

ABSTRACT
The in vitro leishmanicidal activities of a series of 48 recently synthesized selenium derivatives against Leishmania infantum and Leishmania braziliensis parasites were tested using promastigotes and intracellular amastigote forms. The cytotoxicity of the tested compounds for J774.2 macrophage cells was also measured in order to establish their selectivity. Six of the tested compounds (compounds 8, 10, 11, 15, 45, and 48) showed selectivity indexes higher than those of the reference drug, meglumine antimonate (Glucantime), for both Leishmania species; in the case of L. braziliensis, compound 20 was also remarkably selective. Moreover, data on infection rates and amastigote numbers per macrophage showed that compounds 8, 10, 11, 15, 45, and 48 were the most active against both Leishmania species studied. The observed changes in the excretion product profile of parasites treated with these six compounds were also consistent with substantial cytoplasmic alterations. On the other hand, the most active compounds were potent inhibitors of Fe superoxide dismutase (Fe-SOD) in the two parasite species considered, whereas their impact on human CuZn-SOD was low. The high activity, low toxicity, stability, low cost of the starting materials, and straightforward synthesis make these compounds appropriate molecules for the development of affordable antileishmanicidal agents.
KEYWORDS: Leishmania, selenium, superoxide dismutase, glucose metabolism
INTRODUCTION
Leishmaniasis, caused by the intracellular protozoan Leishmania, is transmitted by the bite of phlebotomine sand flies and is endemic in 98 countries, with over 350 million people being at risk (1). Environmental, demographic, and human behavioral factors and coinfections contribute to the changing epidemiology of the disease and to its recent worldwide spread. Clinical manifestations are divided into visceral leishmaniasis (VL; or kala-azar) (2) and the forms cutaneous leishmaniasis (CL) and mucocutaneous leishmaniasis (ML). CL remains the most common form of leishmaniasis both in general and in international travelers (3–5).
The drugs available for the treatment of VL and severe CL include pentavalent antimonials (SbV), deoxycholate, amphotericin B (AMB), and miltefosine (MIL), all of which have high levels of toxicity and/or require long-duration treatment schedules (5, 6). Moreover, with the exception of MIL, all these drugs must be administered by the parenteral route (7).
In the last decade, resistance to SbV has increased mainly due to low rates of compliance with the treatment schedule. In areas where the parasites are resistant to SbV, AMB is commonly used as it is very active but requires a month of hospitalization for monitoring of renal function. MIL is highly active for the treatment of VL and well tolerated, but its potential teratogenicity cannot be ruled out. Paromomycin (PM) is actually in clinical trials for the treatment of leishmaniasis: it is effective, safe, and very cheap, but 21 intramuscular (i.m.) injections are required. Other oral drugs include azoles and allopurinol, but they did not reach the standards of efficacy or safety. On the basis of the available information, nowadays, a single dose of liposomal AMB is the best available therapy for VL in India, as it is more effective and safer than any other treatment. However, liposomal AMB seems to be less effective in Africa and for the treatment of non-self-curing CL and ML (8, 9). Moreover, there are still the high price and heat instability difficulties in its wide and safe distribution to primary health care centers. Therefore, better treatments for leishmaniasis diseases are needed, and they must be safer, cheaper, and easier to administer than the current drugs.
Recently, enzymes that are involved in evading the damage caused by oxidative stress and that present substantial biochemical and structural differences from their human counterparts have been reported to be interesting targets for novel antileishmanial drugs (10). In this sense, the iron superoxide dismutase (Fe-SOD), an exclusive antioxidant defense of trypanosomatids, is of special importance (11).
On the other hand, it is well-known that several of the most effective antiprotozoal agents were originally developed as anticancer drugs. Thus, among the compounds active against leishmaniasis, some antitumor drugs, such as cisplatin (12), miltefosine (13), and tamoxifen (14), can be found.
In addition, the research activity of our group is focused on the synthesis of new selenium (Se) compounds as antitumor agents, and we have intensively studied their mechanisms of action, with some of them being found to be antioxidants (15–20). Moreover, growing evidence suggests a connection between Se and parasites, particularly trypanosomatids (21–24).
In the light of the information mentioned above, different Se chemical entities with proven antiproliferative activity against cancer (16, 25, 26) were chosen for testing as leishmanicidal agents and were found to exhibit potent and selective effects against some Leishmania species (27–30). Due to their antioxidant activity, the Fe-SOD enzyme present in Leishmania spp. might be a plausible and good target for these compounds.
Encouraged by the promising results, we postulated that Se moieties can be central scaffolds for designing new compounds for the treatment of infections caused by leishmania. In this work, we report on the in vitro leishmanicidal activities (against Leishmania infantum and L. braziliensis) of 48 selenocyanates and diselenides, as well as their toxicity against macrophages, using meglumine antimonate (Glucantime) as a reference drug. 1H nuclear magnetic resonance (NMR) analysis of the nature and percentage of the excreted metabolites was performed to obtain information about the inhibitory effect of our compounds on the glycolytic pathway, since this represents the prime energy source of the parasite. Finally, the results of an evaluation of their effectiveness as putative inhibitors of Fe-SOD in relation to human CuZn-SOD are also presented.
RESULTS
Chemistry.
The 48 compounds described in this work were synthesized in the Department of Organic and Pharmaceutical Chemistry using previously published protocols (29–33). Their physical characteristics and spectroscopic data are in agreement with the published data and are available in the supplemental material.
Biological evaluation. (i) In vitro antileishmanial evaluation.
In the first step, we assayed the in vitro biological activities of Se compounds 1 to 48 (Fig. 1) against both the extra- and intracellular forms of two significant species of Leishmania: L. infantum (Table 1) and L. braziliensis (Table 2).
FIG 1.

Chemical structures of the Se compounds presented in this work. Comp., compound.
TABLE 1.
