

ABSTRACT
HIV-1 reverse transcriptase (RT) is targeted by multiple drugs. RT mutations that confer resistance to nucleoside RT inhibitors (NRTIs) emerge during clinical use. Q151M and four associated mutations, A62V, V75I, F77L, and F116Y, were detected in patients failing therapies with dideoxynucleosides (didanosine [ddI], zalcitabine [ddC]) and/or zidovudine (AZT). The cluster of the five mutations is referred to as the Q151M complex (Q151Mc), and an RT or virus containing Q151Mc exhibits resistance to multiple NRTIs. To understand the structural basis for Q151M and Q151Mc resistance, we systematically determined the crystal structures of the wild-type RT/double-stranded DNA (dsDNA)/dATP (complex I), wild-type RT/dsDNA/ddATP (complex II), Q151M RT/dsDNA/dATP (complex III), Q151Mc RT/dsDNA/dATP (complex IV), and Q151Mc RT/dsDNA/ddATP (complex V) ternary complexes. The structures revealed that the deoxyribose rings of dATP and ddATP have 3′-endo and 3′-exo conformations, respectively. The single mutation Q151M introduces conformational perturbation at the deoxynucleoside triphosphate (dNTP)-binding pocket, and the mutated pocket may exist in multiple conformations. The compensatory set of mutations in Q151Mc, particularly F116Y, restricts the side chain flexibility of M151 and helps restore the DNA polymerization efficiency of the enzyme. The altered dNTP-binding pocket in Q151Mc RT has the Q151-R72 hydrogen bond removed and has a switched conformation for the key conserved residue R72 compared to that in wild-type RT. On the basis of a modeled structure of hepatitis B virus (HBV) polymerase, the residues R72, Y116, M151, and M184 in Q151Mc HIV-1 RT are conserved in wild-type HBV polymerase as residues R41, Y89, M171, and M204, respectively; functionally, both Q151Mc HIV-1 and wild-type HBV are resistant to dideoxynucleoside analogs.
KEYWORDS: antivirals, compensatory mutation, DNA polymerase, human immunodeficiency virus, antiviral agents, hepatitis B virus
INTRODUCTION
HIV-1 infections are treated with combinations of multiple drugs. Currently approved anti-HIV-1 drugs target key steps of the viral life cycle, namely, viral entry/fusion, reverse transcription, integration of viral DNA into the chromosome of infected cells, and maturation of newly released immature viral particles. The enzyme reverse transcriptase (RT) of HIV-1 is responsible for copying the viral single-stranded RNA genome into double-stranded DNA (dsDNA) in the cytoplasm after a virus has fused with a host cell. This copying process is accomplished by RNA- and DNA-dependent DNA polymerization, carried out by the polymerase activity and degradation of the RNA strand from an RNA/DNA duplex intermediate by the RNase H activity of RT. The DNA polymerization activity of RT is targeted by 13 approved drugs, of which 8 are nucleoside/nucleotide RT inhibitors (NRTIs) and 5 are nonnucleoside RT inhibitors (NNRTIs); for simplicity, we also refer to the nucleotide analogs as NRTIs in this paper. NNRTIs are allosteric inhibitors, whereas an NRTI is a nucleoside (or nucleotide) analog in which the deoxyribose ring is chemically or structurally altered, and the nucleotide bases are sometimes modified in NRTIs. An NRTI is converted into its triphosphate form (NRTI-TP) intracellularly, and an NRTI-TP binds RT as a deoxynucleoside triphosphate (dNTP) substrate analog. The catalytic reaction of DNA polymerization by RT cleaves off the pyrophosphate (β-γ phosphates) and incorporates the drug into the growing viral DNA primer strand. Once it is incorporated as a nucleotide mimic by RT, an NRTI blocks the addition of the next nucleotide and thereby inhibits the production of viral DNA.
Even though an NRTI functions as a chain terminator rather than a competitive inhibitor of the viral enzyme, RT mutations emerge, resulting in the development of resistance to NRTIs. Following the clinical detection of RT mutations, extensive virology, biochemical, and structural studies have helped provide an understanding of the distinct molecular mechanisms by which mutant RTs confer resistance to NRTIs (1, 2). RT mutations result in the development of resistance to NRTIs either by discrimination (exclusion) or by excision. RT can reverse the direction of the catalytic reaction from polymerization to pyrophosphorolysis for unblocking the 3′ end of the DNA primer (3, 4). This mechanism of resistance by excision is enhanced when RT acquires excision-enhancing mutations (EEMs)/thymidine analog mutations (TAMs) to bind an ATP molecule as the pyrophosphate donor to improve pyrophosphorolysis (5, 6). In contrast, the exclusion mechanisms of resistance by RT are conferred by acquiring mutations primarily in the vicinity of the dNTP-binding site, extending from the flexible β3-β4 fingers loop region to the catalytic YMDD moiety. Among the predominant mutations, (i) M184I/V helps discriminate the β-l-pseudoribose ring of lamivudine triphosphate (3TC-TP) (or emtricitabine triphosphate) (7, 8), (ii) K65R discriminates tenofovir diphosphate from dATP (9, 10), and (iii) a β3-β4 fingers loop insertion or deletion has various effects, such as multidrug resistance and an enhanced excision capability of RT containing exclusion mutations (11, 12).
The mutation Q151M was detected primarily in association with four other RT mutations, A62V, V75I, F77L, and F116Y, in patients failing treatment with dideoxynucleosides (didanosine [ddI], zalcitabine [ddC]) and zidovudine (AZT) (13); the collection of the five mutations is referred to as the Q151M complex (Q151Mc). Subsequently, the Q151M mutation was found to confer high-level resistance to all dideoxynucleoside drugs and lower-level resistance to abacavir (ABC) (14, 15). Some of the major concerns with the Q151M mutation are that (i) first-line stavudine (d4T)-containing antiretroviral therapy selects Q151M as a primary drug resistance mutation (16, 17), (ii) the presence of the Q151M mutation appears to lead to a higher mortality rate (18), and (iii) HIV-1 harboring the Q151M mutation is capable of mother-to-child transmission (19).
The Q151M mutation surfaces first in vivo, followed by the acquisition of the F77L and F116Y mutations, and subsequently, the set of five Q151Mc mutations emerges (13). A single-genome sequencing study suggested the sequential selection of A62V, V75I, and Q151M mutations in patients failing therapy with NRTIs (20). The Q151M mutation alone can confer NRTI resistance; however, viral fitness and drug resistance are enhanced with the coemergence of the F77L/F116Y/Q151M combination. Viral fitness and drug resistance are enhanced further with the acquisition of the set of Q151Mc mutations (21). Q151M in association with other exclusion mutations, as discussed below, enhances the ability of RT to discriminate other NRTIs besides dideoxynucleosides (ddNs). Q151Mc accompanied by the K70S/T/Q mutation confers high-level resistance to tenofovir (22, 23). Q151M with K65R or M184V causes high-level resistance to both lamivudine and zidovudine in patients infected with HIV-2, and the combination of K65R, Q151M, and M184V confers resistance to all NRTIs (24). An investigational nucleotide, GS-9148, selects for the rare Q151L mutation rather than Q151M (25). While the Q151M mutation helps discriminate against NRTIs, the mutant RTs have a decreased ability for NRTI excision (26); the accompanying mutation V75I further reduces the ability of RT for excision (27).
