced an epileptiform EEG activity characterized by biphasic (positive and negative) spikes and spike-wave complexes. The normal electrical activity of EEG recording is shown in Figure 1A from the control group before KA administration, similar to the sham group with saline injection (Figure 1B). EEG recording showed spikes and spike-wave complexes with high frequency and amplitude. Epileptic discharges began at 71.8 ± 23.7 s after KA administration. The time reached the maximum level of spike frequency, and amplitude of epileptiform activity was reached at the 3h time point. Maximum levels of spike frequency and amplitude of epileptiform activity was 13.9 ± 0.3 Hz and 198 ±14.3 mV respectively (Figure 1).
Figure 1.
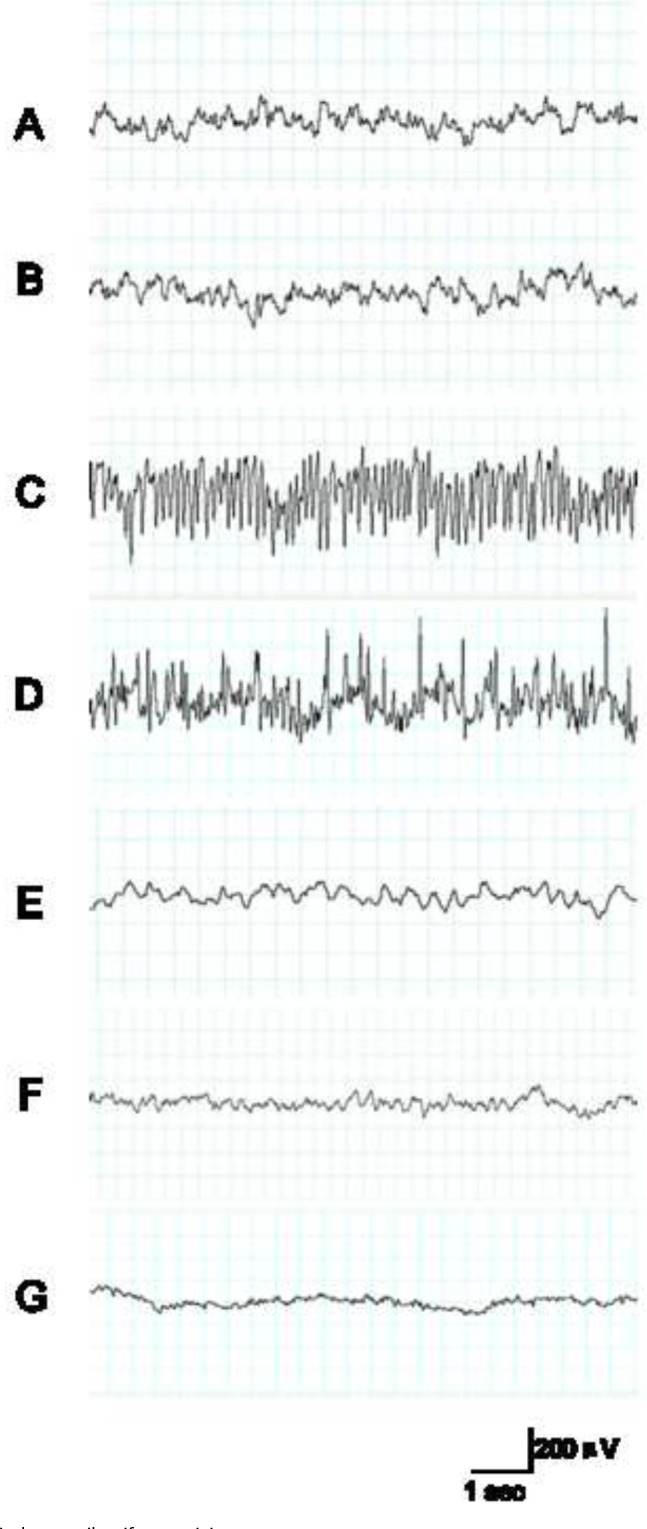
KA induces epileptiform activity.
