

ABSTRACT
We previously reported a series of N2,N4-disubstituted quinazoline-2,4-diamines as dihydrofolate reductase inhibitors with potent in vitro and in vivo antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) strains. In this work, we extended our previous study to the Gram-negative pathogen Acinetobacter baumannii. We determined that optimized N2,N4-disubstituted quinazoline-2,4-diamines are strongly antibacterial against multidrug-resistant A. baumannii strains when the 6-position is replaced with a halide or an alkyl substituent. Such agents display potent antibacterial activity, with MICs as low as 0.5 μM, while proving to be strongly bactericidal. Interestingly, these compounds also possess the potential for antibiofilm activity, eradicating 90% of cells within a biofilm at or near MICs. Using serial passage assays, we observed a limited capacity for the development of resistance toward these molecules (4-fold increase in MIC) compared to existing folic acid synthesis inhibitors, such as trimethoprim (64-fold increase) and sulfamethoxazole (128-fold increase). We also identified limited toxicity toward human cells, with 50% lethal doses (LD50s) of ≤23 μM for lead agents 4 and 5. Finally, we demonstrated that our lead agents have excellent in vivo efficacy, with lead agent 5 proving more efficacious than tigecycline in a murine model of A. baumannii infection (90% survival versus 66%), despite being used at a lower dose (2 versus 30 mg kg−1). Together, our results demonstrate that N2,N4-disubstituted quinazoline-2,4-diamines have strong antimicrobial and antibiofilm activities against both Gram-positive organisms and Gram-negative pathogens, suggesting strong potential for their development as antibacterial agents.
KEYWORDS: Acinetobacter baumannii, quinazoline, biofilm, dihydrofolate reductase inhibitors, Gram negative antibacterial
INTRODUCTION
Acinetobacter baumannii is one of the most successful nosocomial pathogens, causing infections that have over the past few decades become increasingly difficult to treat. The ability of A. baumannii to survive for prolonged periods on abiotic surfaces, alongside broad antimicrobial resistance, allows it not only to survive but also to thrive in hospital settings (1). Consequently, there has been an alarming increase in mortality associated with infections caused by this difficult-to-treat organism (2). In addition to eliciting fatal nosocomial infections, this pathogen is a primary agent of infections in military personnel, often resulting from combat trauma or burns (3, 4). These often result in chronic wound infections and biofilm-mediated disease, with the latter resulting from surgery and implanted devices (4). Such chronic A. baumannii infections lead to complications, extended rehabilitation, increased use of hospital resources, and considerably increased mortality (4).
Drug resistance in A. baumannii has resulted in few antibiotics left to eradicate the infections it causes, with clinicians often turning to last-resort, toxic treatment options (1, 5, 6) The worldwide incidence of pan-drug-resistant (PDR) A. baumannii has spread quickly, at least in part due to its naturally transformable nature, leading to an increased capacity to acquire new determinants of resistance (1, 6) The occurrence of PDR isolates, with no effective treatment options, seemingly marks the beginning of a postantibiotic era for A. baumannii; thus, measures must be taken to identify effective therapeutic options (7).
Quinazolines are an emerging class of compounds that have a broad range of biological activities ranging from anticancer, anti-inflammatory, antipsychotic, antidiabetic, antileishmanial (8, 9), and antibacterial (10–15). Kung et al. discovered a series of 2-substituted quinazolines with broad-spectrum antibacterial activity, inhibiting RNA synthesis and translation in a number of bacterial species (16). More relevant to this study, Harris et al. revealed 5-substituted 2,4-diaminoquinazolines that inhibited the dihydrofolate reductase (DHFR) enzyme of Escherichia coli and Staphylococcus aureus (17). In so doing, they determined that the 5-substituted position of the 2,4-diaminoquinazolines was not as important for enzyme binding affinity as the general structural type of the group. Unfortunately, these molecules were not specific toward the bacterial DHFR enzyme but also inhibited the bovine liver DHFR enzyme (17). Further analysis revealed that smaller substituents created greater activity in bacterial cells, while larger substituents were more active toward the bovine enzyme. However, unlike the quinazolines identified in this study, the 5-substituted 2,4-diaminoquinazolines proved ineffective in animal models of infection (17).
Our group has recently shown the utility of N2,N4-disubstituted quinazoline-2,4-diamines for the treatment of S. aureus infections (18). Specifically, we have shown them to be active against a library of methicillin-resistant S. aureus (MRSA) isolates, displaying strong bactericidal activities, with limited cytotoxic and hemolytic capacities toward human cells. Mechanism-of-action profiling reveals that much like other quinazoline compounds, they appear to function by targeting bacterial dihydrofolate reductase (18–21). We have also shown their potential for antibiofilm activity, low frequencies of mutation, and in vivo efficacy using murine models of infection (18).
In this study, we have further explored the impact of N2,N4-disubstituted quinazoline-2,4-diamines as antibacterial agents, focusing specifically on the Gram-negative species A. baumannii. Using a library of multidrug-resistant isolates, we revealed that these compounds are broadly bactericidal dihydrofolate reductase inhibitors. In addition, we observed that these compounds have low incidences of resistance and possess the potential for antibiofilm activity. Finally, we showed that the compounds are efficacious in vivo using a murine model of A. baumannii infection. Together, our results demonstrate for the first time the very real potential of quinazoline-derived compounds as antibacterial agents against the important human pathogen A. baumannii.
RESULTS AND DISCUSSION
N2,N4-Disubstituted quinazoline-2,4-diamines are active against multidrug-resistant A. baumannii isolates.
We have previously reported the activity of N2,N4-disubstituted quinazoline-2,4-diamines against MRSA strains (18). To determine if our compounds have activity against any other bacterial species, we screened them against the other ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae) pathogens. In so doing, we identified a number of analogues that were effective against A. baumannii but lacked activity toward other members of the ESKAPE pathogen set. To explore these findings more broadly, we expanded our studies to include a clonally diverse collection of A. baumannii isolates (Table 1). Strong activity was found against a number of strains, with single-digit micromolar MICs noted for three benzenoid-substituted N2-benzyl-N4-methylquinazolin-2,4-diamines 1, 2, and 3 against strain 1646.
TABLE 1.