A recent crystal structure of a Q151M mutant RT that contained no nucleic acid did not reveal significant structural perturbations that would account for the resistance mechanism by the Q151M mutation (28). Residue Q151 is located in the palm subdomain of RT, and Q151 is primarily exposed to solvent in all structures of RT except in the structures of RT/nucleic acid/dNTP ternary complexes. Upon binding of a dNTP to the RT/nucleic acid binary complex structure (29), the region undergoes significant rearrangement; Q151 is buried inside as a part of the dNTP-binding pocket in the structure of the RT/DNA/dTTP ternary complex (7). Residue Q151 in the RT/DNA/dTTP ternary complex is surrounded by residues A62, R72, Y115, and F116, as well as the incoming dNTP (Fig. 1, top). The side chain of buried Q151 interacts with a highly conserved residue R72, which is critical for dNTP binding and incorporation (10, 30). Thereby, Q151 is expected to play a critical role in dNTP binding and nucleotide incorporation. For a systematic evaluation of the structural impact of Q151M and Q151Mc mutations, we determined the crystal structures of five ternary complexes of wild-type RT (wtRT), Q151M RT, and Q151Mc RT (Fig. 1, bottom). All of the structures reported here were determined in one crystal form, which minimizes any potential influence of crystal contacts, and thereby, the observed structural differences may be attributed to the molecular mechanisms of Q151M and the accompanying mutations.
FIG 1.
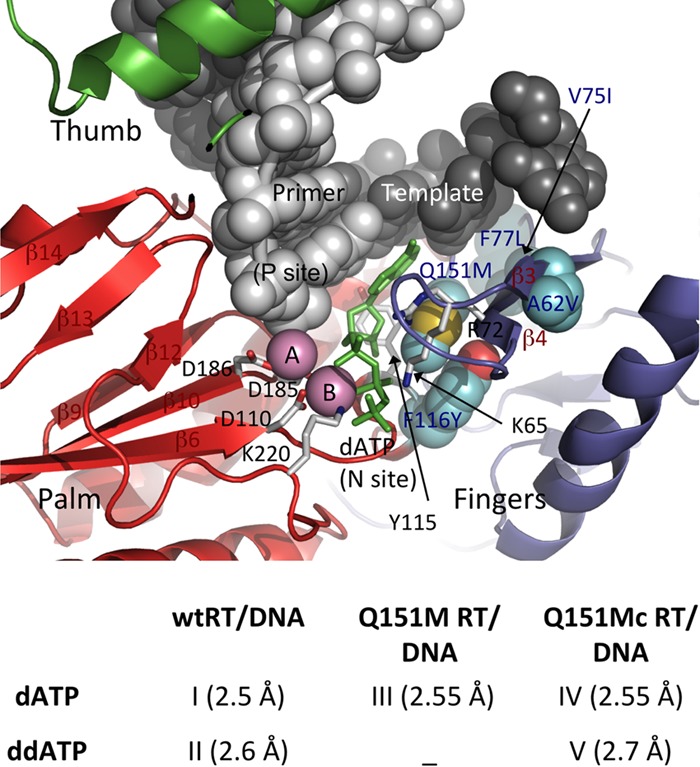
Ternary complexes of wtRT, Q151M RT, and Q151Mc RT in complexes with DNA and dATP (or ddATP). (Top) Relative locations of the Q151Mc mutations with respect to the polymerase active site in RT/DNA/dATP (or analog) ternary structures. (Bottom) Complexes for which the structures are reported in the current study; the resolutions of the structures are in parentheses.
RESULTS AND DISCUSSION
Binding of dATP versus ddATP to wtRT/DNA complex.
At the enzyme level, wild-type RT (wtRT) has comparable binding affinities for both dideoxynucleoside triphosphates (ddNTPs) and dNTPs (26). This pre-steady-state kinetics study found that RT incorporates dideoxynucleoside triphosphates (ddNTPs) at a lower rate (kpol) than dNTPs and the rate is further reduced for Q151M and Q151Mc RTs. This difference in the rate of incorporation appears to be responsible for the discrimination of ddNTPs from dNTPs by RTs. For evaluation of the structural basis for the resistance, comparison of the modes of dNTP and ddNTP binding to wtRT/DNA and the mutant RT/DNA complexes is essential.
We determined the crystal structures of the wtRT/DNA/dATP (complex I) and wtRT/DNA/ddATP (complex II) ternary complexes at 2.5- and 2.7-Å resolutions, respectively (Table 1). A previously studied crystal form of the RT/DNA complexes (31), which allows for considerable flexibility of RT molecules in crystals to permit the binding of a dNTP (or analog) in a catalytically competent mode, was employed for obtaining the structures of both ternary complexes. Crystals of an RT/DNA cross-linked binary complex were grown; the first template overhang was selected as a thymidine for the binding of a dATP as the incoming nucleotide, and dATP (or ddATP) molecules were soaked into the crystals of the RT/DNA complex for the formation of the respective ternary complexes (Fig. 2A and B). The adenine bases of dATP and ddATP superimposed well and maintained the conserved (i) base pairing with the first template overhang, (ii) base stacking with the template-primer, and (iii) interactions with surrounding RT residues, such as R72 and Y115. However, the sugar moiety of ddATP had a conformation that was different from that of dATP (Fig. 2C). The deoxyribose ring of dATP was structurally constrained to 3′-endo, which positions the 3′-OH group appropriately to form a hydrogen bond with the main chain amino group of Y115. Thereby, a dNTP is assumed to bind RT at the N site with its deoxyribose ring in the 3′-endo conformation and maintains the 3′-endo conformation at the P site following incorporation and translocation. The 3′-endo conformation of the deoxyribose ring of the DNA primer 3′-terminal nucleotide is essential for the catalytic incorporation of the next nucleotide; previously reported structures of catalytically active RT/DNA/dNTP ternary complexes confirmed a 3′-endo conformation of the nucleotide at the primer terminus that enabled the incorporation of an incoming nucleotide (32).
TABLE 1.