A: EEG recording shows the electrical activity of the control group before KA administration. B: EEG recording shows the electrical activity of the sham group with saline administration. C: 3 h after administration of KA, EEG recording shows the spikes and spike-wave complexes with high frequency and amplitude in the ictal period; D: 6 h after administration of KA, EEG recording also shows the spikes were during the ictal period. E: 24 h after administration of KA, EEG recording showed spikes but spike-wave complexes were rare. F: 3 d after administration of KA, EEG recording showed no spikes and spike-wave complexes; G: 7 d after administration of KA, EEG recording also showed no spikes.
3.2. Dynamic expression of connexin protein 36 during epileptic period
In order to clarify the dynamic expression of Cx36 during epilepsy, we detected protein expression at different time points including 3 h, 6 h, 24 h, 3 d and 7 d after KA kindling induced epileptic period using Western blotting and immunohistochemistry. We found that hippocampal Cx36 protein expression was significantly increased 6 h after KA kindling compared to the control or sham group using Western blot. However, hippocampal Cx36 protein expression was decreased at 3 d and 7 d after KA kindling compared to control or sham groups (Figure 2, Table 1). To further detect the subcellular expression and confirm our result from the Western blotting experiments, we additionally examined Cx36 protein expression by using immunohistochemistry. We employed MAP-2 as a neuronal specific marker and examined co-expression with Cx36 to determine the subcellular distribution of Cx36 protein. We used DAPI in order to identify nuclei and found that immunofluorescence intensity of Cx36 protein co-localized with MAP-2 in the hippocampus, and was significantly increased at 6 h (Figure 3D) after KA kindling compared to control (Figure 3A) or sham (Figure 3B) groups. However, the expression of Cx36 protein levels in the hippocampus was significantly decreased at 3 d (Figure 3F) and 7 d. (Figure 3G) compared to the sham group (Figure 3H, Table 2).
Figure 2.
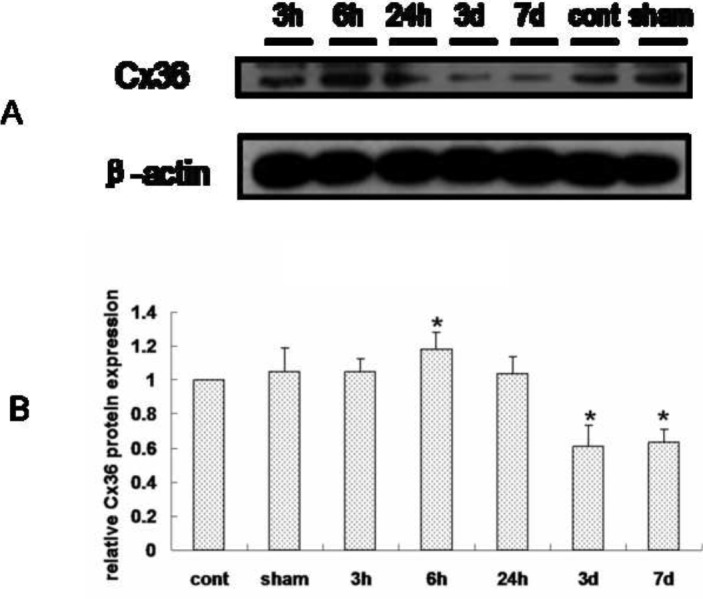
The dynamic expression of Cx36 protein in the hippocampus in KA induced seizures examined by Western blotting.
A: The β-actin protein was used as an endogenous control. The relative Cx36 protein expression was significantly increased in hippocampus 6 h after kindling. B: Quantified analysis of Western blotting data. Data are showed in mean ± sd. (N=6, *: p<0.05.)
Table 1.
The mean ± sd value and p value of quantified data from Western blotting.
| Group | Mean ± sd | P value |
|---|---|---|
| Control | 1 | 1 |
| Sham | 1.049 ± 0.142 | 0.543 |
| 3 h | 1.052 ± 0.072 | 0.249 |
| 6 h | 1.184 ± 0.097 | 0.032* |
| 24 h | 1.04 ± 0.099 | 0.476 |
| 3 d | 0.612 ± 0.124 | 0.008* |
| 7 d | 0.633 ± 0.074 | 0.002* |
(N=6, * : p<0.05.)
Figure 3.