SAR focusing on benzenoid ring substitution of various quinazoline-2,4-diaminesa
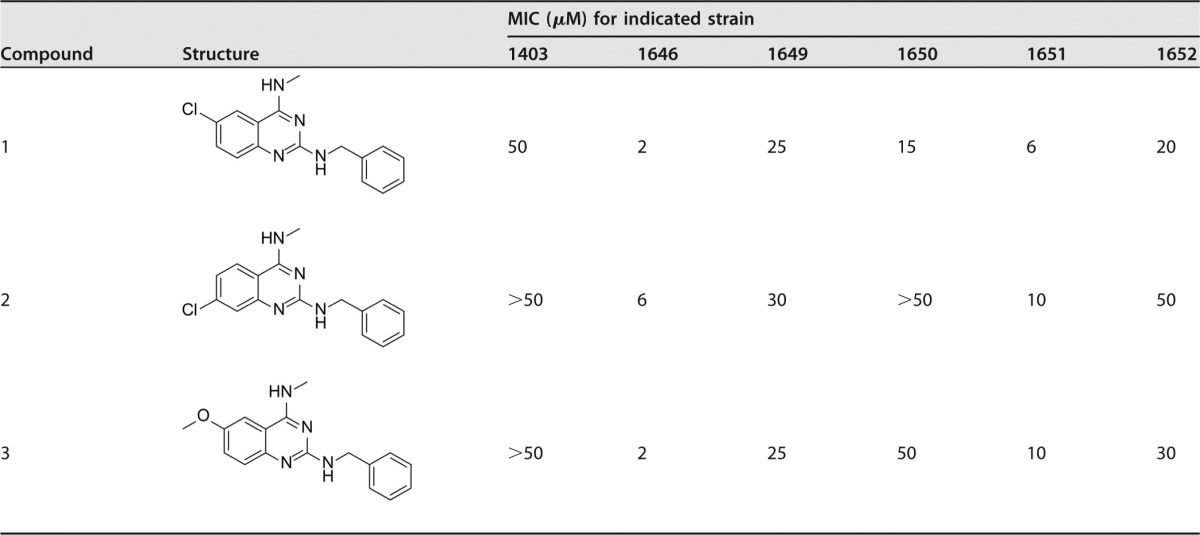
Sulfamethoxazole (SMX) and trimethoprim (TMP) were internal controls for each in vitro MIC assay: SMX, 138 μM (1403), 118 μM (1646), 118 μM (1649), 118 μM (1650), 118 μM (1651), and 118 μM (1652); TMP, 103 μM (1403), 34 μM (1646), 517 μM (1649), 120 μM (1650), 103 μM (1651), and 103 μM (1652).
Following the identification of active quinazolines 1, 2, and 3, additional N2-benzyl-N4-methylquinazolin-2,4-diamines were made with either 6- or 7-substitutions (Table 2). Substitutionat the 6-position with a bromo or a methyl group was found to be more beneficial for activity than substitution at the 7-position when comparing 6-bromoquinazolin-2,4-diamine 4 with its 7-substituted counterpart 6 or the 6-methyl-substituted quinazoline 5 with its 7-substituted analogue 7. Importantly, N2-benzyl-N4-methylquinazolin-2,4-diamine analogue 8, which lacks any substitution at the benzenoid ring, was inactive, with an MIC of ≥50 μM, and therefore demonstrated the importance of a 6- or 7-substituent on the benzenoid ring.
TABLE 2.
Probing benzenoid substitution of N2-benzyl-N4-methylquinazolin-2,4-diaminesa
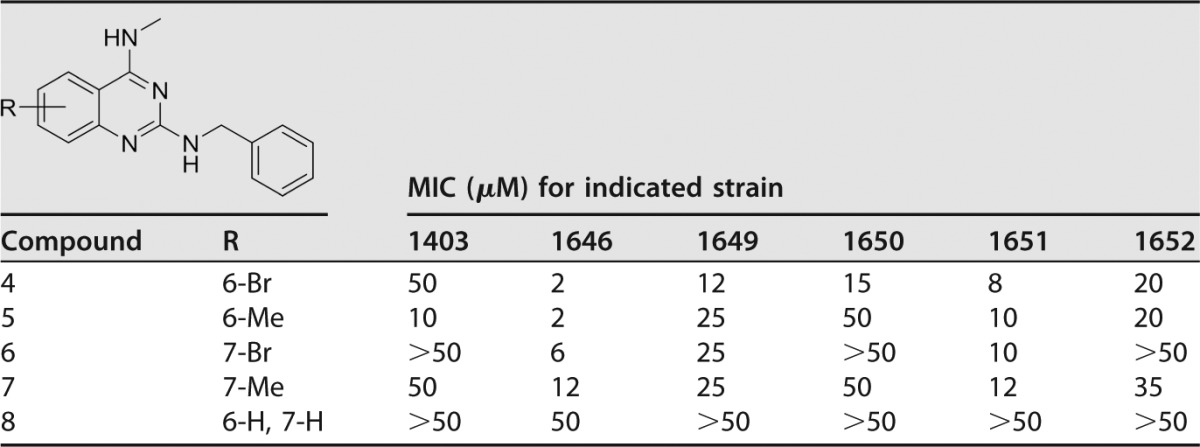
Sulfamethoxazole and trimethoprim were internal controls for each in vitro MIC assay: SMX, 138 μM (1403), 118 μM (1646), 118 μM (1649), 118 μM (1650), 118 μM (1651), and 118 μM (1652); TMP, 103 μM (1403), 34 μM (1646), 517 μM (1649), 120 μM (1650), 103 μM (1651), and 103 μM (1652).
A similar trend was observed with N4-benzyl-N2-methylquinazolin-2,4-diamine analogues when comparing 6-substituted compounds 10 and 11 with the 7-substituted analogues 14 and 15 (Table 3). Furthermore, substitution in 6- or 7-position with an electron withdrawing chloro or a bromo moiety yielded quinazolines 9, 10, 13, and 14, which were more potent than corresponding methyl- or methoxy-substituted analogues 11, 12, and15. Of all the quinazolines of the first two subseries tested, only compound 5 was active against the clinically important strain 1403, with an MIC of 10 μM, leading us to believe that continued work on the benzenoid ring would be highly beneficial.
TABLE 3.