In vitro activities, toxicities, and SIs of the Se derivatives for extra- and intracellular forms of Leishmania infantum
| Compound | IC50a (μM) for: |
SId |
|||
|---|---|---|---|---|---|
|
In vitro activityb |
Macrophage toxicityc | Promastigote forms | Amastigote forms | ||
| Promastigote forms | Amastigote forms | ||||
| Meglumine antimonate | 18.0 ± 3.1 | 24.2 ± 2.6 | 15.2 ± 1.0 | 0.8 | 0.6 |
| 1 | 44.1 ± 5.2 | 31.7 ± 2.0 | 92.6 ± 5.1 | 2 (3) | 3 (5) |
| 2 | 38.7 ± 2.9 | 23.8 ± 2.0 | 5.9 ± 0.6 | 0 (0) | 0 (0) |
| 3 | 36.5 ± 2.7 | 17.9 ± 1.4 | 61.5 ± 3.5 | 2 (2) | 3 (6) |
| 4 | 41.8 ± 4.1 | 16.9 ± 0.9 | 24.7 ± 2.2 | 0 (0) | 1 (2) |
| 5 | 23.9 ± 1.7 | 18.6 ± 1.7 | 19.6 ± 1.7 | 0 (0) | 1 (2) |
| 6 | 19.4 ± 1.6 | 10.4 ± 0.6 | 29.7 ± 2.6 | 1 (2) | 3 (5) |
| 7 | 35.9 ± 3.5 | 28.9 ± 2.2 | 44.8 ± 3.0 | 1 (1) | 2 (3) |
| 8 | 0.7 ± 0.2 | 3.7 ± 0.6 | 132.8 ± 7.5 | 190 (237) | 36 (60) |
| 9 | 13.8 ± 0.9 | 8.3 ± 0.4 | 51.3 ± 4.1 | 4 (5) | 6 (10) |
| 10 | 2.1 ± 0.5 | 4.6 ± 1.0 | 139.9 ± 8.3 | 67 (83) | 30 (51) |
| 11 | 1.9 ± 0.3 | 3.8 ± 0.7 | 62.9 ± 4.4 | 33 (41) | 17 (28) |
| 12 | 23.8 ± 1.1 | 16.4 ± 1.3 | 8.2 ± 0.7 | 0 (0) | 0 (0) |
| 13 | 3.5 ± 0.6 | 6.1 ± 0.3 | 10.1 ± 0.9 | 3 (4) | 1.6 (3) |
| 14 | 28.5 ± 1.9 | 14.8 ± 0.9 | 9.5 ± 1.1 | 0 (0) | 1 (1) |
| 15 | 0.8 ± 0.1 | 2.7 ± 0.3 | 49.4 ± 3.5 | 62 (77) | 18 (30) |
| 16 | 23.7 ± 2.6 | 11.7 ± 0.8 | 19.2 ± 0.7 | 0 (0) | 2 (3) |
| 17 | 16.7 ± 0.8 | 10.3 ± 0.7 | 19.3 ± 1.2 | 1 (1) | 2 (3) |
| 18 | 3.6 ± 0.7 | 5.5 ± 0.6 | 14.7 ± 1.2 | 4 (5) | 2.6 (4) |
| 19 | 41.9 ± 2.5 | 18.7 ± 1.3 | 70.0 ± 4.7 | 2 (2) | 4 (6) |
| 20 | 2.7 ± 0.8 | 3.7 ± 0.4 | 29.5 ± 2.4 | 11 (14) | 8 (13) |
| 21 | 1.2 ± 0.2 | 0.9 ± 0.0 | 8.3 ± 0.4 | 7 (9) | 9 (14) |
| 22 | 37.8 ± 2.5 | 17.5 ± 1.2 | 27.4 ± 0.9 | 0 (0) | 2 (3) |
| 23 | 47.0 ± 5.3 | 19.1 ± 1.6 | 60.3 ± 4.7 | 1 (1) | 3 (5) |
| 24 | 6.5 ± 0.7 | 3.6 ± 0.5 | 11.8 ± 0.8 | 2 (2) | 3.3 (5) |
| 25 | 18.4 ± 0.5 | 14.8 ± 0.7 | 72.4 ± 5.5 | 4 (5) | 5 (8) |
| 26 | 6.6 ± 0.4 | 4.1 ± 0.7 | 47.9 ± 3.1 | 7 (9) | 11.7 (19) |
| 27 | 28.6 ± 2.5 | 20.5 ± 1.7 | 112.3 ± 7.3 | 4 (5) | 11 (18) |
| 28 | 83.6 ± 6.8 | 37.8 ± 2.4 | 74.2 ± 6.6 | 0 (0) | 2 (3) |
| 29 | 7.1 ± 0.8 | 4.2 ± 1.0 | 22.7 ± 1.6 | 3 (4) | 5.4 (9) |
| 30 | 26.7 ± 2.0 | 18.7 ± 1.1 | 64.0 ± 3.0 | 2 (3) | 3 (6) |
| 31 | 16.8 ± 1.2 | 16.8 ± 0.9 | 185.9 ± 81.6 | 11 (14) | 11 (18) |
| 32 | 16.5 ± 2.0 | 13.3 ± 0.8 | 44.6 ± 2.4 | 3 (3) | 3 (6) |
| 33 | 7.9 ± 0.6 | 4.9 ± 0.8 | 44.1 ± 3.7 | 6 (7) | 9.0 (15) |
| 34 | 19.7 ± 1.2 | 15.2 ± 1.1 | 34.8 ± 2.4 | 2 (2) | 2 (4) |
| 35 | 3.9 ± 0.7 | 4.3 ± 0.6 | 41.8 ± 2.7 | 11 (13) | 10 (16) |
| 36 | 23.3 ± 1.7 | 21.6 ± 1.3 | 59.3 ± 3.7 | 2 (3) | 3 (5) |
| 37 | 4.8 ± 1.2 | 2.6 ± 0.2 | 17.3 ± 0.7 | 4 (4) | 6.6 (11) |
| 38 | 13.6 ± 0.8 | 8.9 ± 0.4 | 78.4 ± 5.7 | 6 (7) | 9 (15) |
| 39 | 36.5 ± 3.4 | 30.0 ± 2.2 | 50.7 ± 3.8 | 1 (2) | 2 (3) |
| 40 | 25.8 ± 2.7 | 17.7 ± 1.4 | 36.2 ± 2.4 | 1 (2) | 2 (3) |
| 41 | 19.0 ± 1.1 | 21.8 ± 1.2 | 9.6 ± 1.3 | 0 (0) | 0 (1) |
| 42 | 2.5 ± 0.6 | 3.2 ± 0.3 | 18.7 ± 0.6 | 7 (9) | 5.8 (10) |
| 43 | 8.9 ± 0.7 | 5.8 ± 1.1 | 28.5 ± 3.8 | 3 (4) | 4.9 (8) |
| 44 | 4.4 ± 0.3 | 3.9 ± 0.6 | 33.4 ± 3.0 | 8 (9) | 8.6 (14) |
| 45 | 3.7 ± 0.5 | 2.7 ± 0.3 | 61.1 ± 4.1 | 16 (21) | 23 (38) |
| 46 | 8.6 ± 0.8 | 6.3 ± 1.3 | 29.7 ± 1.8 | 3 (4) | 4.7 (8) |
| 47 | 1.1 ± 0.5 | 2.7 ± 0.5 | 12.6 ± 0.8 | 11 (14) | 5 (8) |
| 48 | 1.6 ± 0.1 | 1.4 ± 0.2 | 52.8 ± 3.61 | 33 (41) | 38 (63) |
Results are the averages from four separate determinations.
IC50, the concentration required to give 50% inhibition, calculated by linear regression analysis from the values of the equilibrium constants (Kc) at the concentrations employed (1, 10, 25, and 200 μM).
Toxicity against J774.2 macrophages after 72 h of culture.
SI, selectivity index, which is calculated as the IC50 for macrophage toxicity/IC50 for activity against the extracellular or intracellular form of the parasite. The number of times that the SI of the compound exceeded the SI of the reference drug is provided in parentheses.
TABLE 2.