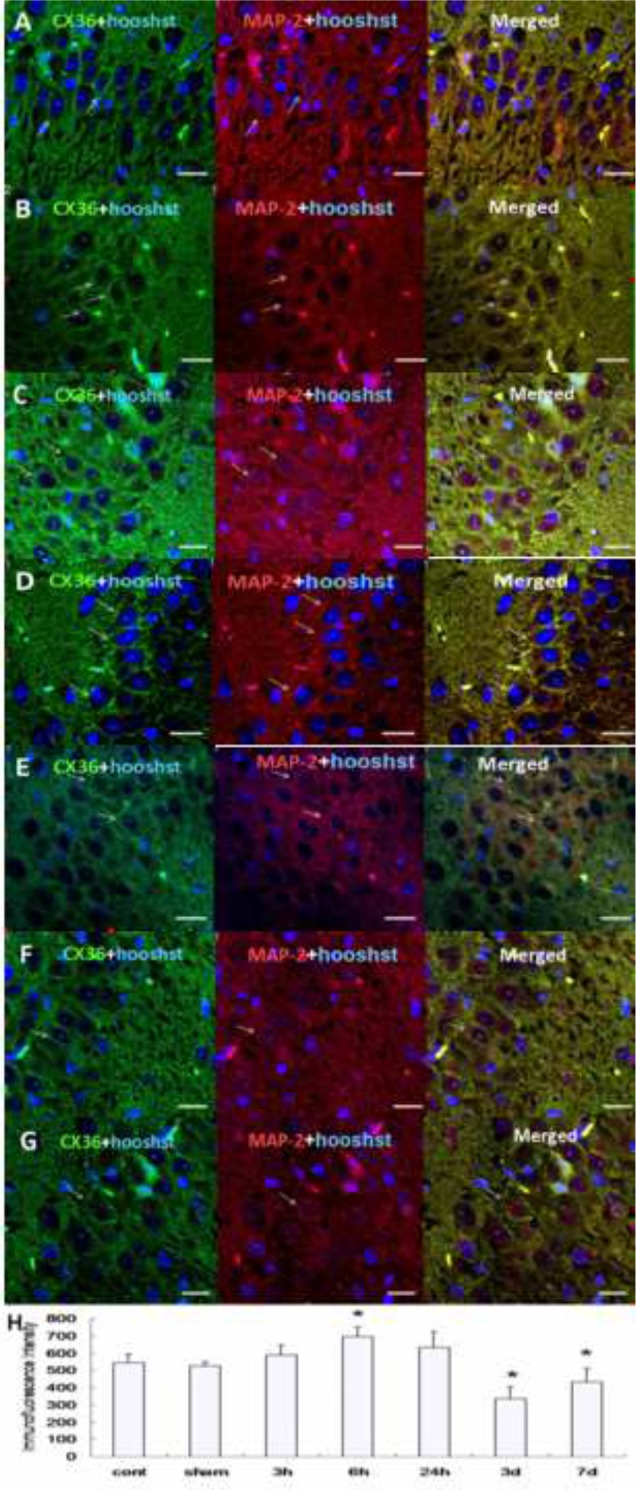
The dynamic expression of Cx36 protein in the hippocampus in KA induced seizures examined by immunohistochemistry double staining with Cx36 and MAP-2.
A: The IHC staining of Cx36 and MAP-2 in the control group. B: The IHC staining of Cx36 and MAP-2 in the sham group. C: The IHC staining of Cx36 and MAP-2 showed that the Cx36 positive expression was increased 3 h after KA induction. D: The IHC staining of Cx36 and MAP-2 for 6 h group. E: The IHC staining of Cx36 and MAP-2 for 24 h group. F: The IHC staining of Cx36 and MAP-2 for 3 d group. G: The IHC staining of Cx36 and MAP-2 for 7 d group. H: Quantified analysis of A-G. Images were recorded by a 40x lens. Scale bars: 25 µm. Blue staining is DAPI positive showing cell nucleus. Cx36 positive expression is shown in green.
Table 2.