Probing benzenoid ring substitution of N4-benzyl-N2-methylquinazolin-2,4-diaminesa
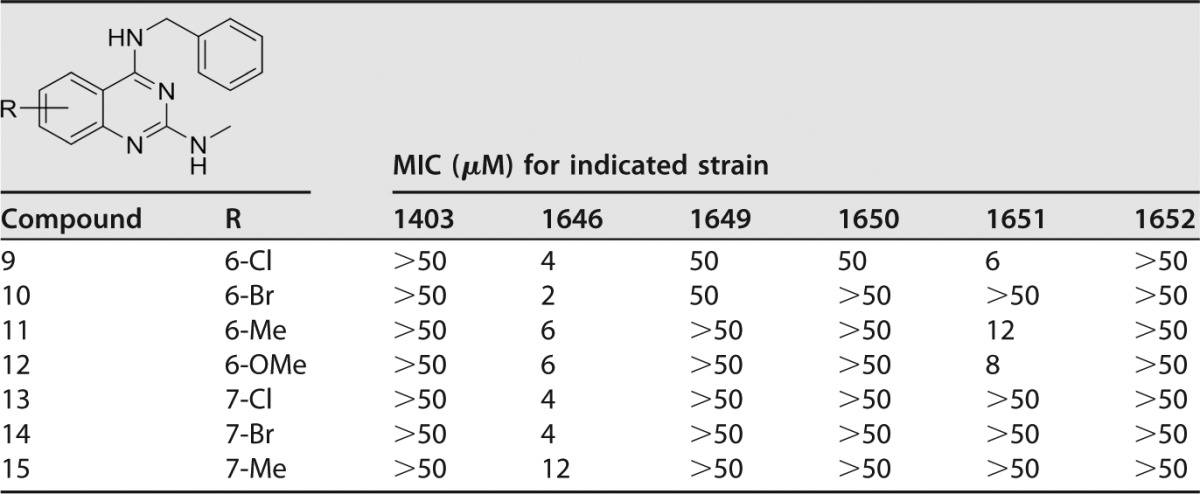
Sulfamethoxazole and trimethoprim were internal controls for each in vitro MIC assay: SMX, 138 μM (1403), 118 μM (1646), 118 μM (1649), 118 μM (1650), 118 μM (1651), and 118 μM (1652); TMP, 103 μM (1403), 34 μM (1646), 517 μM (1649), 120 μM (1650), 103 μM (1651), and 103 μM (1652).
Extending the N2-benzyl chain to an N2-phenethyl was investigated to see if an increase in activity would be found (Table 4). Compound 16, with no benzenoid substitution, was 4-fold more active than the benzyl analogue 8 (Table 1). Compounds 17 to 20 were also found to be slightly more potent than the benzyl analogues 9 to 12 (Table 3), with MICs of 2 or 4 μM.
TABLE 4.
Benzenoid ring substitutions of N4-methyl-N2-phenethylquinazolin-2,4-diaminesa
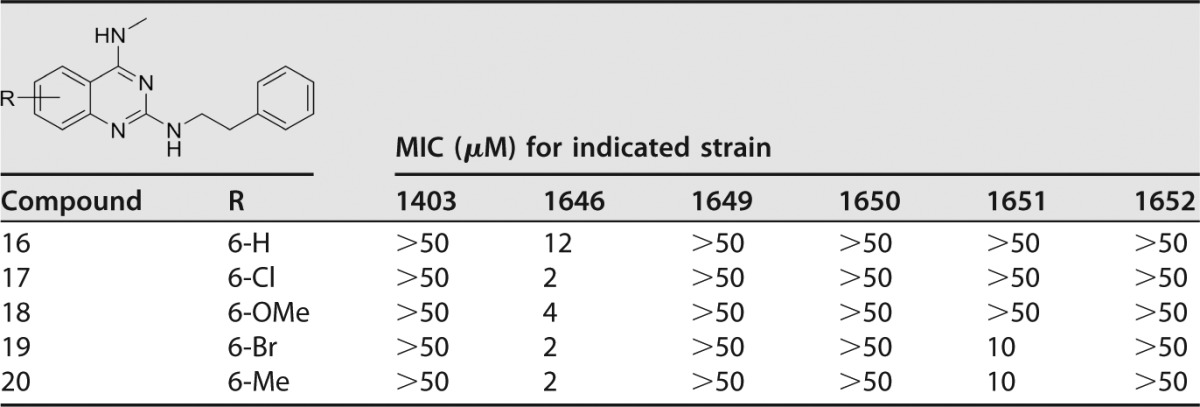
Sulfamethoxazole and trimethoprim were internal controls for each in vitro MIC assay: SMX, 138 μM (1403), 118 μM (1646), 118 μM (1649), 118 μM (1650), 118 μM (1651), and 118 μM (1652); TMP, 103 μM (1403), 34 μM (1646), 517 μM (1649), 120 μM (1650), 103 μM (1651), and 103 μM (1652).
With the importance of substitution at the 6-position identified, new analogues were evaluated with vinyl, alkyl, or aryl substitutions (Table 5; see also Fig. S1 in the supplemental material). While the MIC barrier of 2 μM against the most susceptible strain (1646) was not broken, major advances were seen in activity against the most resistant isolate (1403). In particular, n-pentyl-, cyclohexenyl-, and cyclohexyl-substituted quinazolines 27, 29, and 30 had MICs of 2 μM against most isolates besides strain 1652, for which they had MICs of 10 μM and 30 μM. These three compounds revealed that large, bulky, and lipophilic groups at the 6-position are not only tolerated but also beneficial for inhibiting the growth of A. baumannii. Phenyl- and furanyl-substituted quinazolines 31 and 32 were less active, as were the vinyl and ethyl analogues 22 and 23, the isopropenyl and isopropyl analogues 24 and 25, and the cyclopentenyl-quinazoline 28.
TABLE 5.