Crystallographic data and refinement statistics
| Parametera | Value(s) forb: |
||||
|---|---|---|---|---|---|
| wtRT/DNA/dATP | wtRT/DNA/ddATP | Q151M RT/DNA/dATP | Q151Mc RT/DNA/dATP | Q151Mc RT/DNA/ddATP | |
| Complex | I | II | III | IV | V |
| PDB accession no. | 5TXL | 5TXM | 5TXN | 5TXO | 5TXP |
| Data collection date | September 2010 | September 2010 | March 2013 | June 2013 | February 2013 |
| Data collection source | NSLS X25 | NSLS X25 | CHESS F1 | CHESS F1 | CHESS F1 |
| Data collection statistics | |||||
| Space group | P21 | P21 | P21 | P21 | P21 |
| Unit cell dimensions | |||||
| a, b, c (Å) | 90.38, 133.85, 139.34 | 90.34, 133.95, 139.40 | 90.04, 133.63, 139.20 | 89.99, 132.93, 131.12 | 89.95, 133.27, 139.06 |
| α, β, γ (°) | 90, 97.3, 90 | 90, 97.71, 90 | 90, 97.88, 90 | 90, 97.38, 90 | 90, 97.59, 90 |
| Resolution (Å) | 50–2.5 | 50–2.7 | 50–2.55 | 50–2.55 | 40–2.7 |
| Highest-resolution cell (Å) | 2.54–2.5 | 2.75–2.7 | 2.59–2.55 | 2.59–2.55 | 2.75–2.7 |
| Rmerge | 0.067 (0.53) | 0.065 (0.570) | 0.095 (0.746) | 0.091 (0.646) | 0.098 (0.687) |
| Rmeas | 0.076 (0.634) | 0.074 (0.679) | 0.106 (0.855) | 0.104 (0.765) | 0.112 (0.831) |
| No. of unique reflections | 111,859 (4,966) | 87,794 (4,104) | 106,245 (5,251) | 104,998 (5,256) | 89,232 (4,362) |
| Completeness | 98.7 (88.1) | 97.4 (91.1) | 98.1 (97.0) | 99.2 (99.0) | 99.4 (97.8) |
| Multiplicity | 4.1 (2.7) | 4.2 (3.1) | 5.0 (4.1) | 3.9 (3.3) | 3.9 (2.9) |
| I/σ (I) | 15.6 (1.6) | 12.9 (1.6) | 9.0 (1.7) | 9.3 (1.8) | 8.8 (1.4) |
| Refinement statistics | |||||
| Resolution (Å) | 48.1–2.5 | 46.0–2.7 | 36.35–2.55 | 46–2.55 | 37.6–2.7 |
| Cutoff criteria | F < 1.37 σ(F) | F < 1.38 σ(F) | F < 1.39 σ(F) | F < 0.0 | F < 1.37 σ(F) |
| No. of reflections (Rfree set) | 111,811 (3,349) | 87,934 (2,621) | 104,270 (3,048) | 104,909 (3,135) | 89,184 (1,788) |
| Rwork/Rfree | 0.193/0.225 | 0.184/0.223 | 0.196/0.226 | 0.191/0.228 | 0.195/0.238 |
| No. of atoms | |||||
| Nonsolvent | 17,761 | 17,626 | 17,745 | 17,759 | 17,767 |
| Solvent | 219 | 187 | 195 | 169 | 101 |
| Stereochemistry (RMSDs) | |||||
| Bond length (Å) | 0.006 | 0.009 | 0.007 | 0.007 | 0.007 |
| Bond angle (°) | 0.899 | 1.020 | 0.882 | 0.956 | 0.769 |
I, intensity of a reflection; RMSD, root mean square deviation.
The values in parentheses represent the highest-resolution shell.
FIG 2.

Binding of dATP versus ddATP to the HIV-1 RT/DNA complex. The positioning of dATP (green) (A) and ddATP (blue) (B), the surrounding amino acid residues (gray carbon), and the DNA template/primer (yellow carbon) in the crystal structures were ascertained by difference Fourier (Fo − Fc) maps calculated prior to inclusion of the dATP (or ddATP) into the refinement and are displayed at 3σ (Fo and Fc are the amplitudes of observed and calculated structure factors, respectively). The chelation of the catalytic metal ion B is shown as thin solid lines, and hydrogen bonds are shown as dashed lines. Like in most published RT/DNA ternary complexes (10, 31, 51, 52), dATP (or ddATP) molecules in the current structures chelate one Mg2+ ion (metal B) with a superimposable coordination geometry involving one oxygen each from the three (α, β, and γ) phosphates. Ion B also chelates the main chain carbonyl oxygen of V111 and one carboxyl oxygen of the catalytic residues D110 and D185 to complete an octahedral coordination. This coordination environment is invariant in all available structures of RT/DNA/dNTP ternary complexes (7). The images in panels A and B are in stereo. (C) An active-site superposition of the two structures reveals 3′-endo and 3′-exo conformations of RT-bound dATP and ddATP, respectively. (D and E) The hydrophobic interactions of dATP (D) and ddATP (E) with Y115 shows that the lack of the 3′-OH in ddATP is partly compensated for by the 3′-exo conformation of the sugar ring.
The conformation of the sugar ring of a ddATP that lacks the 3′-OH group is relatively flexible compared to that of dNTP. In the current RT/DNA/ddATP structure (complex II), the dideoxyribose ring of ddATP at the N site and of the terminal ddGMP at the P site have 3′-exo conformations (Fig. 2C), even though a 3′-endo conformation of a dideoxyribose ring can be accommodated at both sites. In fact, the 3′-terminal nucleotide of the DNA primer in almost all reported RT/DNA cross-linked ternary complexes terminated with a ddGMP, which was catalytically incorporated into the primer strand by RT (7, 33); the terminal ddGMP assumed either of the conformations. The difference of the 3′-exo versus the 3′-endo conformations of the sugar rings of ddATP versus dATP apparently was not discriminatory at the dNTP-binding state; however, the difference may be imperative for the slower incorporation of ddATP by RT. There is the possibility that the more flexible sugar ring of ddATP may either (i) maintain the 3′-exo conformation or (ii) switch between the 3′-exo and 3′-endo conformation, whereas the ring of dATP has a single 3′-endo conformation. wtRT has a slightly (∼5-fold) decreased rate of incorporation (kpol) for ddNTP than for dNTP, as observed by pre-steady-state kinetic studies (25, 34, 35). Further, in agreement with the kinetic data demonstrating that the binding constant (Kd) of ddNTP versus that of dNTP for binding to RT is not significantly altered, both ddATP and dATP in the respective ternary complex structures have highly superimposable binding locations and interactions with RT, nucleic acid, and metal ions. The loss of the 3′-OH interaction with the main chain amino group of Y115 for ddATP, which is not lost in the complex with dATP, appears to be partly compensated for by the enhanced hydrophobic contacts between the 3′-exo sugar ring of ddATP and the aromatic side chain of Y115 (Fig. 2D and E).
Structure of the Q151M RT/DNA/dATP complex.