The mean ± sd value and p value of quantified data from immunohistochemistry.
| Group | Mean ± sd | P value |
|---|---|---|
| Control | 545.21 ±49.88 | 1 |
| Sham | 526.67 ± 23.70 | 0.549 |
| 3 h | 589.79 ± 58.92 | 0.572 |
| 6 h | 696.96 ± 55.94 | 0.001* |
| 24 h | 632.65 ± 91.53 | 0.183* |
| 3 d | 336.23 ±69.65 | 0.001 |
| 7 d | 433.51 ±81.27 | 0.038* |
(N=6, * : p<0.05.)
4. Discussion
It has been previously demonstrated that Cx36 is the first connexin protein located in neurons, more specifically, in intermediate neurons without expression in astrocytes and oligodendrocytes [5, 17]. Some studies have suggested that given Cx36 is the only preferentially expressed protein in the nervous system, which may be implicated in epilepsy [9]. In this study, we employed MAP-2 as a neuronal specific marker to determine co-expression with Cx36.
EEG recording is an important diagnostic tool in epilepsy. Spikes and sharp waves are the most important diagnostic indicators of seizures in EEG readings. We found that when seizures began, the number of abnormal discharging neurons, spikes, sharp waves, as well as other abnormal waves gradually increased, consistent with EEG recordings in other reports using kindled rats [18]. The fundamental pathogenic mechanisms of epilepsy involve the abnormal discharge of neurons [19]. Therefore, the EEG recording is the optimal method to study neuronal bioelectrical activity [20]. During ictal and interictal periods, there are over-synchronized discharges in the cerebral cortex [21]. Furthermore, it has been shown that EEG recordings demonstrate that kainic acid-induced epilepsy is a useful model for epilepsy research [22].
From our findings utilizing Western blotting and immunohistochemistry, we found that Cx36 protein expression in the hippocampus was increased 3 h after KA administration and reached a peak at 6 h, then gradually decreased and returned to baseline from 3 d to 7 d. However, the level of Cx36 protein during the epileptic period remains controversial based on previous findings [8-11]. The expression pattern of Cx36 is poorly understood to date. Some studies have shown that Cx36 protein expression does not change in epileptic patients who undergo amygdalo-hippocampectomy for the treatment of intractable seizures [23]. Studies have also shown that Cx36 expression levels was decreased 3 h after a single brief electrically evoked epileptic seizure and 72 h after bilateral intracerebroventricular injections of kainic acid [24, 25]. Here we analyzed five time points during the epileptic period to study the dynamic expression of Cx36 in the hippocampus after KA induction. We found that Cx36 protein expression was decreased after an initial increase at the first 6 h time window, a different time course to previous studies, which may explain the aforementioned conflicting findings. The differences between the above studies may result from the time at which samples were collected and tested.
Gap junctions are dynamic structures that can be modulated by increased neuronal activity and a number of both intracellular and extracellular factors. Intercellular communication at gap junctions can be affected by the number, distribution, change in structure and internalization of gap junctions [26]. Connexin may enhance electrical conductivity, possibly by increasing the number of electrical synapses, which promote synchronous firing of neurons in the generation of seizures [27]. Synchronous electrical activity of neurons may be promoted by the increasing number of electrical synapses and coupling, which may have important significance in the generation and propagation of epilepsy.
Down-regulation of Cx36 protein may be induced by the phosphorylation of Cx36 [28], which has been demonstrated to be involved in the regulation of transmission, assembly, disintegration, degradation, opening and closing of gap junction channels [29]. However, there are no commercially available antibodies specific for phosphorylated Cx36, preventing further study.
5. Conclusions
Gap junctions play important roles in epileptogenesis. Modification of Cx36-associated channels may alter the effects and propagation of epilepsy. Therefore, more studies examining the role of gap junction functions are warranted. The development of new drugs to inhibit gap junctions may provide novel treatment targets for epilepsy. Our preliminary, yet novel findings suggest a possible role of Cx36 channel inhibitors as a potential anti-epileptogenic agent, which require further investigation.
Acknowledgement
This work was founded by the National Natural Science Foundation of China(No. 81401068)
Footnotes
Conflict of interest The authors declare no competing financial or personal interests.
Ethical statement The experimental protocol was established, according to the ethical guidelines of the Helsinki Declaration and was approved by the Human Ethics Committee of Department of Neurology, First Hospital of Jilin University, Changchun 130021, China. Written informed consent was obtained from individual participants.