Extension of the 6-position of N2-benzyl-N4-methylquinazolin-2,4-diaminesa
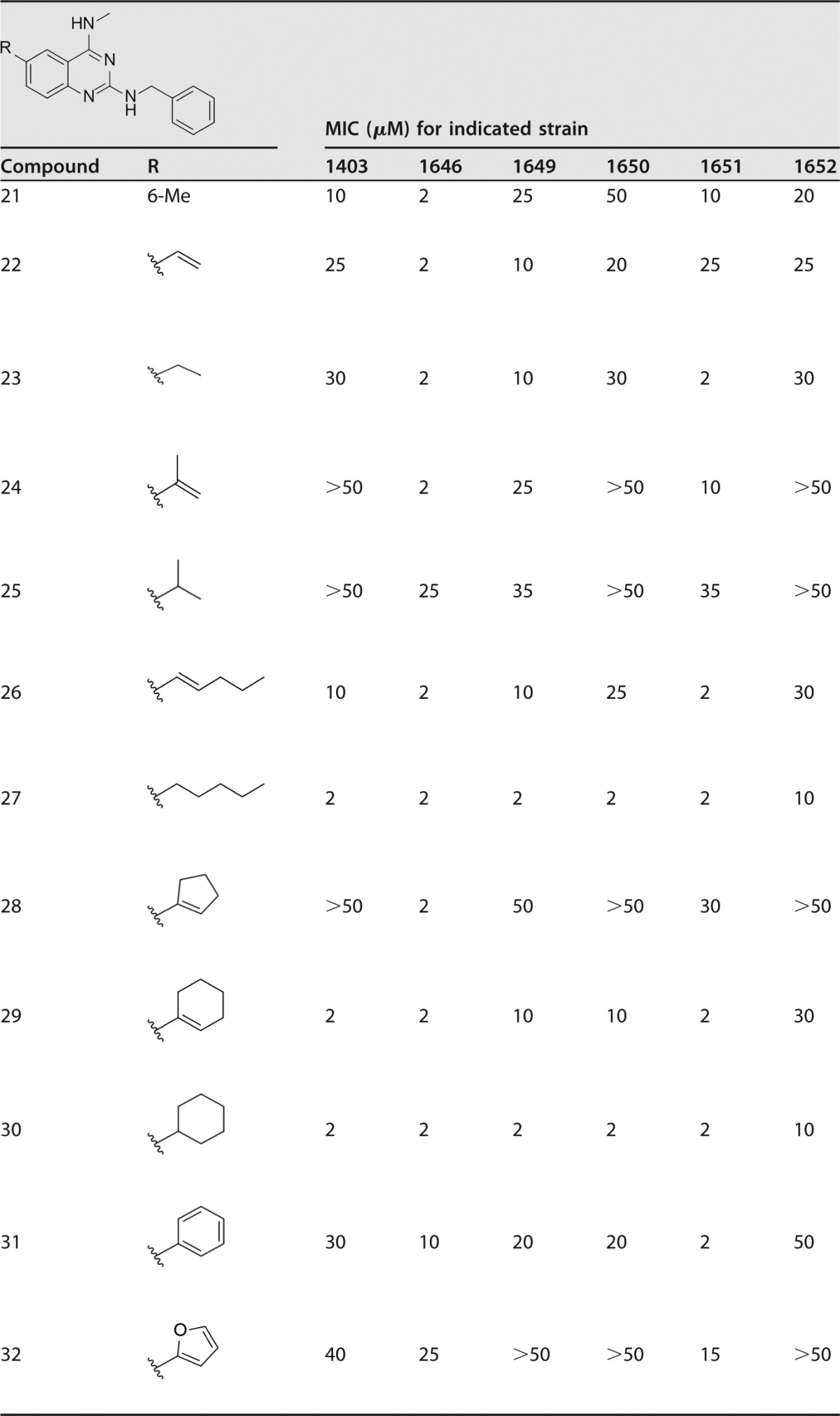
Sulfamethoxazole and trimethoprim were internal controls for each in vitro MIC assay: SMX, 138 μM (1403), 118 μM (1646), 118 μM (1649), 118 μM (1650), 118 μM (1651), and 118 μM (1652); TMP, 103 μM (1403), 34 μM (1646), 517 μM (1649), 120 μM (1650), 103 μM (1651), and 103 μM (1652).
Lead quinazolines have bactericidal activity.
Lead quinazolines 4, 5, 26, 29, 27, and 30 were selected to be further evaluated for antimicrobial effects. The first assay utilized was a minimal bactericidal concentration (MBC) assay, to assess whether leads compounds were bacteriostatic or bactericidal. The six lead agents were screened to identify their MBC90 toward each of the six A. baumannii isolates used in the SAR studies (Table S2), with data for isolate 1646 detailed in Table 6. Lead agents were all found to be broadly bactericidal, with MBC90 values ranging from 0.8 μM to 1.8 μM. Compounds 4 and 5 were found to be the most efficacious at eliminating bacterial growth, with an MBC90 value of 0.8 μM. Further to this, we were able to obtain complete eradication of bacterial growth for these two compounds at 1 μM for 4 and 5 μM for 5. Although marginally less effective, compounds 26, 29, 30, and 27 all still efficiently reduced bacterial viability, with MBC90 values of 1.8 μM, 1.5 μM, 1.1 μM, and 1.1 μM, respectively. Moreover, compound 26 resulted in complete bacterial eradication at 5 μM, which is only 5 times its MIC.
TABLE 6.
In vitro antibacterial assessment of front-runner quinazoline compounds against multidrug-resistant A. baumannii
| Compound | MIC (μM) | MBC90 (μM) | MBEC90 (μM) | LD50 (HepG2) (μM) | Activity indexa |
|---|---|---|---|---|---|
| 4 | 0.5 | 0.8 | 3.3 | 23 | 46 |
| 5 | 1 | 0.8 | 2.8 | 22 | 22 |
| 26 | 1 | 1.8 | 8.9 | 16 | 22 |
| 29 | 2 | 1.5 | 8.9 | 23 | 12 |
| 27 | 1 | 1.1 | 11.5 | 12 | 12 |
| 30 | 2 | 1.1 | 41.2 | 11 | 6 |
Activity index = LD50/MIC.
Front-runner agents impact the viability of cells within a biofilm.
A. baumannii, like many nosocomial pathogens, utilizes biofilm formation to increase persistence and decrease sensitivity to the action of antibiotics. Accordingly, the ability to impact cell viability within a biofilm is an important attribute for novel antimicrobial compounds. Therefore, we next tested our isolates for this activity, again using our library of multidrug-resistant strains (Table S2), with data from strain 1646 shown in Table 6. As with our bactericidal profiling, lead quinazolines 4 and 5 again had the most promising activity, with 90% biofilm eradication (MBEC90) seen at 3.3 μM and 2.8 μM, respectively (Table 6). Further to this, analogue 5 was the most effective lead agent, with a 3-log reduction in biofilm viability observed at 10 μM. Compound 4 reduced biofilm viability by 3.6 logs, but not until at a concentration of 50 μM. Lead quinazoline 30 had biofilm eradication potential similar to 4, reducing viability by 4.2 logs at 50 μM, although its MBEC90 (1-log reduction) was found to be close to this value at 41 μM. Compounds 26 and 29 also had promising activity, with both displaying MBEC90 at a concentration of 8.9 μM. Extended testing with these two quinazolines revealed that compound 29 reduced biofilm viability by 1.6 logs at 50 μM, while compound 26 resulted in a 1.4-log reduction in biofilm viability at the same concentration.