In vitro activities, toxicities, and SIs for the Se derivatives on extra- and intracellular forms of Leishmania braziliensis
| Compound | IC50a (μM) for: |
SId |
|||
|---|---|---|---|---|---|
|
In vitro activityb |
Macrophage toxicityc | Promastigote forms | Amastigote forms | ||
| Promastigote forms | Amastigote forms | ||||
| Meglumine antimonate | 25.6 ± 1.7 | 30.4 ± 2.6 | 15.2 ± 1.0 | 0.6 | 0.6 |
| 1 | 43.9 ± 3.7 | 31.8 ± 2.5 | 92.6 ± 5.1 | 2 (3) | 3 (5) |
| 2 | 21.7 ± 2.3 | 16.8 ± 1.1 | 5.9 ± 0.6 | 0 (0) | 0 (0) |
| 3 | 20.8 ± 1.2 | 17.9 ± 1.3 | 61.5 ± 3.5 | 3 (5) | 3 (6) |
| 4 | 37.53.5 | 26.4 ± 1.6 | 24.7 ± 2.2 | 0 (0) | 1 (2) |
| 5 | 15.8 ± 0.6 | 9.6 ± 0.4 | 19.6 ± 1.7 | 1 (2) | 2 (3) |
| 6 | 16.8 ± 1.1 | 16.8 ± 0.9 | 29.7 ± 2.6 | 2 (3) | 2 (3) |
| 7 | 30.7 ± 1.6 | 22.7 ± 1.6 | 44.8 ± 3.0 | 1 (2) | 2 (3) |
| 8 | 1.3 ± 0.3 | 3.8 ± 0.6 | 132.8 ± 7.5 | 102 (170) | 35 (58) |
| 9 | 10.8 ± 0.7 | 9.4 ± 1.4 | 51.3 ± 4.1 | 5 (8) | 5.4 (9) |
| 10 | 4.4 ± 0.1 | 5.7 ± 0.8 | 139.9 ± 8.3 | 32 (53) | 24 (41) |
| 11 | 1.6 ± 0.4 | 2.8 ± 0.5 | 62.9 ± 4.4 | 39 (65) | 22 (37) |
| 12 | 22.7 ± 1.8 | 15.8 ± 0.8 | 8.2 ± 0.7 | 0 (0) | 0 (1) |
| 13 | 4.5 ± 0.4 | 5.8 ± 1.5 | 10.1 ± 0.9 | 2 (3) | 1.7 (3) |
| 14 | 20.7 ± 1.3 | 19.8 ± 1.3 | 9.5 ± 1.1 | 0 (0) | 0 (0) |
| 15 | 0.6 ± 0.1 | 1.4 ± 0.3 | 49.4 ± 3.5 | 82 (137) | 35 (59) |
| 16 | 16.9 ± 1.2 | 11.7 ± 0.6 | 19.2 ± 0.7 | 1 (2) | 2 (3) |
| 17 | 10.5 ± 1.0 | 8.5 ± 2.0 | 19.3 ± 1.2 | 2 (3) | 2.3 (4) |
| 18 | 2.1 ± 0.1 | 1.9 ± 0.1 | 14.7 ± 1.2 | 7 (12) | 7.7 (13) |
| 19 | 8.9 ± 0.6 | 10.4 ± 1.3 | 70.0 ± 4.7 | 8 (13) | 7 (11) |
| 20 | 1.3 ± 0.3 | 1.1 ± 0.2 | 29.5 ± 2.4 | 23 (38) | 27 (45) |
| 21 | 2.0 ± 0.3 | 1.3 ± 0.2 | 8.3 ± 0.4 | 4 (7) | 6.4 (11) |
| 22 | 9.6 ± 1.2 | 7.5 ± 1.2 | 27.4 ± 0.9 | 3 (5) | 3.6 (6) |
| 23 | 20.8 ± 2.5 | 17.7 ± 0.8 | 60.3 ± 4.7 | 3 (5) | 3 (6) |
| 24 | 5.7 ± 0.4 | 4.3 ± 0.9 | 11.8 ± 0.8 | 2 (3) | 2.7 (5) |
| 25 | 13.9 ± 2.0 | 10.6 ± 1.7 | 72.4 ± 5.5 | 5 (9) | 7 (11) |
| 26 | 4.3 ± 0.3 | 4.8 ± 0.3 | 47.9 ± 3.1 | 11 (19) | 10 (17) |
| 27 | 18.4 ± 1.2 | 12.8 ± 0.4 | 112.3 ± 7.3 | 6 (10) | 9 (15) |
| 28 | 37.4 ± 3.4 | 23.6 ± 1.8 | 74.2 ± 6.6 | 2 (3) | 3 (5) |
| 29 | 6.6 ± 0.5 | 4.2 ± 0.8 | 22.7 ± 1.6 | 3 (6) | 5.4 (9) |
| 30 | 20.5 ± 0.9 | 13.8 ± 0.8 | 64.0 ± 3.0 | 3 (5) | 5 (8) |
| 31 | 20.9 ± 2.6 | 19.7 ± 1.2 | 185.9 ± 81.6 | 9 (15) | 9 (16) |
| 32 | 13.7 ± 0.7 | 6.8 ± 0.2 | 44.6 ± 2.4 | 3 (5) | 6 (11) |
| 33 | 6.9 ± 0.6 | 4.9 ± 0.7 | 44.1 ± 3.7 | 6 (11) | 9.0 (15) |
| 34 | 14.5 ± 1.8 | 10.6 ± 1.7 | 34.8 ± 2.4 | 2 (4) | 3 (5) |
| 35 | 2.5 ± 0.7 | 2.4 ± 0.6 | 41.8 ± 2.7 | 17 (28) | 17 (29) |
| 36 | 17.8 ± 0.7 | 12.2 ± 0.4 | 59.3 ± 3.7 | 3 (5) | 5 (8) |
| 37 | 3.7 ± 0.5 | 2.4 ± 0.4 | 17.3 ± 0.7 | 5 (8) | 7.2 (12) |
| 38 | 15.8 ± 0.9 | 9.9 ± 0.5 | 78.4 ± 5.7 | 5 (8) | 8 (13) |
| 39 | 31.5 ± 3.5 | 17.6 ± 1.4 | 50.7 ± 3.8 | 2 (3) | 3 (5) |
| 40 | 24.7 ± 1.7 | 20.1 ± 2.3 | 36.2 ± 2.4 | 2 (2) | 2 (3) |
| 41 | 11.9 ± 1.3 | 10.8 ± 0.5 | 9.6 ± 1.3 | 0 (0) | 0 (0) |
| 42 | 4.7 ± 0.2 | 3.3 ± 0.6 | 18.7 ± 0.6 | 4 (7) | 5.7 (9) |
| 43 | 4.7 ± 0.6 | 3.7 ± 0.5 | 28.5 ± 3.8 | 6 (10) | 7.7 (13) |
| 44 | 2.8 ± 0.8 | 4.3 ± 0.8 | 33.4 ± 3.0 | 8 (13) | 10 (17) |
| 45 | 1.7 ± 0.1 | 2.0 ± 0.5 | 61.1 ± 4.1 | 36 (60) | 30 (51) |
| 46 | 3.3 ± 0.4 | 3.4 ± 0.7 | 29.7 ± 1.8 | 9 (15) | 9 (15) |
| 47 | 0.8 ± 0.2 | 1.0 ± 0.1 | 12.6 ± 0.8 | 16 (26) | 13 (21) |
| 48 | 0.9 ± 0.1 | 1.8 ± 0.5 | 52.8 ± 3.6 | 59 (98) | 29 (49) |
Results are the averages from four separate determinations.