References
- [1].Beghi E, Hesdorffer D. Prevalence of epilepsy—An unknown quantity. Epilepsia. 2014;55:963–967. doi: 10.1111/epi.12579. [DOI] [PubMed] [Google Scholar]
- [2].Mac TL, Tran DS, Quet F, Odermatt P, Preux PM, Tan CT. Epidemiology, aetiology, and clinical management of epilepsy in Asia: a systematic review. The Lancet Neurology. 2007;6:533–543. doi: 10.1016/S1474-4422(07)70127-8. [DOI] [PubMed] [Google Scholar]
- [3].Connors BW. Tales of a dirty drug: carbenoxolone, gap junctions, and seizures. Epilepsy currents / American Epilepsy Society. 2012;12:66–68. doi: 10.5698/1535-7511-12.2.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Söhl G, Willecke K. Gap junctions and the connexin protein family. Cardiovascular Research. 2004;62:228–232. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- [5].Condorelli DF, Parenti R, Spinella F, Salinaro AT, Belluardo N, Cardile V. et al. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. European Journal of Neuroscience. 1998;10:1202–1208. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- [6].Stafstrom CE. Epileptogenesis Beyond the Hippocampus. Epilepsy currents / American Epilepsy Society. 2003;3:66–67. doi: 10.1046/j.1535-7597.2003.03212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Passouant P, Cadilhac J. The hippocampus in epilepsy. The relation between hippocampus and reticular formation, World Neurology. 1960;1:50–59. [PubMed] [Google Scholar]
- [8].Söhl G, Güldenagel M, Beck H, Teubner B, Traub O, Gutiérrez R. et al. Expression of connexin genes in hippocampus of kainate-treated and kindled rats under conditions of experimental epilepsy. Molecular Brain Research. 2000;83:44–51. doi: 10.1016/s0169-328x(00)00195-9. [DOI] [PubMed] [Google Scholar]
- [9].Beheshti S, Sayyah M, Golkar M, Sepehri H, Babaie J, Vaziri B. Changes in hippocampal connexin 36 mRNA and protein levels during epileptogenesis in the kindling model of epilepsy. Progress in Neuro Psychopharmacology and Biological Psychiatry. 2010;34:510–515. doi: 10.1016/j.pnpbp.2010.02.006. [DOI] [PubMed] [Google Scholar]
- [10].Gajda Z, Gyengesi E, Hermesz E, Ali KS, Szente M. Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia. 2003;44:1596–1600. doi: 10.1111/j.0013-9580.2003.25803.x. [DOI] [PubMed] [Google Scholar]
- [11].Gajda Z, Hermesz E, Gyengesi E, Szupera Z, Szente M. The functional significance of gap junction channels in the epileptogenicity and seizure susceptibility of juvenile rats. Epilepsia. 2006;47:1009–1022. doi: 10.1111/j.1528-1167.2006.00573.x. [DOI] [PubMed] [Google Scholar]
- [12].Brandt C, Ebert U, Löscher W. Epilepsy induced by extended amygdala kindling in rats: lack of clear association between development of spontaneous seizures and neuronal damage. Epilepsy Res. 2004;62:135–156. doi: 10.1016/j.eplepsyres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- [13].Diviney M, Reynolds JP, Henshall DC. Comparison of short-term effects of midazolam and lorazepam in the intra-amygdala kainic acid model of status epilepticus in mice. Epilepsy Behav. 2015;51:191–198. doi: 10.1016/j.yebeh.2015.07.038. [DOI] [PubMed] [Google Scholar]
- [14].Michalak Z, Sano T, Engel T, Miller-Delaney SF, Lerner-Natoli M, Henshall DC. Spatio-temporally restricted blood-brain barrier disruption after intra-amygdala kainic acid-induced status epilepticus in mice. Epilepsy Res. 2013;103:167–179. doi: 10.1016/j.eplepsyres.2012.10.006. [DOI] [PubMed] [Google Scholar]