Lead quinazoline derivatives appear to function by targeting DHFR.
To determine if lead quinazolines inhibit the A. baumannii dihydrofolate reductase (DHFR) enzyme, similar to that seen for sister compounds in our work with MRSA, an in vitro rescue assay was performed (18). Accordingly, the viability of A. baumannii cells was tested using lead quinazoline 5 in the presence or absence of tetrahydrofolic acid (THF; 0 μM to 225 μM), the end product produced by DHFR. After 24 h of incubation, we determined that only 10 μM THF was sufficient to rescue bacterial growth from the inhibitory effects of lead agent 5. These data suggest that the potential mechanism of action for our compounds is perhaps via inhibition of tetrahydrofolic acid production. While this finding supports data generated by us and others regarding the impact of quinazoline molecules on bacterial cells (18–21), we cannot discount the possibility of other potential targets within for these compounds within A. baumannii.
Quinazoline-derived compounds induce limited capacity for resistance.
An important attribute of novel antibiotics is the ability to fend off the development of resistance toward their effects. To assess this capacity, A. baumannii strains were incubated overnight with 0.5× MIC of each of the front runners. The next day, cells were washed and used to inoculate fresh medium that contained a 2-fold increase in drug. This was repeated for a total of 8 days, alongside sulfamethoxazole (SMX) and trimethoprim (TMP) controls, both of which target the same pathway as our lead agents (Fig. 1), as well as an unrelated agent, tetracycline (TET) (22). Upon analysis, we determined that all of our front-runner compounds outperformed SMX, TMP, and TET, generating much lower incidences of resistance. Specifically, lead agents 4 and 5 had the most striking effects, with MICs increasing over the 8-day test period only 4-fold, compared to 64-fold (TMP) and 128-fold (SMX and TET) for the control agents. All of the other 4 agents were similarly impressive in their ability to limit resistance development, resulting in an increased MIC of only 16-fold, which, while not as promising as the results for agents 4 and 5, is still profoundly reduced compared to our controls.
FIG 1.
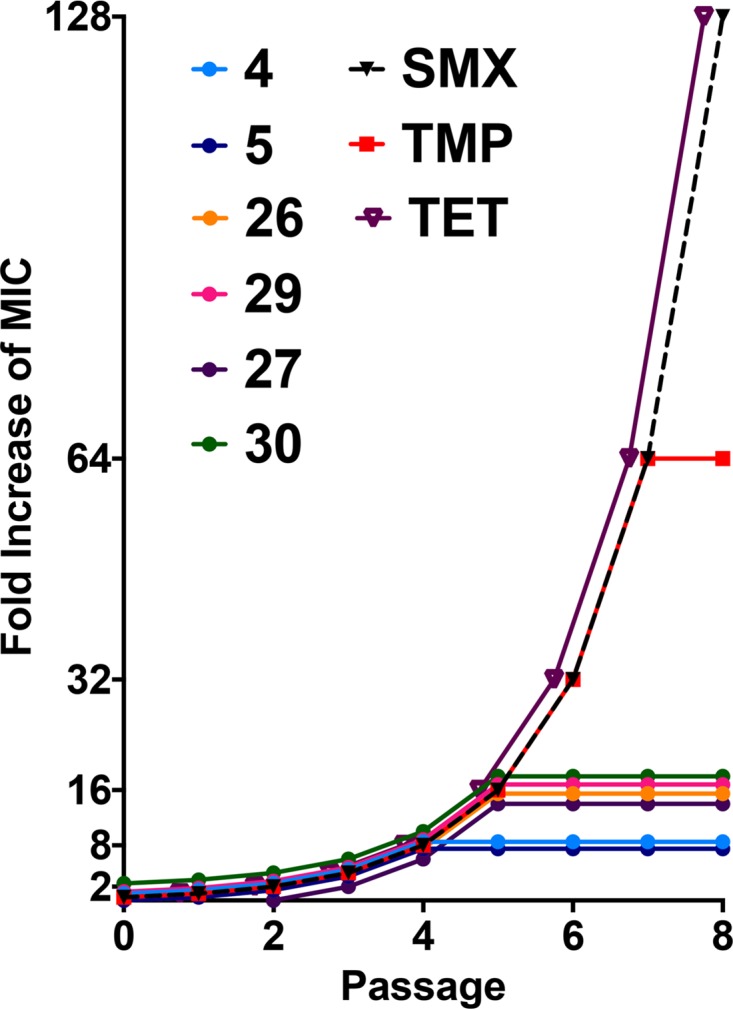
Lead quinazolines generate limited resistance by A. baumannii isolates. The six lead quinazolines were tested in a stepwise serial passage assay, alongside sulfamethoxazole (SMX), trimethoprim (TMP), and tetracycline (TET) controls. The first passage began with 0.5× the MIC, and with each passage, the concentrations of all compounds were increased 2-fold. The graph displays the fold increase in the MIC over the course of 8 days. Representative data generated using strain 1403 are shown.
Front-runner quinazolines have limited toxicity toward human cells.
In order to gain a sense of the toxicity of lead quinazolines toward eukaryotic cells, we determined 50% lethal doses (LD50s) for human HepG2 cells (Table 6; Fig. 2A). Importantly, we observed >50% cell viability for all compounds at concentrations up to 6 μM. Furthermore, 4 of our 6 lead agents returned >50% viability at 12 μM, while compounds 26 and 29 were only marginally less promising, returning HepG2 cell viabilities of 49% and 42%, respectively, at this concentration. When we used a 25 μM concentration of each lead quinazoline or control antibiotic, we observed only fractionally less than 50% recovery. Importantly, lead agents 4 and 5 at 25 μM performed the best, with 43% and 44% viability observed, respectively. Similarly, lead agents 26 and 30 allowed for 41% and 39% respective viability at this concentration, while treatment with lead agents 27 and 29 resulted in 31% and 32% viability, respectively. The control compounds sulfamethoxazole and trimethoprim returned 46% and 45% viability at the highest concentration tested, which is in line with data generated from our front-runners. To place lead compound data in context, lead agents 4 and 5 have the greatest therapeutic window for infection treatment. Specifically, lead agent 4 possesses a 46-fold preference in specificity toward bacteria, with an MIC (0.5 μM) much lower than the LD50 (23 μM) toward human liver cells. Similarly, lead agent 5 displayed a 22-fold activity index (AI; LD50/MIC), which is a measure of specificity toward bacterial cells (Table 6). As an additional measure of toxicity, we next tested the hemolytic capacity of the front-runners using whole human blood (Fig. 2B). Importantly, we observed negligible capacity of our lead quinazolines to lyse human red blood cells (hRBCs) when incubated for 1 h at a concentration of 10 μM. Specifically, we observed average hemolysis well below 1% (range = 0.24% to 0.47%), while the positive control (1% Triton X-100) produced 100% lysis during a similar time frame.