We determined the crystal structure of the Q151M RT/DNA/dATP complex (complex III) at a 2.55-Å resolution (Table 1). The metal chelation, base pairing, and deoxyribose ring conformation of dATP are highly superimposable with those in the wtRT/DNA/dATP complex (complex I). In wtRT, residue Q151 is a part of the dNTP-binding pocket, and Q151 forms a hydrogen bond with the guanidinium moiety of the highly conserved residue R72 (Fig. 2A and B); R72 interacts with the base and α-phosphate of dNTP and apparently plays a critical role in the process of dNTP binding and incorporation (10, 30). Q151 in coordination with R72 seemingly assists the binding and positioning of a dNTP for catalysis. The Q151M mutation breaks the hydrogen bond with R72, and the mutation introduces the flexible unbranched side chain of M151. Similarly, as with Q151, the side chain of M151 is buried, and thereby, the rotameric state of the residue is somewhat constrained.
In the structure of complex III (Fig. 3A), the side chain conformation of R72 is altered compared to that in the structures of complexes I and II with wtRT; the guanidinium group of R72 is switched via alteration of the side chain torsions χ3 and χ4. The side chain torsion angles χ1, χ2, and χ3 of Q151 in the structures of wtRT ternary complexes I and II are about 60, 180, and 180°, respectively, and Q151 forms a hydrogen bond with R72 (Fig. 3B). In the structure of complex III, the side chain of M151 remains buried analogously to that of Q151 in the structures of complexes I and II, and the χ angles are very similar for both M151 and Q151. The Q151M substitution eliminates the branching at C-δ and the hydrogen bond with R72. The flexible unbranched side chain of M151 and the conformational change of R72 may permit M151 to have more than one conformation via variation of χ3 (Fig. 3C). This possibility of the existence of multiple conformational states in the Q151M mutant RT compared to a single conformation of the pocket in wtRT may be a factor contributing to the reduced polymerase activity of the Q151M mutant (26).
FIG 3.
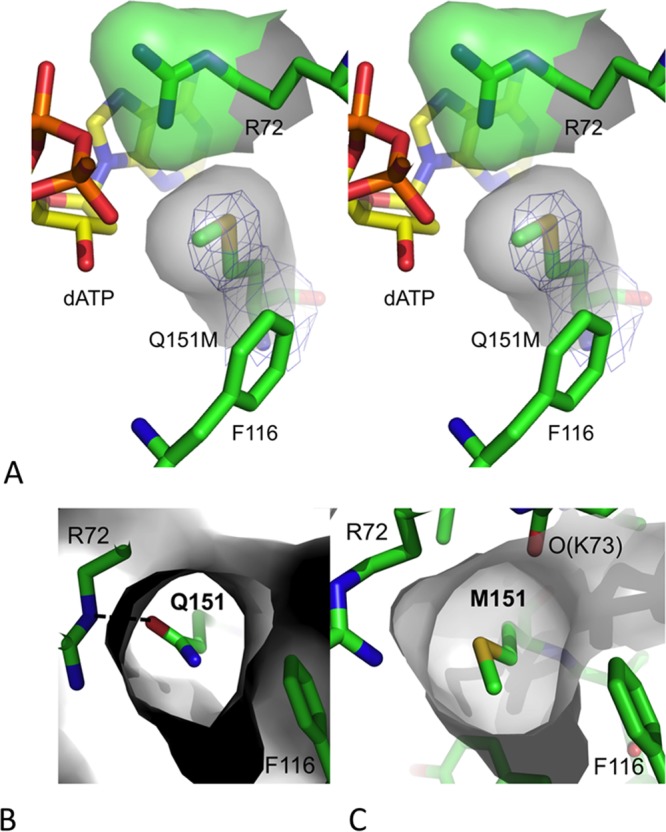
Conformation of M151 in the Q151M RT/DNA/dATP complex. (A) A stereo view showing the position and location of mutated residue Q151M in the structure of complex III defined by electron density (blue mesh). The Q151M mutation altered the side chain torsion of conserved residue R72. The van der Waals surfaces of R72 and M151 show the complementarity in the positioning of the two residues in the dNTP-binding site of the Q151M ternary structure. (B and C) van der Waals surfaces (in gray) surrounding Q151 (B) and M151 (C) in the structures of wtRT and Q151M complexes indicate that the flexible M151 side chain, unlike the Q151 side chain in wtRT, may alter the position of its C-ε atom by varying the torsion χ3 and perturbing the conformation of dNTP binding.
Q151Mc RT/DNA ternary complexes.
The crystal structures of the Q151Mc RT/DNA complexes with dATP and ddATP were determined at 2.55- and 2.7-Å resolutions, respectively, and are referred to as the structures of complexes IV and V, respectively (Table 1). As in the wtRT complexes, the deoxyribose and dideoxyribose rings maintained 3′-endo and 3′-exo conformations for dATP and ddATP, respectively, and the remaining parts of both complexes were also highly superimposable.
The structures reveal that the Q151Mc mutation F116Y extends the aromatic side chain by the addition of an O-η atom, which introduces an O-H…O-type hydrogen bond with the main chain carbonyl of K73 (Fig. 4A). The structural restraint from the F116Y mutation in Q151Mc restricts the M151 side chain to only one conformation, whereas there is the possibility of the existence of multiple conformations in Q151M RT. This structural finding is consistent with the findings of previously reported biochemical studies and explains that the accompanying mutation (F116Y) improves the polymerization fitness by restricting the dNTP-binding pocket to a single conformational state (Fig. 4B). The sites of compensatory mutations V75, F77, and F116 and residue Y146 are parts of a hydrophobic core responsible for the folding of the fingers subdomain, and the hydrophobicity of this core is preserved in the Q151Mc RT. The mutations V75I and F77L appear to help stabilize the positioning of the mutated side chain of F116Y to maintain the hydrogen bond with the carbonyl of K73 (Fig. 4C). Conceivably, the compensatory mutations V75I, F77L, and F116Y help in defining and stabilizing the dNTP-binding site in the complexes containing Q151Mc (complexes IV and V), whereas there is the possibility that the Q151M RT/DNA/dATP complex (complex III) has additional structural states. This difference may explain a role of the compensatory mutations in improving the fitness of RT or virus containing Q151Mc over that of RT or virus containing Q151M. The mutation A62V is located at a distance of ∼10 Å away from the Q151M site. A62 is located at the base of the fingers β3-β4 hairpin and is proximal to the template strand. Presumably, the A62V mutation helps to reposition the template strand, and the repositioned template might help in discriminating a ddNTP from dNTP in the process of polymerization by Q151Mc mutant RT.
FIG 4.