IC50, the concentration required to give 50% inhibition, calculated by linear regression analysis from the values of the equilibrium constants (Kc) at the concentrations employed (1, 10, 25, and 200 μM).
Toxicity against J774.2 macrophages after 72 h of culture.
SI, selectivity index, which is calculated as the IC50 for macrophage toxicity/IC50 for activity against the extracellular or intracellular form of the parasite. The number of times that the compound SI exceeded the reference drug SI is provided in parentheses.
Extracellular forms are more commonly used due to the ease of working with them, but the activities of compounds against this form are less indicative of leishmanicidal activity. The use of intracellular forms is more cumbersome but provides more accurate results, as promastigotes are converted to amastigotes in vertebrate host cells (34, 35). Intracellular assays were performed by infecting macrophage cells with promastigotes, which transformed into amastigotes within 1 day after infection.
Table 1 shows the 50% inhibitory concentration (IC50) values obtained after 72 h of exposure when the activities of compounds 1 to 48 against extra- and intracellular forms of L. infantum were tested. IC50 values for toxicity against J774.2 macrophages after 72 h were also calculated to establish the selectivity indexes (SIs). The results obtained for the reference drug, meglumine antimonate, were included in all cases for comparison. Biological data evidenced that half of the screened compounds (compounds 8, 9, 10, 11, 13, 15, 17, 18, 20, 21, 24, 26, 29, 33, 35, 37, 38, 42, 43, 44, 45, 46, 47, and 48) showed high levels of bioactivity against L. infantum, presenting greater potency than the reference drug, meglumine antimonate, against both forms (IC50s, 18.0 μM for promastigotes and 24.2 μM for amastigotes).
In order to obtain a more accurate picture of the features commented on above, we show in Table 1 the SI values calculated from the IC50 data, since they are very illustrative of the in vitro potential of the tested compounds with respect to that of the reference drug. The number of times that the SI of each compound exceeded the SI of meglumine antimonate is also shown in parentheses. The differences between meglumine antimonate and the tested compounds are clearly revealed. Twelve compounds (compounds 8, 10, 11, 15, 20, 21, 26, 31, 35, 44, 45, and 48) presented notable selectivity index values (SIs > 7) for both forms. These derivatives exhibited substantially better SI values than the reference drug for L. infantum, and in the most remarkable case, the SI of compound 8 for the intracellular form of L. infantum exceeded that of meglumine antimonate by 237-fold, a relevant data point, which was by far the best SI value.
Six of the compounds (compounds 8, 10, 11, 15, 45, and 48) stood out as the most active and selective molecules, showing IC50s lower than 5 μM along with SIs higher than 15, and were selected for further studies.
Considering their activity against L. braziliensis (Table 2), the results recurred in the two species and in both extra- and intracellular forms, since compounds 8, 10, 11, 15, 45, and 48 showed substantially better results, in terms of activity and selectivity, than the rest of the tested compounds in all cases. For example, the SI of compound 8 exceeded that of the reference drug by 237- and 60-fold for the extra- and intracellular forms of L. infantum, respectively, and by 170- and 58-fold for the extra- and intracellular forms of L. braziliensis, respectively (Table 2). Compound 10 had an SI similar to that of derivative 8, with the respective values obtained being 83- and 51-fold for L. infantum (Table 1) and 53- and 41-fold for L. braziliensis (Table 2); compounds 11 and 15 were equally effective against the two species of Leishmania and against both the extra- and intracellular forms. Compound 11 had 41- and 28-fold increases in the SIs for the extra- and intracellular forms ofL. infantum, respectively (Table 1), along with 65- and 37-fold in SIs for the extra- and intracellular forms L. braziliensis, respectively (Table 2). On the other hand, compounds 45 and 48 were more selective for the extracellular form of L. braziliensis (Table 2). The rest of the tested compounds presented similar values in both species, except compound 20, which was effective against L. braziliensis but not against L. infantum.
(ii) Leishmanicidal activity in infected macrophages.
In order to gain better insight into the activities of the most active compounds, their effects on the infectivity and intracellular replication of amastigotes were subsequently determined. Macrophage cells were grown and infected with promastigotes in the stationary phase. The parasites invaded cells and underwent morphological conversion to amastigotes within 1 day after infection. On day 10, the rate of host cell infection reached its maximum (control experiment). We used the IC25 of each product as the test dosage. Different authors have claimed that compounds with SI values less than 20-fold those of the reference drug should be discarded as candidates for more advanced leishmanicidal tests due to their poor selectivity against mammalian cells (36). Therefore, we selected the compounds that fulfilled this requirement (compounds 8, 10, 11, 15, 45, and 48 for L. infantum and those compound plus compound 20 for L. braziliensis).
As shown in Fig. 2A and B, when the selected compounds were added to macrophages infected with L. infantum promastigotes, the infection rate decreased significantly with respect to that for the control; furthermore, the six compounds (compounds 8, 10, 11, 15, 45, and 48) were also remarkably more effective in decreasing infectivity than meglumine antimonate. A measure of the average number of amastigotes per infected macrophage (Fig. 2C and D) led to the same conclusion: all six compounds were more effective than meglumine antimonate.
FIG 2.

Effects of Se derivatives 8, 10, 11, 15, 45, and 48 on the rates of infection by and growth rates of L. infantum. (A, B) Rates of infection; (C, D) mean numbers of amastigotes of L. infantum per infected J774.2 macrophage cell (when the compounds were tested at the IC25). Values are the means from three separate experiments. Gluc., Glucantime (meglumine antimonate).
Infection rates (Fig. 3A and B) and the reduction in amastigote numbers (Fig. 3C and D) obtained with L. braziliensis also showed that, in both cases, these six compounds along with compound 20 were clearly more effective than meglumine antimonate, and also in both cases, the order of effectiveness of the compounds was 48 > 45 > 8 > 11 > 10 > 15 > 20 > meglumine antimonate, since the reductions in the infectivity rates calculated were 71%, 62%, 60%, 53%, 50%, 47%, 40%, and 28%, respectively. The decreases in amastigote numbers were 70%, 66%, 60%, 53%, 48%, 48%, 43%, and 30% for compounds 48, 45, 8, 11, 10, 15, and 20 and meglumine antimonate, respectively. When these results are taken into account, it can be concluded that all compounds analyzed were shown to be substantially more active than meglumine antimonate against the two Leishmania species tested.