FIG 2.

Cytotoxicity toward human cells. (A) Lead quinazolines were tested at 25 μM, 12 μM, 6 μM, and 3 μM against human HepG2 cells, compared to solvent-only controls. The known antibiotics sulfamethoxazole and trimethoprim, which target the same pathway as our quinazolines, were also tested in parallel. (B) The six lead quinazolines were tested at 10 μM against whole human blood for the ability to lyse RBCs. Shown is the percent lysis of human red blood cells compared to a Triton X-100 (T) control at a concentration of 1%.
N2,N4-Disubstituted quinazoline-2,4-diamines are efficacious in vivo.
As a final assessment, we used a murine model of lethal A. baumannii infection to determine the efficacy of quinazolines in vivo. This was performed using front-runner 5, which had the most promising properties from all of our biological testing. Accordingly, mice were inoculated with a lethal dose of A. baumannii via intraperitoneal (i.p.) injection on the right side of the abdomen. One hour postchallenge, mice were treated with an i.p. injection of 2 mg kg−1 of front-runner 5 on the left side of the abdomen. As a control, we also performed similar testing using 30 mg kg−1 of tigecycline, which we already know our test strain to be susceptible to in vitro. In so doing, we determined that quinazoline 5 resulted in a statistically significant survival rate of 83% of infected animals, compared to only 17% for vehicle-only controls (Fig. 3). We also saw significant survival of animals injected with tigecycline, although this was at a rate of 66%, which is inferior to that of our front-runner agent. Consequently, this would suggest that our class of N2,N4-disubstitutedquinazoline-2,4-diamines have excellent potential for development as antibacterial agents targeting multidrug-resistant A. baumannii infections.
FIG 3.

Lead quinazolines are efficacious in vivo. Mice were administered a lethal dose of A. baumannii 1646. They were then given lead quinazoline 5 at 2 mg kg−1, tigecycline at 30 mg kg−1, or a vehicle. Survival was monitored over a 5-day period. Statistical significance was determined using a log rank (Mantel-Cox) test. *, P < 0.05.
Concluding remarks.
A library of N2,N4-disubstituted quinazoline-2,4-diamines, which was previously shown to have antibacterial activity against MRSA (18), was also found to have potent effects toward the multidrug-resistant Gram-negative species A. baumannii. We assessed 73 N2,N4-disubstituted quinazoline-2,4-diamines and found that 6- or 7-substituted N2-benzyl-N4-methylquinazoline-2,4-diamines displayed promising activity, with MICs ranging from 0.5 to 30 μM against the six strains of A. baumannii tested. Over 30 molecules were designed and synthesized to conduct a structure-activity relationship study to systematically probe the substituents in the N2-, N4-, 6-, and 7-positions. The most potent in vitro activities were obtained with quinazoline-2,4-diamines bearing an N2-benzyl moiety and an N4-methyl group. Furthermore, quinazolines with substitutions in the 6-position with a halide or alkyl group were more potent than analogues with substitutions at the 7-position. 6-n-Pentyl- and 6-cyclohexyl-substituted quinazolines 27 and 30 were among the most effective agents, since they were equipotent with single-digit micromolar MICs against the six tested A. baumannii strains. Following this, front-runner compounds 4, 5, 26, 29, 27, and 30 were tested for bactericidal activities and biofilm eradication. We found that the lead quinazolines 4 and 5 displayed the strongest bactericidal and biofilm activity toward A. baumannii, with MBC90 values of <1 μM and MBEC90 values of <4 μM. These compounds also allowed for limited resistance development, displaying only a 4-fold increase in MIC against A. baumannii over an 8-day period, which was only a fraction of that observed for control compounds. Using a murine model of infection, we determined that lead agent 5 was more effective, and at lower concentrations, in rescuing mice from a lethal dose of A. baumannii than our control agent tigecycline. Our results reveal the potent antibacterial activities of N2-benzyl-N4-methylquinazoline-2,4-diamines against A. baumannii and show their potential for development to treat both Gram-positive and Gram-negative multidrug-resistant infections.
MATERIALS AND METHODS
General.
All strains used in this study are listed in Table S1 in the supplemental material.
Synthetic protocols and compound characterization.
Full details of compound synthesis and characterization can be found in the supplemental material.
Antibacterial activity assessment.
MIC and minimal bactericidal concentration (MBC) assays were performed in this study as documented by us previously (18, 23–25). Briefly, A. baumannii strains were grown in tryptic soy broth overnight cultures at 37°C with shaking. MIC determination was performed in a 96-well plate by diluting overnight cultures 1:1,000 in Mueller-Hinton broth (MHB) and adding 195 μl to each well. Subsequently, 5 μl of quinazoline (or control compound) was added before incubation for 24 h at 37°C. Following this, MICs were determined as the lowest concentration to produce a complete absence of growth. All compounds were diluted prior to testing in dimethyl sulfoxide (DMSO) to assess multiple concentrations with the addition of the same volume of solvent. MBC assays were performed in an identical manner to MIC experiments; however, after 24 h of incubation, bacterial cells were serially diluted in phosphate-buffered saline (PBS) and recovered on antibiotic-free tryptic soy agar (TSA) for 24 h at 37°C. MBC90 values were calculated using linear regression of the percent recovery compared to no-treatment controls.
Biofilm eradication determination assay.