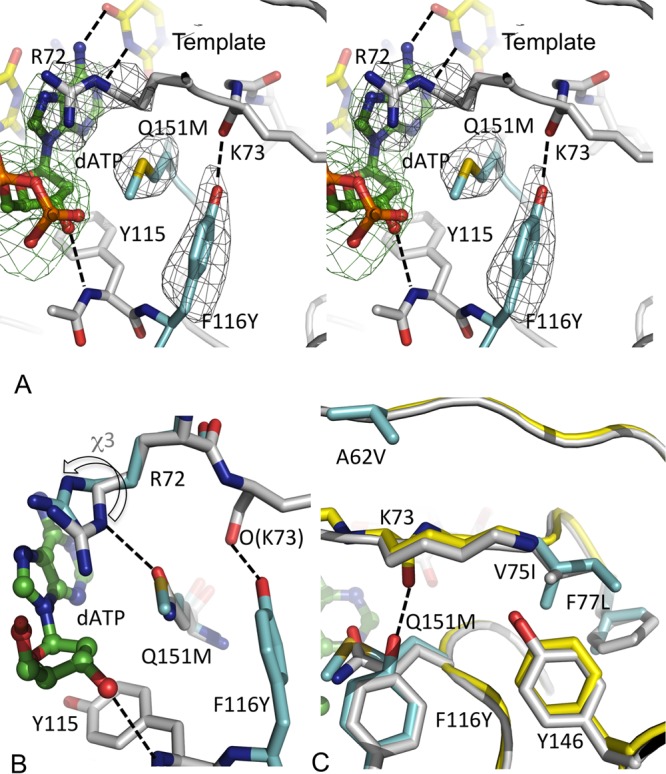
Ternary structures of Q151Mc RT complexes with dATP and ddATP. (A) A stereo view of dATP binding to the Q151Mc RT/DNA complex (complex IV); the two mutated residues F116Y and Q151M are in cyan, dATP is in green, and some of the key hydrogen-bonding interactions are shown as dotted lines. The hydrogen bond between F116Y and K73 appears to stabilize an alternate conformation of the dNTP-binding pocket in Q151Mc RT. (B) A comparison of the dNTP-binding pocket conformations of Q151Mc RT (cyan) and wtRT (gray). The side chain orientations of Q151 and M151 are very similar; however, the chemical alteration of the side chain by the Q151M mutation repositions the guanidinium group of R72 (indicated by the arrow). (C) A structural superposition of wtRT (gray) and Q151Mc RT (yellow and cyan for mutated side chains) structures. The mutated side chains are in cyan. All of the mutated side chains except A62V are parts of a hydrophobic core of the fingers subdomain, and the mutations maintain the hydrophobicity of the core.
Implications for HBV polymerase.
Hepatitis B virus (HBV) polymerase appears to contain a dNTP-binding site/polymerase active-site architecture similar to that of HIV-1 RT (36). The nucleoside analog anti-HIV-1 drugs, such as the β-l-oxathiolane ring-containing lamivudine (3TC) and acyclic tenofovir (or adefovir), are effective inhibitors of HBV polymerase and are used clinically to treat HBV-infected individuals (37, 38). Analogous nucleoside drug resistance mutations emerge in HBV polymerase and in HIV-1 RT under specific drug pressure; e.g., HIV-1 RT mutation M184V and HBV polymerase mutation M204V emerge in the YMDD loop in response to 3TC treatments (39). Interestingly, the positional analog for HIV-1 RT Q151 is a methionine (M171) in wild-type HBV (wtHBV) polymerase; residues R72, Y116, and M151, which are observed to be responsible for altering and stabilizing the conformational state of the dNTP-binding pocket of Q151Mc HIV-1 RT, are also conserved as amino acid residues R41, Y89, and M171, respectively, in wtHBV polymerase. Thereby, it may be conceivable that wtHBV polymerase has a dNTP-binding site conformation closely related to that observed in Q151Mc structures rather than the conformation observed in wtHIV-1 RT structures. Mechanistically, dideoxynucleoside drugs like dideoxyinosine (ddI), dideoxycytosine (ddC), and stavudine (d4T), which have reduced efficacy against Q151Mc mutant HIV-1 RT, are also not used to treat HBV infections; l-nucleosides are relatively effective against HBV polymerase (40).
Conclusions.
In dNTP-bound ternary complexes, the dNTP-binding pocket of Q151Mc RT has structural differences from wtRT, particularly the side chain conformations of R72 and Q151M (Fig. 5). The RT-bound ddATP and dATP have 3′-exo versus 3′-endo conformations for their respective sugar rings; however, the flexible sugar ring of a ddNTP also has a 3′-endo conformation, particularly in high-resolution RT/nucleic acid complexes in which both of the catalytic Mg2+ ions are present at the polymerase active site (41, 42). The dNTP-binding pocket would undergo a series of structural transitions in the process of nucleotide incorporation starting from the structurally observed dNTP-bound state. Presumably, the altered conformation of the dNTP-binding pocket in Q151Mc is able to discriminate the flexible sugar ring of a ddNTP from the 3′-endo dNTP during the process of catalytic incorporation. Recent kinetic studies revealed that the lower rate of pyrophosphate (PPi) release by RT could decrease the rate of polymerization (43). Our recent structure of the drug foscarnet (PFA), a pyrophosphate analog, in complex with RT/DNA showed that R72 directly interacts with PFA (32), and the interaction likely plays an important role in PPi release. Thereby, residue R72 in Q151M (or Q151Mc) RT may slow down the rate of release of PPi for ddNTP incorporation versus that for dNTP incorporation and contribute to ddNTP discrimination. While biochemical and structural information helps provide an understanding of the phenotypic characteristics of drug resistance mutations, there are several complex factors, such as the distribution of infected cell types and evolutionary dynamics, that influence infectivity, the effects of drug resistance, and compensatory mutations (44); statistical analysis of the fitness of a large number of HIV variants demonstrated the existence of epistasis in HIV (45). The Q151M mutation provides a strong example of a primary mutation that leads to fitness defects in HIV-1 RT, as reflected by deleterious effects on polymerase catalysis and the viral replication rate, that become entrenched upon accompaniment by the compensatory mutations A62V, V75I, F77L, and F116Y in Q151Mc to restore enzymatic and viral fitness. Our results provide an explanation of the structural basis for entrenchment of the multidrug-resistant Q151M mutant by the emergence of the Q151Mc set of mutations in the dNTP-binding region of HIV-1 RT through essentially an evolutionary annealing of the local environment of M151.
FIG 5.
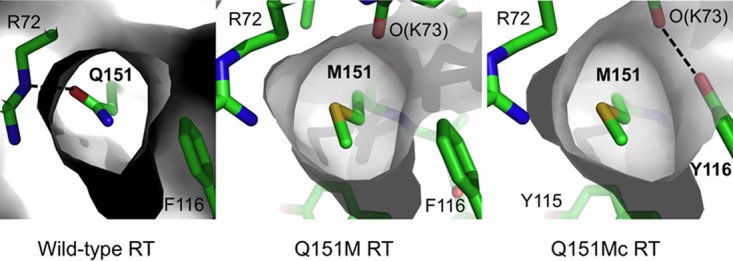
The Q151M mutation alters the dNTP-binding pocket by switching the side chain conformation of the conserved residue R72, and the altered conformation is further stabilized in the Q151Mc ternary complex.