FIG 3.

Effects of Se derivatives 8, 10, 11, 15, 20, 45, and 48 on the rates of infection by and growth rates of Leishmania braziliensis. (A, B) Rates of infection; (C, D) mean numbers of amastigotes of L. braziliensis per infected J774 A.2 macrophage cell (when the compounds were tested at the IC25). Values are the means from three separate experiments.
(iii) Studies on the mechanism of action. (a) Metabolite excretion effect.
Since trypanosomatids are unable to completely degrade glucose to CO2 under aerobic conditions, they excrete much of the hexose skeleton into the medium as partially oxidized fragments in the form of fermented metabolites. The nature and percentages of those excretion products depend on the pathway used for glucose metabolism by each species (37).
The final products of glucose catabolism in Leishmania are usually CO2, succinate, acetate, d-lactate, l-alanine, and, in some cases, ethanol (37). Among them, succinate is the most relevant, because its main role is to maintain the glycosomal redox balance, allowing the reoxidation of NADH produced in the glycolytic pathway. Succinic fermentation requires only half of the phosphoenolpyruvate produced to maintain the NAD+/NADH balance, and the remaining pyruvate is converted inside the mitochondrion and the cytosol into acetate, d-lactate, l-alanine, or ethanol, according to the degradation pathway followed by a specific species (38).
We report below that the most active compounds caused great damage to the mitochondria of the parasites of both Leishmania species, so that their highly disruptive action presumably affected their glucose metabolism and, consequently, modified the percentages of the final excretion products formed. Therefore, we registered the 1H NMR spectra of promastigotes from the L. infantum and L. braziliensis species after treatment with the selected compounds, and the final excretion products were identified qualitatively and quantitatively. The spectra obtained were compared with those from promastigotes maintained in cell-free medium (control) for 4 days after inoculation with the parasites. The variations in the areas of the signals corresponding to the most significant catabolites are displayed. The expected presence of acetate, d-lactate, l-alanine, and succinate was confirmed in the control experiments performed on both species, and the major metabolite was succinate in all cases, in agreement with data reported in the literature (38). However, relevant differences were found in parasites treated with the derivatives, whereas the presence of meglumine antimonate did not lead to significant alterations in the energetic metabolism (data not shown).
Compounds 10, 11, and 20 induced increases in the levels of succinate production in L. braziliensis (Fig. 4 and Table 3), as can be seen by measuring the area under the succinate peak determined by 1H NMR. In the case of L. infantum, all the compounds increased the level of succinate production, although product 8 did so to a lesser extent than the other compounds (Fig. 5 and Table 4). In fact, this succinate augmentation is in agreement with transmission electron microscopy (TEM) evidence, indicating that these derivatives severely damage the mitochondrial structures of the parasites (data not shown).
FIG 4.
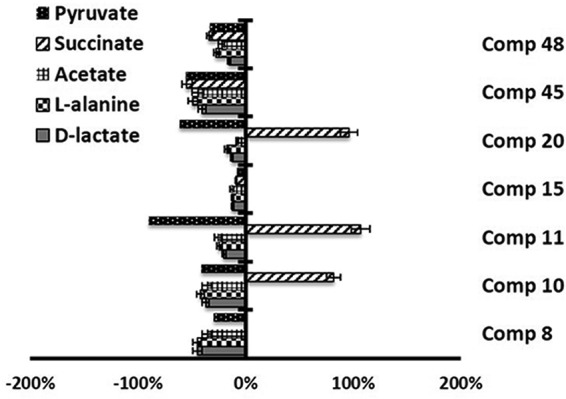
Variation in the area of the peaks (in percent) corresponding to metabolites excreted by L. braziliensis forms in the presence of compounds 8, 10, 11, 15, 20, 45, and 48 at their IC25s compared to the area for a control sample, determined by 1H NMR.
TABLE 3.
Variation in areas of peaks corresponding to catabolites excreted by L. braziliensis promastigotes in presence of compounds with respect to those in control tests
| Compound | % variation in area of peaks |
||||
|---|---|---|---|---|---|
| d-Lactate | l-Alanine | Acetate | Pyruvate | Succinate | |
| Control | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 8 | −44.7 | −44.9 | −36.2 | −29.4 | −0.2 |
| 10 | −36.9 | −41.7 | −36.1 | −40.8 | 81.7 |
| 11 | −20.0 | −24.2 | −25.7 | −89.6 | 107.1 |
| 15 | −12.1 | −11.9 | −12.7 | −7.9 | −8.6 |
| 20 | −12.7 | −17.6 | −7.7 | −61.5 | 96.2 |
| 45 | −40.4 | −49.0 | −44.5 | −55.1 | −54.4 |
| 48 | −15.5 | −27.4 | −22.4 | −33.3 | −33.6 |
FIG 5.
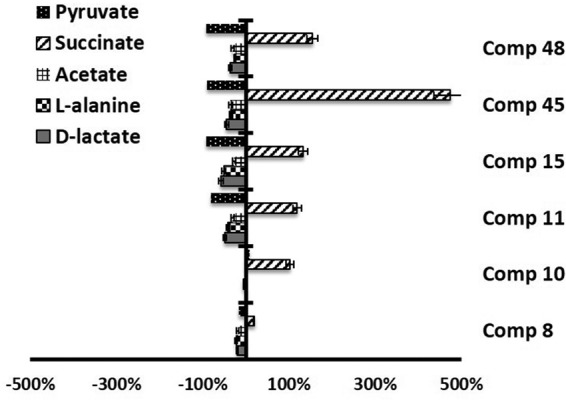
Variation in the area of the peaks (in percent) corresponding to metabolites excreted by L. infantum promastigote forms in the presence of compounds 8, 10, 11, 15, 45, and 48 at their IC25s compared to the area for a control sample, determined by 1H NMR.
TABLE 4.
Variation in areas of peaks corresponding to catabolites excreted by L. infantum promastigotes in presence of compounds with respect to those in control tests
| Compound | % variation in area of peaks |
||||
|---|---|---|---|---|---|
| d-Lactate | l-Alanine | Acetate | Pyruvate | Succinate | |
| Control | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 8 | −20.4 | −22.5 | −18.8 | −17.0 | 18.6 |
| 10 | 1.8 | −4.8 | 1.1 | 8.3 | 102.1 |
| 11 | −48.8 | −41.8 | −30.6 | −80.7 | 118.9 |
| 15 | −58.1 | −50.7 | −27.8 | −91.3 | 132.1 |
| 45 | −44.6 | −34.2 | −35.4 | −90.3 | 474.1 |
| 48 | −36.0 | −25.7 | −30.3 | −91.1 | 153.6 |
(b) SOD enzymatic inhibition in Leishmania parasites and in human erythrocytes.
These results prompted us to evaluate the inhibitory effect of the most promising compounds on the activity of Fe-SOD from L. infantum and L. braziliensis at concentrations ranging from 0.5 to 100 μM.