Biofilm eradication determination assays were performed as described by us previously (25, 26), as follows. Each of the A. baumannii strains was grown overnight in MHB. The next day, these were used to seed fresh MHB to an optical density at 600 nm (OD600) of 0.5, with 150 μl then added to the wells of a 96-well plate and grown for 24 h at 37°C. After 24 h, the planktonic bacteria were carefully removed and fresh MHB was added with increasing concentrations of lead quinazolines. After incubation at 37°C for 24 h, planktonic cells were removed and biofilms were washed three times with PBS. Biofilms were then resuspended in PBS and plated for cell viability on TSA. Biofilm recovery was assessed compared to that with no-drug controls and determined as percent eradication. This was used to determine MBEC90 values (minimal biofilm eradication concentration), where the viability of cells within the biofilm was reduced by 90%.
Investigating the mechanism of action of quinazoline-based compounds.
To evaluate the effect quinazolines have on DHFR reduction of dihydrofolic acid, a tetrahydrofolic acid rescue assay was performed as described by us previously (18). A. baumannii strain 1403 was grown overnight in LB and then diluted 1:1,000 into fresh medium. These cultures were then seeded into a sterile 96-well plate with tetrahydrofolic acid added at concentrations ranging from 0 to 225 μM. Lead quinazoline 25 was then added at 1×, 2×, and 5× the MIC, and cultures were incubated at 37°C for 18 h. MICs were determined and used to assess whether the addition of tetrahydrofolic acid rescued A. baumannii growth from quinazoline inhibition. Assays were repeated in triplicate, alongside trimethoprim and sulfamethoxazole controls.
Serial passage assay.
In order to test potential resistance toward the quinazolines, a serial passage assay was performed alongside control compounds (sulfamethoxazole and trimethoprim), as described by us previously (25). A. baumannii strain 1403 was grown overnight in LB medium at 37°C. The next day, cultures were diluted 1:100 in fresh medium and seeded into a 96-well plate. Lead quinazolines or control agents were added to respective wells at half MICs. Plates were then incubated for 24 h at 37°C, followed by the removal of aliquots from these cultures to inoculate fresh medium (1:100 dilution) containing compounds at 2-fold higher concentrations. These were then grown overnight, and the procedure was repeated for a total of 8 days. The cultures were observed for a lack of growth, indicating that strains were no longer able to resist the action of a given compound. Each experiment was performed in triplicate, yielding identical results.
HepG2 cytotoxicity.
Cytotoxicity assays were performed using human HepG2 cells (human liver epithelial with hepatocellular carcinoma), as described by us previously (18, 25). Cells were cultured in Dulbecco's modified Eagle medium (DMEM), supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, for 3 days at 37°C and 5% CO2. Cells were then diluted to 1 × 105 ml−1 using fresh DMEM and added to 96-well tissue culture plates at a volume of 100 μl. Plates were incubated for 24 h at 37°C and 5% CO2, allowing the cells to adhere to the plastic. After this time, medium was carefully removed and 200 μl of fresh DMEM was added with test compounds at concentrations of 0, 1, 2, 5, 10, 15, 30, and 50 μM. Plates were then incubated for 48 h at 37°C and 5% CO2. After 48 h, the DMEM was removed and 100 μl of new medium containing 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was added, followed by incubation for 4 h at 37°C and 5% CO2. After 4 h, 75 μl of medium was removed and replaced with 50 μl of 16% (wt/vol) SDS and DMSO, followed by incubation for 10 min at 37°C to solubilize any formazan produced. A BioTek plate reader was used to measure the absorbance of formazan production at 540 nM. Lead compounds were solvated in 100% DMSO for these studies, which served as the negative control. LD50s were determined for each compound by comparison to vehicle-only controls.
Hemolysis assay.
A hemolysis assay was performed using whole human blood (Bioreclamation), as described previously (25). Briefly, human red blood cells (hRBCs) were resuspended in 20% vol/vol 1× HA buffer (4.25 ml of 10% NaCl and 1 ml of CaCl2 in 50 ml of sterile water), before lead compounds were added at 2 μM, 10 μM, and 20 μM, in a final volume of 100 μl. Cells were incubated for 15 min at 37°C before being centrifuged at 5,500 × g for 1 min to pellet nonlysed hRBCs. The supernatant was removed and added to a 96-well microtiter plate, and the OD543 was read using a BioTek Synergy2 plate reader. The negative control was vehicle only (DMSO), and the positive control was 1% Triton X-100. Assays were performed in triplicate, with data displayed as percent hemolysis compared to controls, defined as percent hemolysis = (OD543 of test sample − OD543 of no-drug control)/(OD543 of Triton X-100 − OD543 of no-drug control) × 100.
In vivo efficacy testing using a murine model of lethal peritonitis.
A murine model of lethal peritonitis was used to demonstrate the effectiveness of the lead quinazolines to clear bacterial infections, as described by us previously (25). Six mice per group were infected via intraperitoneal (i.p.) injection (right side) with 7.5 × 108 CFU ml−1 of A. baumannii 1646 in PBS containing 5% mucin. After 1 h, mice were inoculated by i.p. injection to the left side of the abdomen with either 2 mg/kg of lead agent 25 (test group), 30 mg kg−1 of tigecycline (positive control), or a vehicle alone [45% (wt/vol) (2-hydroxypropyl)-β-cyclodextrin in water (negative control)]. Mice were monitored twice daily for 5 days to assess mortality. All animal studies received written approval after review by the Institutional Animal Care and Use Committee in the Division of Comparative Medicine and Division of Research Integrity and Compliance at the University of South Florida. The clinical endpoint was reached for this study when the mice reached a premoribund state. The numbers of mice surviving in control and treatment groups were compared and analyzed for statistical significance using a log rank (Mantel-Cox) test.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by the University of South Florida, Northeastern University, and grants AI103715 and AI80626 (both to L.N.S.) from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00059-17.