The structure of HBV polymerase has not been available, and the structure of HIV-1 RT has been used as a surrogate for understanding the structure, function, and drug resistance of HBV polymerase (36). The Q151Mc HIV-1 RT and wtHBV polymerase share key conserved structural elements that are involved in dNTP binding and incorporation and in reducing the sensitivity to dideoxynucleoside analogs. The current study provides a structural basis for understanding the molecular mechanism of Q151M and Q151Mc resistance mutations and the resilience of the mutant HIV-1 RT and wtHBV polymerase to dideoxynucleoside drugs. This information may be exploited in designing new inhibitors of viral DNA polymerases, including those of HIV and HBV.
MATERIALS AND METHODS
RT expression, RT/DNA cross-linking, purification, and crystallization.
A previously described wtRT construct, RT127A, was used in the structural studies of complexes I and II (31); the construct has a D498N mutation added to inactivate the RNase H activity. The D498N mutant RT has polymerase activity comparable to that of wtRT (46). The Q151M and Q151Mc mutations (A62V, V75I, F77L, F116Y, and Q151M) were introduced using the methods described for RT127A (31). All three RT constructs were expressed and purified using a previously described protocol (47).
The 27-mer DNA template (5′-ATGGTCGGCGCCCGAACAGGGACTGTG-3′) was synthesized by Integrated DNA Technologies. The 20-mer primer (5′-ACAGTCCCTGTTCGGGCGCC-3′) with a cross-linkable thiopropyl group (at N-2 on G in the primer strand) was custom synthesized, annealed, and cross-linked to the three RT constructs to generate the posttranslocation complex, which was purified and crystallized as reported previously (31). During the cross-linking reaction, the primer was extended with a 2′,3′-dideoxyguanosine at the 3′ end. For growing the crystal of Q151Mc in complex with bound ddATP, drops were set up with 1 μl of 10 mg/ml RT/DNA, 1 μl of well solution, and 1 μl of 2 mM ddATP. After 3.5 weeks, the drops were seeded and crystals grew; the microseeds were of the RT127A binary complex, as described above, except that the fifth and sixth template bases were TT and the primer was terminated with dTdT to generate the pretranslocation complex.
Crystal soaking and freezing. (i) dATP (complex I) or ddATP (complex II) soak.
An RT/DNA (ddG-terminated primer) binary complex crystal with a size of 240 by 80 by 50 μm3 was transferred to 50 μl of a stabilization solution containing 2 mM dATP, 12% (wt/vol) polyethylene glycol 8000, 5% (vol/vol) glycerol, 5% (wt/vol) sucrose, 100 mM (NH4)2SO4, 20 mM MgCl2, and 50 mM bis-Tris propane, pH 7.2, for 4 min. The crystal was soaked for 30 s in 50 μl cryoprotective solution in which the amount of glycerol was raised to 20% (vol/vol) and flash-cooled in liquid N2. The 2 mM ddATP soak was prepared similarly with a crystal with dimensions of 160 by 80 by 30 μm3.
(ii) dATP soak and RT127A with Q151M (complex III) or Q151Mc (complex IV).
For the single Q151M mutation, an RT/DNA (ddG-terminated primer) binary complex crystal with a size of 160 by 120 by 60 μm3 was frozen as described above but at pH 7.0 with 2 mM dATP and 10% (vol/vol) glycerol in stabilization solution for 24 min and 1 min in the 20% (vol/vol) glycerol-containing cryoprotective solution. For the Q151M complex, a crystal with a size of 320 by 120 by 80 μm3 was prepared similarly at pH 7.4 with a 13-min soak in 10% (vol/vol) glycerol and 1 min in the 20% (vol/vol) glycerol-containing cryoprotective solution.
(iii) ddATP soak to Q151Mc (complex V).
An RT/DNA (ddG-terminated primer) ternary complex crystal with a size of 160 by 120 by 80 μm3 was cryo-cooled as described above for the Q151M single mutant but with 2 mM ddATP and for 7 min in the stabilization solution and 1 min in the cryoprotective solution.
X-ray crystallography.
X-ray diffraction data sets were collected from the crystals of complexes I to V using synchrotron sources: the F1 beamline at the Cornell High Energy Synchrotron Source (CHESS) and the X25 beamline at Brookhaven National Laboratory (BNL). The data were processed and scaled using the HKL2000 software application (48), as summarized in Table 1. The structures were solved by molecular replacement, using protein atoms in the crystal structure of the RT/DNA/AZT triphosphate complex (PDB accession no. 3V4I) (31), and the subdomains were positioned by rigid-body refinements in which each RT molecule was divided into 13 segments. Each crystal structure had two RT complexes per asymmetric unit; however, the first copy (chains A and B) was used in the analysis, unless otherwise specified. Individual structures were refined using the Phenix system (49), and the model building was carried out using Coot software (50). The figures showing structural information were generated using the PyMOL molecular graphics system (http://www.pymol.org/).
Accession number(s).
The coordinates and structure factors for the crystal structures of wild-type RT/dsDNA/dATP (complex I), wild-type RT/dsDNA/ddATP (complex II), Q151M RT/dsDNA/dATP (complex III), Q151Mc RT/dsDNA/dATP (complex IV), and Q151Mc RT/dsDNA/ddATP (complex V) ternary complexes are deposited in the Protein Data Bank (PDB) under accession numbers 5TXL, 5TXM, 5TXN, 5TXO, and 5TXP, respectively.
ACKNOWLEDGMENTS
We are grateful to the CHESS F1 and NSLS X25 beamline facilities for data collection.
E.A. acknowledges National Institutes of Health merit award R37 AI027690 for support.