To do so, we used the promastigote forms of both species, which excrete Fe-SOD when cultured in a medium lacking inactive fetal bovine serum (39). The inhibition data obtained are shown in Fig. 6A to D, and the corresponding IC50s are included for easier evaluation of the graphs displayed. For comparison, the graphs in Fig. 7 show the effects of the same compounds on CuZn-SOD obtained from human erythrocytes. The most remarkable result was the significant inhibitory effect of the highly leishmanicidal compounds 8, 10, and 11 found on Fe-SOD from both species and compound 20 in the case of L. braziliensis, whereas their inhibition of human CuZn-SOD was clearly lower.
FIG 6.

In vitro inhibition (in percent) of Fe-SOD from L. infantum (A and B) and L. braziliensis (C and D) promastigote forms by the Se derivatives. The differences between the activities of the control homogenate and homogenates incubated with the indicated compounds were obtained according to the Newman-Keuls test. The concentrations in the keys are IC50s, which are the concentrations required to give 50% inhibition and which were calculated by linear regression analysis from the values of the equilibrium constants (Kc) at the concentrations employed (0.5 to 100 μM). The values are averages from three different determinations.
FIG 7.

In vitro inhibition (in percent) of CuZn-SOD from human erythrocytes by the Se derivatives. The differences between the activities of the control homogenate and homogenates incubated with the indicated compounds were obtained according to the Newman-Keuls test. The concentrations in the keys are IC50s, which are the concentrations required to give 50% inhibition and which were calculated by linear regression analysis from the values of the equilibrium constants (Kc) at the concentrations employed (0.5 to 100 μM). The values are averages from three different determinations.
If we consider the IC50 calculated for L. infantum, the levels of inhibition of Fe-SOD by compounds 8, 10, and 11 were, respectively, 5-, 19- and 16-fold higher than the levels of inhibition of CuZn-SOD, and in the case of L. braziliensis, the levels of inhibition by compounds 8, 10, and 11 were 4-, 34-, and 0-fold higher, respectively, and the level of inhibition by compound 20 was 9-fold higher.
DISCUSSION
The present results document the leishmanicidal effects of 48 Se derivatives. After taking into consideration the IC50 data shown in Tables 1 and 2, it can be seen that compounds 8, 10, 11, 15, 45, and 48 stand out as the molecules most active and selective against the two Leishmania spp. studied (L. infantum and L. braziliensis). Furthermore, they were much more active than meglumine antimonate against both the extra- and intracellular forms of the parasite. Regarding the evaluation of cytotoxicity against macrophage cells, it was also shown that these six compounds along with compound 20 were clearly more selective than meglumine antimonate against L. infantum and L. braziliensis. Attending to other studies to elucidate the possible mechanisms of action, it should be noted that these compounds produced great alterations in glucose metabolism in the studied species, according to the 1H NMR data for catabolites. It is feasible that the small size of these compounds allows them to easily penetrate the cristae (tubular invaginations of the inner mitochondrial membrane), leading to subsequent changes in the metabolic pathway, although further studies are required to prove this point. In addition, these compounds led to important levels of Fe-SOD inhibition. All these data seem to confirm some kind of relation between parasiticidal activity and inhibition of Fe-SOD, in accordance with results described in a previous work (39). At another level, Fe-SOD inhibition could also be related to the catabolic changes discussed above, since a mitochondrial malfunction might originate from the redox stress produced by inhibition of the mitochondrion-resident Fe-SOD enzyme (40). Since Fe-SOD is a specific chemotherapy target also present in mitochondria, this fact would agree with the hypothesis presented above that these compounds might have a greater ability to pass through the mitochondrial membrane.
From all the results detailed above, we conclude that the Se derivatives (compounds 8, 10, 11, 15, 45, and 48) show interesting in vitro leishmanicidal activity which seems to be related to their ability to inhibit the parasite Fe-SOD. Interestingly, the 1H NMR data for the catabolites are indicative of substantial alterations in glucose metabolism in the studied species. In fact, this succinate augmentation is in agreement with transmission electron microscopy (TEM) evidence indicating that these derivatives severely damage the mitochondrial structures of the parasites (data not shown). A graphical summary of the conclusions drawn from this work is presented in Fig. 8. On that basis, we think that the activities of these compounds fulfill the requirements needed to justify a more detailed investigation of the nature of the mechanisms involved in their patterns of activity and, furthermore, that they could be considered candidates for the study of their antiparasitic activities at a higher level. Finally, it is worth mentioning that due to their different mechanisms of action, combined therapies should be considered to obtain improved efficacy.
FIG 8.

Schematic illustration of conclusions.
MATERIALS AND METHODS
Chemistry.
The Se compounds reported herein can be categorized into three groups (Fig. 1): selenocyanate derivatives, or group I (compounds 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27, 28, 29, 30, and 32); the diselenide analogs of the selenocyanate derivatives mentioned above, or group II (compounds 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, and 31); and the diphenyldiselenide-derived compounds bearing different sulfonamides substituted on the para position, or group III (compounds 33 to 48).
The group I compounds were obtained by the nucleophilic substitution of the halogen atom of the corresponding alkyl or benzyl halides by a selenocyanate group using potassium selenocyanate (KSeCN) under reflux conditions according to previously reported methods (29, 30). Thus, the corresponding organic halide was refluxed for 2 to 4 h in acetone with potassium selenocyanate. The resulting precipitate was filtered off, and the filtrate was evaporated under vacuum. Then, the residue was treated with water (2 times with 50 ml each time), filtered, and dried. The solid obtained was purified either by recrystallization or by washing with different solvents.
Group II derivatives were synthesized by reduction of the corresponding selenocyanate from group I using sodium borohydride following the previously described procedure (29, 30). In order to do so, sodium borohydride was added with caution to a stirring solution of the corresponding selenocyanate from group I in absolute ethanol. After 2 h of reaction, the solvent was removed under reduced pressure and the residue was treated with water (50 ml). The mixture was extracted with dichloromethane (3 times with 50 ml each time); the organic fractions were collected, dried over sodium sulfate, and filtered off; and the solvent was removed under vacuum by rotary evaporation. The resulting solid was recrystallized using ethanol.
Finally, targeted compounds of group III were prepared from 4,4′-diselanediylaniline coupled to different substituted sulfonyl chlorides (31). Briefly, the corresponding sulfonyl chloride was added to a mixture of 4,4′-diselanediylaniline and dry ether, and the reaction mixture was stirred at room temperature for 48 to 72 h under a nitrogen atmosphere. The precipitate formed in the reaction mixture was filtered, dried, and purified accordingly by recrystallization or washing with different solvents.
Biological evaluation. (i) Determination of the potential leishmanicidal activity and toxicity of new compounds. (a) Axenic cultures.
The parasites were cultivated in axenic cultures using standard conditions for the promastigote and amastigote forms of L. infantum (MCAN/ES/2001/UCM-10) and L. braziliensis (MHOM/BR/1975/M2904) strains (41, 42).