- [15].Myers MH, Threatt M, Solies KM, McFerrin BM, Hopf LB, Birdwell JD. et al. Ambulatory Seizure Monitoring: From Concept to Prototype Device. Annals of Neurosciences. 2016;23:100–111. doi: 10.1159/000443567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gigout S, Deisz R, Dehnicke C, Turak B, Devaux B, Pumain R. et al. Role of gap junctions on synchronization in human neocortical networks. Brain Res. 2016;1637:14–21. doi: 10.1016/j.brainres.2016.02.005. [DOI] [PubMed] [Google Scholar]
- [17].Belluardo N, Trovato-Salinaro A, Mudo G, Hurd YL, Condorelli DF. Structure, chromosomal localization, and brain expression of human Cx36 gene. Journal of neuroscience research. 1999;57:740–752. [PubMed] [Google Scholar]
- [18].Gajda Z, Szupera Z, Blazso G, Szente M. Quinine, a blocker of neuronal cx36 channels, suppresses seizure activity in rat neocortex in vivo. Epilepsia. 2005;46:1581–1591. doi: 10.1111/j.1528-1167.2005.00254.x. [DOI] [PubMed] [Google Scholar]
- [19].Tankus A. Exploring human epileptic activity at the single-neuron level. Epilepsy & behavior : E&B. 2016;58:11–17. doi: 10.1016/j.yebeh.2016.02.014. [DOI] [PubMed] [Google Scholar]
- [20].Tan G, Thornby J, Hammond DC, Strehl U, Canady B, Arnemann K. et al. Meta-analysis of EEG biofeedback in treating epilepsy. Clinical EEG and neuroscience. 2009;40:173–179. doi: 10.1177/155005940904000310. [DOI] [PubMed] [Google Scholar]
- [21].Umeoka SC, Luders HO, Turnbull JP, Koubeissi MZ, Maciunas RJ. Requirement of longitudinal synchrony of epileptiform discharges in the hippocampus for seizure generation: a pilot study. Journal of neurosurgery. 2012;116:513–524. doi: 10.3171/2011.10.JNS11261. [DOI] [PubMed] [Google Scholar]
- [22].Van Nieuwenhuyse B, Raedt R, Sprengers M, Dauwe I, Gadeyne S, Carrette E. et al. The systemic kainic acid rat model of temporal lobe epilepsy: Long-term EEG monitoring. Brain research. 2015;1627:1–11. doi: 10.1016/j.brainres.2015.08.016. [DOI] [PubMed] [Google Scholar]
- [23].Collignon F, Wetjen NM, Cohen-Gadol AA, Cascino GD, Parisi J, Meyer FB. et al. Altered expression of connexin subtypes in mesial temporal lobe epilepsy in humans. Journal of neurosurgery. 2006;105:77–87. doi: 10.3171/jns.2006.105.1.77. [DOI] [PubMed] [Google Scholar]
- [24].McCracken CB, Roberts DC. A single evoked afterdischarge produces rapid time-dependent changes in connexin36 protein expression in adult rat dorsal hippocampus. Neuroscience letters. 2006;405:84–88. doi: 10.1016/j.neulet.2006.06.025. [DOI] [PubMed] [Google Scholar]
- [25].Condorelli DF, Trovato-Salinaro A, Mudo G, Mirone MB, Belluardo N. Cellular expression of connexins in the rat brain: neuronal localization, effects of kainate-induced seizures and expression in apoptotic neuronal cells. The European journal of neuroscience. 2003;18:1807–1827. doi: 10.1046/j.1460-9568.2003.02910.x. [DOI] [PubMed] [Google Scholar]
- [26].Mylvaganam S, Ramani M, Krawczyk M, Carlen PL. Roles of gap junctions, connexins, and pannexins in epilepsy. Frontiers in physiology. 2014;5:172. doi: 10.3389/fphys.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Haupt C, Tolner EA, Heinemann U, Witte OW, Frahm C. The combined use of non-radioactive in situ hybridization and real-time RT-PCR to assess gene expression in cryosections. Brain research. 2006;1118:232–238. doi: 10.1016/j.brainres.2006.08.037. [DOI] [PubMed] [Google Scholar]
- [28].Ivanova E, Yee CW, Sagdullaev BT. Increased phosphorylation of Cx36 gap junctions in the AII amacrine cells of RD retina. Frontiers in cellular neuroscience. 2015;9:390. doi: 10.3389/fncel.2015.00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochimica et biophysica acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]