REFERENCES
- 1.Menendez-Arias L. 2008. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res 134:124–146. doi: 10.1016/j.virusres.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 2.Das K, Arnold E. 2013. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr Opin Virol 3:111–118. doi: 10.1016/j.coviro.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer PR, Matsuura SE, So AG, Scott WA. 1998. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc Natl Acad Sci U S A 95:13471–13476. doi: 10.1073/pnas.95.23.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arion D, Parniak MA. 1999. HIV resistance to zidovudine: the role of pyrophosphorolysis. Drug Resist Updat 2:91–95. doi: 10.1054/drup.1999.0076. [DOI] [PubMed] [Google Scholar]
- 5.Boyer PL, Sarafianos SG, Arnold E, Hughes SH. 2001. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J Virol 75:4832–4842. doi: 10.1128/JVI.75.10.4832-4842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tu X, Das K, Han Q, Bauman JD, Clark AD Jr, Hou X, Frenkel YV, Gaffney BL, Jones RA, Boyer PL, Hughes SH, Sarafianos SG, Arnold E. 2010. Structural basis of HIV-1 resistance to AZT by excision. Nat Struct Mol Biol 17:1202–1209. doi: 10.1038/nsmb.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang H, Chopra R, Verdine GL, Harrison SC. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 8.Sarafianos SG, Das K, Clark AD Jr, Ding J, Boyer PL, Hughes SH, Arnold E. 1999. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with β-branched amino acids. Proc Natl Acad Sci U S A 96:10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White KL, Chen JM, Margot NA, Wrin T, Petropoulos CJ, Naeger LK, Swaminathan S, Miller MD. 2004. Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob Agents Chemother 48:992–1003. doi: 10.1128/AAC.48.3.992-1003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das K, Bandwar RP, White KL, Feng JY, Sarafianos SG, Tuske S, Tu X, Clark AD Jr, Boyer PL, Hou X, Gaffney BL, Jones RA, Miller MD, Hughes SH, Arnold E. 2009. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J Biol Chem 284:35092–35100. doi: 10.1074/jbc.M109.022525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mas A, Parera M, Briones C, Soriano V, Martinez MA, Domingo E, Menendez-Arias L. 2000. Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J 19:5752–5761. doi: 10.1093/emboj/19.21.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imamichi T, Berg SC, Imamichi H, Lopez JC, Metcalf JA, Falloon J, Lane HC. 2000. Relative replication fitness of a high-level 3′-azido-3′-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr → Gly) at codon 69. J Virol 74:10958–10964. doi: 10.1128/JVI.74.23.10958-10964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirasaka T, Kavlick MF, Ueno T, Gao WY, Kojima E, Alcaide ML, Chokekijchai S, Roy BM, Arnold E, Yarchoan R, Mitsuya H. 1995. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc Natl Acad Sci U S A 92:2398–2402. doi: 10.1073/pnas.92.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kavlick MF, Wyvill K, Yarchoan R, Mitsuya H. 1998. Emergence of multi-dideoxynucleoside-resistant human immunodeficiency virus type 1 variants, viral sequence variation, and disease progression in patients receiving antiretroviral chemotherapy. J Infect Dis 177:1506–1513. doi: 10.1086/515324. [DOI] [PubMed] [Google Scholar]
- 15.Melikian GL, Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia-Cancio PV, Zolopa A, Robbins GK, Kagan R, Israelski D, Shafer RW. 2012. Standardized comparison of the relative impacts of HIV-1 reverse transcriptase (RT) mutations on nucleoside RT inhibitor susceptibility. Antimicrob Agents Chemother 56:2305–2313. doi: 10.1128/AAC.05487-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang MW, Rhee SY, Bertagnolio S, Ford N, Holmes S, Sigaloff KC, Hamers RL, de Wit TF, Fleury HJ, Kanki PJ, Ruxrungtham K, Hawkins CA, Wallis CL, Stevens W, van Zyl GU, Manosuthi W, Hosseinipour MC, Ngo-Giang-Huong N, Belec L, Peeters M, Aghokeng A, Bunupuradah T, Burda S, Cane P, Cappelli G, Charpentier C, Dagnra AY, Deshpande AK, El-Katib Z, Eshleman SH, Fokam J, Gody JC, Katzenstein D, Koyalta DD, Kumwenda JJ, Lallemant M, Lynen L, Marconi VC, Margot NA, Moussa S, Ndung'u T, Nyambi PN, Orrell C, Schapiro JM, Schuurman R, Sirivichayakul S, Smith D, Zolfo M, Jordan MR, Shafer RW. 2013. Nucleoside reverse transcriptase inhibitor resistance mutations associated with first-line stavudine-containing antiretroviral therapy: programmatic implications for countries phasing out stavudine. J Infect Dis 207(Suppl 2):S70–S77. doi: 10.1093/infdis/jit114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nouhin J, Madec Y, Ngo-Giang-Huong N, Ferradini L, Nerrienet E. 2013. Increased risk of Q151M and K65R mutations in patients failing stavudine-containing first-line antiretroviral therapy in Cambodia. PLoS One 8:e73744. doi: 10.1371/journal.pone.0073744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherrer AU, von Wyl V, Joos B, Klimkait T, Burgisser P, Yerly S, Boni J, Ledergerber B, Gunthard HF. 2011. Predictors for the emergence of the 2 multi-nucleoside/nucleotide resistance mutations 69 insertion and Q151M and their impact on clinical outcome in the Swiss HIV cohort study. J Infect Dis 203:791–797. doi: 10.1093/infdis/jiq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henry M, Thuret I, Solas C, Genot S, Colson P, Tamalet C. 2008. Vertical transmission of multidrug-resistant Q151M human immunodeficiency virus type 1 strains. Pediatr Infect Dis J 27:278–280. doi: 10.1097/INF.0b013e31815db4c6. [DOI] [PubMed] [Google Scholar]
- 20.Mbisa JL, Gupta RK, Kabamba D, Mulenga V, Kalumbi M, Chintu C, Parry CM, Gibb DM, Walker SA, Cane PA, Pillay D. 2011. The evolution of HIV-1 reverse transcriptase in route to acquisition of Q151M multi-drug resistance is complex and involves mutations in multiple domains. Retrovirology 8:31. doi: 10.1186/1742-4690-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeda Y, Venzon DJ, Mitsuya H. 1998. Altered drug sensitivity, fitness, and evolution of human immunodeficiency virus type 1 with pol gene mutations conferring multi-dideoxynucleoside resistance. J Infect Dis 177:1207–1213. doi: 10.1086/515282. [DOI] [PubMed] [Google Scholar]
- 22.Van Laethem K, Pannecouque C, Vandamme AM. 2007. Mutations at 65 and 70 within the context of a Q151M cluster in human immunodeficiency virus type 1 reverse transcriptase impact the susceptibility to the different nucleoside reverse transcriptase inhibitors in distinct ways. Infect Genet Evol 7:600–603. doi: 10.1016/j.meegid.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Hachiya A, Kodama EN, Schuckmann MM, Kirby KA, Michailidis E, Sakagami Y, Oka S, Singh K, Sarafianos SG. 2011. K70Q adds high-level tenofovir resistance to “Q151M complex” HIV reverse transcriptase through the enhanced discrimination mechanism. PLoS One 6:e16242. doi: 10.1371/journal.pone.0016242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith RA, Anderson DJ, Pyrak CL, Preston BD, Gottlieb GS. 2009. Antiretroviral drug resistance in HIV-2: three amino acid changes are sufficient for classwide nucleoside analogue resistance. J Infect Dis 199:1323–1326. doi: 10.1086/597802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scarth BJ, White KL, Chen JM, Lansdon EB, Swaminathan S, Miller MD, Gotte M. 2011. Mechanism of resistance to GS-9148 conferred by the Q151L mutation in HIV-1 reverse transcriptase. Antimicrob Agents Chemother 55:2662–2669. doi: 10.1128/AAC.01738-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deval J, Selmi B, Boretto J, Egloff MP, Guerreiro C, Sarfati S, Canard B. 2002. The molecular mechanism of multidrug resistance by the Q151M human immunodeficiency virus type 1 reverse transcriptase and its suppression using alpha-boranophosphate nucleotide analogues. J Biol Chem 277:42097–42104. doi: 10.1074/jbc.M206725200. [DOI] [PubMed] [Google Scholar]