REFERENCES
- 1.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Necati Hakyemez I, Kucukbayrak A, Tas T, Burcu Yikilgan A, Akkaya A, Yasayacak A, Akdeniz H. 2013. Nosocomial Acinetobacter baumannii infections and changing antibiotic resistance. Pak J Med Sci 29:1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebeny PJ, Riddle MS, Petersen K. 2008. Acinetobacter baumannii skin and soft-tissue infection associated with war trauma. Clin Infect Dis 47:444–449. doi: 10.1086/590568. [DOI] [PubMed] [Google Scholar]
- 4.Akers KS, Mende K, Cheatle KA, Zera WC, Yu X, Beckius ML, Aggarwal D, Li P, Sanchez CJ, Wenke JC, Weintrob AC, Tribble DR, Murray CK. 2014. Biofilms and persistent wound infections in United States military trauma patients: a case-control analysis. BMC Infect Dis 14:190. doi: 10.1186/1471-2334-14-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J.. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 6.Manchanda V, Sanchaita S, Singh N. 2010. Multidrug resistant Acinetobacter. J Glob Infect Dis 2:291–304. doi: 10.4103/0974-777X.68538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falagas ME, Bliziotis IA. 2007. Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era? Int J Antimicrobial Agents 29:630–636. doi: 10.1016/j.ijantimicag.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Van Horn KS, Zhu X, Pandharkar T, Yang S, Vesely B, Vanaerschot M, Dujardin J-C, Rijal S, Kyle DE, Wang MZ, Werbovetz KA, Manetsch R. 2014. Antileishmanial activity of a series of N2,N4-disubstituted quinazoline-2,4-diamines. J Med Chem 57:5141–5156. doi: 10.1021/jm5000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X, Van Horn KS, Barber MM, Yang S, Wang MZ, Manetsch R, Werbovetz KA. 2015. SAR refinement of antileishmanial N2,N4-disubstituted quinazoline-2,4-diamines. Bioorg Med Chem 23:5182–5189. doi: 10.1016/j.bmc.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alagarsamy V, Raja Solomon V, Dhanabal K. 2007. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3H-quinazolin-4-one as analgesic, anti-inflammatory agents. Bioorg Med Chem 15:235–241. doi: 10.1016/j.bmc.2006.09.065. [DOI] [PubMed] [Google Scholar]
- 11.Alvarado M, Barceló M, Carro L, Masaguer CF, Raviña E. 2006. Synthesis and biological evaluation of new quinazoline and cinnoline derivatives as potential atypical antipsychotics. Chem Biodivers 3:106–117. doi: 10.1002/cbdv.200690001. [DOI] [PubMed] [Google Scholar]
- 12.Chandregowda V, Kush AK, Chandrasekara Reddy G. 2009. Synthesis and in vitro antitumor activities of novel 4-anilinoquinazoline derivatives. Eur J Med Chem 44:3046–3055. doi: 10.1016/j.ejmech.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Malamas MS, Millen J. 1991. Quinazolineacetic acids and related analogs as aldose reductase inhibitors. J Med Chem 34:1492–1503. doi: 10.1021/jm00108a038. [DOI] [PubMed] [Google Scholar]
- 14.Rohini R, Muralidhar Reddy P, Shanker K, Hu A, Ravinder V. 2010. Antimicrobial study of newly synthesized 6-substituted indolo[1,2-c]quinazolines. Eur J Med Chem 45:1200–1205. doi: 10.1016/j.ejmech.2009.11.038. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Gao F. 2013. Quinazoline derivatives: synthesis and bioactivities. Chem Cent J 7:95. doi: 10.1186/1752-153X-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kung PP, Casper MD, Cook KL, Wilson-Lingardo L, Risen LM, Vickers TA, Ranken R, Blyn LB, Wyatt JR, Cook PD, Ecker DJ. 1999. Structure-activity relationships of novel 2-substituted quinazoline antibacterial agents. J Med Chem 42:4705–4713. doi: 10.1021/jm9903500. [DOI] [PubMed] [Google Scholar]
- 17.Harris NV, Smith C, Bowden K. 1990. Antifolate and antibacterial activities of 5-substituted 2,4-diaminoquinazolines. J Med Chem 33:434–444. doi: 10.1021/jm00163a067. [DOI] [PubMed] [Google Scholar]
- 18.Van Horn KS, Burda WN, Fleeman R, Shaw LN, Manetsch R. 2014. Antibacterial activity of a series of N2,N4-disubstituted quinazoline-2,4-diamines. J Med Chem 57:3075–3093. doi: 10.1021/jm500039e. [DOI] [PubMed] [Google Scholar]
- 19.Blaney JM, Hansch C, Silipo C, Vittoria A. 1984. Structure-activity relationships of dihydrofolate reductase inhibitors. Chem Rev 84:333–407. [Google Scholar]
- 20.DeGraw JI, Brown VH, Colwell WT, Morrison NE. 1974. Potential antileprotic agents. 3. Inhibition of mycobacterial dihydrofolic reductase by 2,4-diamino-5-methyl-6-alkylquinazolines. J Med Chem 17:762–764. [DOI] [PubMed] [Google Scholar]
- 21.Lam T, Hilgers MT, Cunningham ML, Kwan BP, Nelson KJ, Brown-Driver V, Ong V, Trzoss M, Hough G, Shaw KJ, Finn J. 2014. Structure-based design of new dihydrofolate reductase antibacterial agents: 7-(benzimidazol-1-yl)-2,4-diaminoquinazolines. J Med Chem 57:651–668. doi: 10.1021/jm401204g. [DOI] [PubMed] [Google Scholar]
- 22.Clinical and Laboratory Standards Institute. 2005. M100-S15; NCCLS document 0896-6443 (DLC)sf 88020146 (OCoLC)17367266. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 23.Burda WN, Fields KB, Gill JB, Burt R, Shepherd M, Zhang XP, Shaw LN. 2012. Neutral metallated and meso-substituted porphyrins as antimicrobial agents against gram-positive pathogens. Eur J Clin Microbiol Infect Dis 31:327–335. doi: 10.1007/s10096-011-1314-y. [DOI] [PubMed] [Google Scholar]
- 24.Cormier R, Burda WN, Harrington L, Edlinger J, Kodigepalli KM, Thomas J, Kapolka R, Roma G, Anderson BE, Turos E, Shaw LN. 2012. Studies on the antimicrobial properties of N-acylated ciprofloxacins. Bioorg Med Chem Lett 22:6513–6520. doi: 10.1016/j.bmcl.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleeman R, LaVoi TM, Santos RG, Morales A, Nefzi A, Welmaker GS, Medina-Franco JL, Giulianotti MA, Houghten RA, Shaw LN. 2015. Combinatorial libraries as a tool for the discovery of novel, broad-spectrum antibacterial agents targeting the ESKAPE pathogens. J Med Chem 58:3340–3355. doi: 10.1021/jm501628s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Salm JL, Witowski CG, Fleeman RM, McClintock JB, Amsler CD, Shaw LN, Baker BJ. 2016. Darwinolide, a new diterpene scaffold that inhibits methicillin-resistant Staphylococcus aureus biofilm from the Antarctic sponge Dendrilla membranosa. Org Lett 18:2596–2599. doi: 10.1021/acs.orglett.6b00979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.