(b) Cytotoxicity tests.
The viability of J774.2 macrophages and Vero cells was determined by use of a Coulter cell counter using previously published standardized methods (39).
(c) In vitro activity assays.
For extracellular forms (promastigote assay), promastigotes were collected in the exponential growth phase and distributed in culture trays (with 24 wells) at a final concentration of 5 × 104 parasites/well according to previously reported methods (39).
For intracellular forms (amastigote assay), macrophages of the J774.2 macrophage line (European Collection of Cell Cultures [ECACC] number 91051511) were cultured in 24-well microplates with round coverslips. The assays were performed as previously described (39).
(d) Infectivity assay.
The mammalian cells were infected in vitro with metacyclic forms of Leishmania spp. at a ratio of 10:1 according to information presented in the literature (39). At least three different assays were performed to calculate the SI of each compound, which was determined as the ratio between the IC50 obtained in macrophages and the corresponding IC50 in promastigotes.
(ii) Studies on the mechanism of action. (a) Metabolite excretion.
According to previous reports (39), cultures of extracellular forms received the selected compounds at a dose of the IC25 and were then cultured for 72 h at 28°C. Then, the supernatant was collected to detect the excreted metabolites by 1H NMR.
(b) Enzymatic inhibition of Fe-SOD.
For SOD inhibition studies, the parasites cultured were centrifuged and processed as described previously (34, 35). Iron, manganese, and copper-zinc SOD activities were determined using the method described by Beyer and Fridovich (43).
Supplementary Material
ACKNOWLEDGMENTS
We express our gratitude to the Spanish MINECO, FEDER (Consolider-Ingenio projects CSD2010-00065-2010 and CTQ2013-48917-C3-1-P), and FIMA (Fundación para la Investigación Médica Aplicada) project ISTUN-API-2011/02 for financial support. R.M.-E. is grateful for an FPU grant (FPU14/01537) from the Ministry of Education of Spain.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02546-16.
REFERENCES
- 1.Seifert K. 2011. Structures, targets and recent approaches in anti-leishmanial drug discovery and development. Open Med Chem J 5:31–39. doi: 10.2174/1874104501105010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2014. WHO fact sheet no. 375. World Health Organization, Geneva, Switzerland: Accessed 9 September 2016. [Google Scholar]
- 3.Kenneth JR, Ray CG, Ahmad N, Drew WL, Plorde JJ. 2010. Sherris medical microbiology: an introduction to infectious diseases, 5th ed McGraw-Hill, New York, NY. [Google Scholar]
- 4.Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, Alvar J, Boelaert M. 2007. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol 5:873–882. [DOI] [PubMed] [Google Scholar]
- 5.Lukes J, Mauricio IL, Schonian G, Dujardin JC, Soteriadou K, Dedet JP, Kuhls K, Tintaya KW, Jirku M, Chocholova E, Haralambous C, Pratlong F, Obornik M, Horak A, Ayala FJ, Miles MA. 2007. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc Natl Acad Sci U S A 104:9375–9380. doi: 10.1073/pnas.0703678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croft SL, Sundar S, Fairlamb AH. 2006. Drug resistance in leishmaniasis. Clin Microbiol Rev 19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouellette M, Drummelsmith J, Papadopoulou B. 2004. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug Resist Updat 7:257–266. doi: 10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Freitas-Junior LH, Chatelain E, Kim HA, Siqueira-Neto JL. 2012. Visceral leishmaniasis treatment: what do we have, what do we need and how to deliver it? Int J Parasitol Drugs Drug Resist 2:11–19. doi: 10.1016/j.ijpddr.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loiseau PM, Cojean S, Schrevel J. 2011. Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite 18:115–119. doi: 10.1051/parasite/2011182115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menna-Barreto RF, de Castro SL. 2014. The double-edged sword in pathogenic trypanosomatids: the pivotal role of mitochondria in oxidative stress and bioenergetics. Biomed Res Int 2014:614014. doi: 10.1155/2014/614014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh K, Garg G, Ali V. 2016. Current therapeutics, their problems and thiol metabolism as potential drug targets in leishmaniasis. Curr Drug Metab 17:897–919. doi: 10.2174/1389200217666160819161444. [DOI] [PubMed] [Google Scholar]
- 12.Tavares J, Ouaissi M, Ouaissi A, Cordeiro-da-Silva A. 2007. Characterization of the anti-Leishmania effect induced by cisplatin, an anticancer drug. Acta Trop 103:133–141. doi: 10.1016/j.actatropica.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Castelo Branco PV, Soares RE, de Jesus LC, Moreira VR, Alves HJ, de Castro Belfort MR, Silva VL, Ferreira Pereira SR. 2016. The antileishmanial drug miltefosine (Impavido(R)) causes oxidation of DNA bases, apoptosis, and necrosis in mammalian cells. Mutat Res Genet Toxicol Environ Mutagen 806:34–39. doi: 10.1016/j.mrgentox.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Doroodgar M, Delavari M, Doroodgar M, Abbasi A, Taherian AA, Doroodgar A. 2016. Tamoxifen induces apoptosis of Leishmania major promastigotes in vitro. Korean J Parasitol 54:9–14. doi: 10.3347/kjp.2016.54.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominguez-Alvarez E, Plano D, Font M, Calvo A, Prior C, Jacob C, Palop JA, Sanmartin C. 2014. Synthesis and antiproliferative activity of novel selenoester derivatives. Eur J Med Chem 73:153–166. doi: 10.1016/j.ejmech.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 16.Plano D, Baquedano Y, Ibanez E, Jimenez I, Palop JA, Spallholz JE, Sanmartin C. 2010. Antioxidant-prooxidant properties of a new organoselenium compound library. Molecules 15:7292–7312. doi: 10.3390/molecules15107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romano B, Plano D, Encio I, Palop JA, Sanmartin C. 2015. In vitro radical scavenging and cytotoxic activities of novel hybrid selenocarbamates. Bioorg Med Chem 23:1716–1727. doi: 10.1016/j.bmc.2015.02.048. [DOI] [PubMed] [Google Scholar]
- 18.Shaaban S, Arafat MA, Hamama WS. 2014. Vistas in the domain of organoselenocyanates. Arkivoc 2014:470–505. doi: 10.3998/ark.5550190.p008.763. [DOI] [Google Scholar]
- 19.Mecklenburg S, Shaaban S, Ba LA, Burkholz T, Schneider T, Diesel B, Kiemer AK, Roseler A, Becker K, Reichrath J, Stark A, Tilgen W, Abbas M, Wessjohann LA, Sasse F, Jacob C. 2009. Exploring synthetic avenues for the effective synthesis of selenium- and tellurium-containing multifunctional redox agents. Org Biomol Chem 7:4753–4762. doi: 10.1039/b907831b. [DOI] [PubMed] [Google Scholar]