- 27.Matamoros T, Nevot M, Martinez MA, Menendez-Arias L. 2009. Thymidine analogue resistance suppression by V75I of HIV-1 reverse transcriptase: effects of substituting valine 75 on stavudine excision and discrimination. J Biol Chem 284:32792–32802. doi: 10.1074/jbc.M109.038885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura A, Tamura N, Yasutake Y. 2015. Structure of the HIV-1 reverse transcriptase Q151M mutant: insights into the inhibitor resistance of HIV-1 reverse transcriptase and the structure of the nucleotide-binding pocket of hepatitis B virus polymerase. Acta Crystallogr F Struct Biol Commun 71:1384–1390. doi: 10.1107/S2053230X15017896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding J, Das K, Hsiou Y, Sarafianos SG, Clark AD Jr, Jacobo-Molina A, Tantillo C, Hughes SH, Arnold E. 1998. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J Mol Biol 284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- 30.Sarafianos SG, Pandey VN, Kaushik N, Modak MJ. 1995. Site-directed mutagenesis of arginine 72 of HIV-1 reverse transcriptase. Catalytic role and inhibitor sensitivity. J Biol Chem 270:19729–19735. [DOI] [PubMed] [Google Scholar]
- 31.Das K, Martinez SE, Bauman JD, Arnold E. 2012. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat Struct Mol Biol 19:253–259. doi: 10.1038/nsmb.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das K, Balzarini J, Miller MT, Maguire AR, DeStefano JJ, Arnold E. 2016. Conformational states of HIV-1 reverse transcriptase for nucleotide incorporation vs pyrophosphorolysis-binding of foscarnet. ACS Chem Biol 11:2158–2164. doi: 10.1021/acschembio.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarafianos SG, Clark AD Jr, Tuske S, Squire CJ, Das K, Sheng D, Ilankumaran P, Ramesha AR, Kroth H, Sayer JM, Jerina DM, Boyer PL, Hughes SH, Arnold E. 2003. Trapping HIV-1 reverse transcriptase before and after translocation on DNA. J Biol Chem 278:16280–16288. doi: 10.1074/jbc.M212911200. [DOI] [PubMed] [Google Scholar]
- 34.Feng JY, Anderson KS. 1999. Mechanistic studies comparing the incorporation of (+) and (−) isomers of 3TCTP by HIV-1 reverse transcriptase. Biochemistry 38:55–63. doi: 10.1021/bi982340r. [DOI] [PubMed] [Google Scholar]
- 35.Selmi B, Boretto J, Sarfati SR, Guerreiro C, Canard B. 2001. Mechanism-based suppression of dideoxynucleotide resistance by K65R human immunodeficiency virus reverse transcriptase using an alpha-boranophosphate nucleoside analogue. J Biol Chem 276:48466–48472. [DOI] [PubMed] [Google Scholar]
- 36.Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG, Arnold E. 2001. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J Virol 75:4771–4779. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Clercq E, Ferir G, Kaptein S, Neyts J. 2010. Antiviral treatment of chronic hepatitis B virus (HBV) infections. Viruses 2:1279–1305. doi: 10.3390/v2061279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones SA, Hu J. 2013. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect 2:e56. doi: 10.1038/emi.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ling R, Mutimer D, Ahmed M, Boxall EH, Elias E, Dusheiko GM, Harrison TJ. 1996. Selection of mutations in the hepatitis B virus polymerase during therapy of transplant recipients with lamivudine. Hepatology 24:711–713. doi: 10.1002/hep.510240339. [DOI] [PubMed] [Google Scholar]
- 40.Bryant ML, Bridges EG, Placidi L, Faraj A, Loi AG, Pierra C, Dukhan D, Gosselin G, Imbach JL, Hernandez B, Juodawlkis A, Tennant B, Korba B, Cote P, Marion P, Cretton-Scott E, Schinazi RF, Sommadossi JP. 2001. Antiviral l-nucleosides specific for hepatitis B virus infection. Antimicrob Agents Chemother 45:229–235. doi: 10.1128/AAC.45.1.229-235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das K, Martinez SE, Bandwar RP, Arnold E. 2014. Structures of HIV-1 RT-RNA/DNA ternary complexes with dATP and nevirapine reveal conformational flexibility of RNA/DNA: insights into requirements for RNase H cleavage. Nucleic Acids Res 42:8125–8137. doi: 10.1093/nar/gku487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salie ZL, Kirby KA, Michailidis E, Marchand B, Singh K, Rohan LC, Kodama EN, Mitsuya H, Parniak MA, Sarafianos SG. 2016. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proc Natl Acad Sci U S A 113:9274–9279. doi: 10.1073/pnas.1605223113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li A, Gong S, Johnson KA. 2016. Rate-limiting pyrophosphate release by HIV reverse transcriptase improves fidelity. J Biol Chem 291:26554–26565. doi: 10.1074/jbc.M116.753152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Kleist M, Metzner P, Marquet R, Schutte C. 2012. HIV-1 polymerase inhibition by nucleoside analogs: cellular- and kinetic parameters of efficacy, susceptibility and resistance selection. PLoS Comput Biol 8:e1002359. doi: 10.1371/journal.pcbi.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonhoeffer S, Chappey C, Parkin NT, Whitcomb JM, Petropoulos CJ. 2004. Evidence for positive epistasis in HIV-1. Science 306:1547–1550. doi: 10.1126/science.1101786. [DOI] [PubMed] [Google Scholar]
- 46.DeStefano JJ, Wu W, Seehra J, McCoy J, Laston D, Albone E, Fay PJ, Bambara RA. 1994. Characterization of an RNase H deficient mutant of human immunodeficiency virus-1 reverse transcriptase having an aspartate to asparagine change at position 498. Biochim Biophys Acta 1219:380–388. doi: 10.1016/0167-4781(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 47.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. 2008. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res 36:5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. 2001. DENZO and SCALEPACK, vol F. Crystallography of biological macromolecules. Kluwer Academic Publishers, Boston, MA. [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Tuske S, Sarafianos SG, Clark AD Jr, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, Wang G, Gaffney BL, Jones RA, Jerina DM, Hughes SH, Arnold E. 2004. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat Struct Mol Biol 11:469–474. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- 52.Lansdon EB, Samuel D, Lagpacan L, Brendza KM, White KL, Hung M, Liu X, Boojamra CG, Mackman RL, Cihlar T, Ray AS, McGrath ME, Swaminathan S. 2010. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J Mol Biol 397:967–978. doi: 10.1016/j.jmb.2010.02.019. [DOI] [PubMed] [Google Scholar]