- 20.Shaaban S, Negm A, Ashmawy AM, Ahmed DM, Wessjohann LA. 2016. Combinatorial synthesis, in silico, molecular and biochemical studies of tetrazole-derived organic selenides with increased selectivity against hepatocellular carcinoma. Eur J Med Chem 122:55–71. doi: 10.1016/j.ejmech.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Cassago A, Rodrigues EM, Prieto EL, Gaston KW, Alfonzo JD, Iribar MP, Berry MJ, Cruz AK, Thiemann OH. 2006. Identification of Leishmania selenoproteins and SECIS element. Mol Biochem Parasitol 149:128–134. doi: 10.1016/j.molbiopara.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Lobanov AV, Gromer S, Salinas G, Gladyshev VN. 2006. Selenium metabolism in Trypanosoma: characterization of selenoproteomes and identification of a Kinetoplastida-specific selenoprotein. Nucleic Acids Res 34:4012–4024. doi: 10.1093/nar/gkl541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noguchi N. 2016. Ebselen, a useful tool for understanding cellular redox biology and a promising drug candidate for use in human diseases. Arch Biochem Biophys 595:109–112. doi: 10.1016/j.abb.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 24.Souza CC, Barreto TDO, da Silva SM, Pinto AW, Figueiredo MM, Rocha OG, Cangussu SD, Tafuri WL. 2014. A potential link among antioxidant enzymes, histopathology and trace elements in canine visceral leishmaniasis. Int J Exp Pathol 95:260–270. doi: 10.1111/iep.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alcolea V, Plano D, Encio I, Palop JA, Sharma AK, Sanmartin C. 2016. Chalcogen containing heterocyclic scaffolds: new hybrids with antitumoral activity. Eur J Med Chem 123:407–418. doi: 10.1016/j.ejmech.2016.07.042. [DOI] [PubMed] [Google Scholar]
- 26.Plano D, Ibanez E, Calvo A, Palop JA, Sanmartin C. 2011. Novel library of selenocompounds as kinase modulators. Molecules 16:6349–6364. doi: 10.3390/molecules16086349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno D, Plano D, Baquedano Y, Jimenez-Ruiz A, Palop JA, Sanmartin C. 2011. Antileishmanial activity of imidothiocarbamates and imidoselenocarbamates. Parasitol Res 108:233–239. doi: 10.1007/s00436-010-2073-x. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez-Rubio C, Campbell D, Vacas A, Ibanez E, Moreno E, Espuelas S, Calvo A, Palop JA, Plano D, Sanmartin C, Nguewa PA. 2015. Leishmanicidal activities of novel methylseleno-imidocarbamates. Antimicrob Agents Chemother 59:5705–5713. doi: 10.1128/AAC.00997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plano D, Baquedano Y, Moreno-Mateos D, Font M, Jimenez-Ruiz A, Palop JA, Sanmartin C. 2011. Selenocyanates and diselenides: a new class of potent antileishmanial agents. Eur J Med Chem 46:3315–3323. doi: 10.1016/j.ejmech.2011.04.054. [DOI] [PubMed] [Google Scholar]
- 30.Baquedano Y, Alcolea V, Toro MA, Gutierrez KJ, Nguewa P, Font M, Moreno E, Espuelas S, Jimenez-Ruiz A, Palop JA, Plano D, Sanmartin C. 2016. Novel heteroaryl selenocyanates and diselenides as potent antileishmanial agents. Antimicrob Agents Chemother 60:3802–3812. doi: 10.1128/AAC.02529-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baquedano Y, Moreno E, Espuelas S, Nguewa P, Font M, Gutierrez KJ, Jimenez-Ruiz A, Palop JA, Sanmartin C. 2014. Novel hybrid selenosulfonamides as potent antileishmanial agents. Eur J Med Chem 74:116–123. doi: 10.1016/j.ejmech.2013.12.030. [DOI] [PubMed] [Google Scholar]
- 32.Shaaban S, Negm A, Sobh MA, Wessjohann LA. 2016. Expeditious entry to functionalized pseudo-peptidic organoselenide redox modulators via sequential Ugi/SN methodology. Anticancer Agents Med Chem 16:621–632. doi: 10.2174/1871520615666150916092035. [DOI] [PubMed] [Google Scholar]
- 33.Shaaban S, Negm A, Sobh MA, Wessjohann LA. 2015. Organoselenocyanates and symmetrical diselenides redox modulators: design, synthesis and biological evaluation. Eur J Med Chem 97:190–201. doi: 10.1016/j.ejmech.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Cortazar TM, Coombs GH, Walker J. 2007. Leishmania panamensis: comparative inhibition of nuclear DNA topoisomerase II enzymes from promastigotes and human macrophages reveals anti-parasite selectivity of fluoroquinolones, flavonoids and pentamidine. Exp Parasitol 116:475–482. doi: 10.1016/j.exppara.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 35.Marin C, Boutaleb-Charki S, Diaz JG, Huertas O, Rosales MJ, Perez-Cordon G, Guitierrez-Sanchez R, Sanchez-Moreno M. 2009. Antileishmaniasis activity of flavonoids from Consolida oliveriana. J Nat Prod 72:1069–1074. doi: 10.1021/np8008122. [DOI] [PubMed] [Google Scholar]
- 36.Nwaka S, Ramirez B, Brun R, Maes L, Douglas F, Ridley R. 2009. Advancing drug innovation for neglected diseases—criteria for lead progression. PLoS Negl Trop Dis 3:e440. doi: 10.1371/journal.pntd.0000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michels PA, Bringaud F, Herman M, Hannaert V. 2006. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta 1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 38.Cazzulo JJ. 1992. Aerobic fermentation of glucose by trypanosomatids. FASEB J 6:3153–3161. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez P, Marin C, Rodriguez-Gonzalez I, Hitos AB, Rosales MJ, Reina M, Diaz JG, Gonzalez-Coloma A, Sanchez-Moreno M. 2005. In vitro activity of C20-diterpenoid alkaloid derivatives in promastigotes and intracellular amastigotes of Leishmania infantum. Int J Antimicrob Agents 25:136–141. doi: 10.1016/j.ijantimicag.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 40.Bringaud F, Riviere L, Coustou V. 2006. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol 149:1–9. doi: 10.1016/j.molbiopara.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 41.Mpamhanga CP, Spinks D, Tulloch LB, Shanks EJ, Robinson DA, Collie IT, Fairlamb AH, Wyatt PG, Frearson JA, Hunter WN, Gilbert IH, Brenk R. 2009. One scaffold, three binding modes: novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J Med Chem 52:4454–4465. doi: 10.1021/jm900414x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piacenza L, Zago MP, Peluffo G, Alvarez MN, Basombrio MA, Radi R. 2009. Enzymes of the antioxidant network as novel determiners of Trypanosoma cruzi virulence. Int J Parasitol 39:1455–1464. doi: 10.1016/j.ijpara.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beyer WF Jr, Fridovich I. 1987. Assaying for superoxide dismutase activity: some large consequences of minor changes in conditions. Anal Biochem 161:559–566. doi: 10.1016/0003-2697(87)90489-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.