

Abstract
Lipotoxicity induced by saturated fatty acids (SFAs) plays a pathological role in the development of non-alcoholic fatty liver disease (NAFLD); however, the exact mechanism remains to be clearly elucidated. Palmitate is the most abundant SFA in the circulation and major lipotoxic inducer. Accumulating evidence supports that autophagy induction is protective against palmitate-induced cell death in a variety of cell types, including hepatocytes. Nicotinamide is the amide form of nicotinic acid (vitamin B3, Niacin) and a dietary supplementation as a source of vitamin B3. We previously reported that nicotinamide endowed hepatocytes resistance to palmitate-induced ER stress via upregulating SIRT1, with cAMP/PKA/CREB pathway activation being a fundamental mechanism. This study was undertaken to investigate the potential anti-lipotoxic effect of nicotinamide and to elucidate underlying mechanism(s). Our data demonstrated that nicotinamide supplementation protected hepatocytes against palmitate-induced cell death. Mechanistic investigations revealed that nicotinamide supplementation activated autophagy in hepatocytes. Importantly, we showed that the anti-lipotoxic property of nicotinamide was abolished by autophagy inhibitors, suggesting that autophagy induction plays a mechanistic role in nicotinamide's anti-lipotoxic effect. Furthermore, we showed that SIRT1 inhibition blunted autophagy induction in response to nicotinamide supplementation and similarly abrogated the anti-lipotoxic effect conferred by nicotinamide supplementation. In conclusion, our data suggest that nicotinamide protects against palmitate-induced hepatotoxicity via SIRT1-dependent autophagy induction and that nicotinamide supplementation may represent a therapeutic choice for NAFLD.
Keywords: Nicotinamide, palmitate, lipotoxicity, SIRT1, autophagy
1. Introduction
Lipotoxicity refers to the toxic effect of lipid over-accumulation in non-lipid storage tissues. Liver, muscle, heart, and pancreas, are all among the target tissues of lipotoxic injury [1]. Lipotoxicity plays an important role in the initiation and progression of non-alcoholic fatty liver disease (NAFLD) [2]. Although much progress has been made over the last several decades, the exact mechanism underlying lipotoxicity remains to be clearly defined.
Accumulating evidence supports that autophagy induction confers protection against lipotoxicity induced by palmitate, a 16-carbon saturated fatty acid and one of the most abundant fatty acids in human circulation [3, 4]. Autophagy, a lysosomal degradative pathway, plays a pivotal role in maintaining cellular homeostasis via eliminating old/damaged proteins/organelles. In addition, autophagy also has an important role in promoting cell survival by providing energy via recycling cellular components during long-term fasting [5, 6]. Impaired autophagy induction is associated with conditions that predispose to NAFLD in both experimental and clinical settings [7, 8]. Although the exact mechanism is not clear, several potential actions underlying NAFLD-associated autophagy dysfunction have been proposed, including both inhibitory expression of autophagy related genes and reduced production of degradative lysosomal enzymes [9]. The effect of palmitate on autophagy remains controversial and may be cell-type dependent [10-12]; however, it is unequivocal that autophagy induction confers protective effect against palmitate-induced cytotoxicity [3-4].
Sirtuins (SIRTs), a family of proteins that possess either deacetylase or mono-ADP transferase activity, are found in mammalian cells. Seven sirtuins have been identified (SIRT1 to SIRT7). These proteins regulate a variety of intracellular events, ranging from metabolism, inflammation, cell cycle, insulin secretion, to DNA repair [13, 14]. SIRT1 is the founding member in the sirtuin family, and localized mainly in the nucleus. It plays an important role in regulating cellular functions by modifying acetylation status of a wide range of molecules, including enzymes, histones and transcriptional regulators. As an NAD+-dependent deacetylase, SIRT1 catalyzes the deacetylation of protein targets at a specific lysine residue, accompanied by the hydrolysis of NAD+ and producing a de-acetylated protein, acetyl-ADP-ribose, and nicotinamide [15]. The discovery that SIRT1 exclusively utilizes NAD+ as a co-substrate raises the possibility that its activity is strongly correlated with energy status. Specifically a low-energy state such as fasting and caloric restriction would likely result in an increased SIRT1 activity due to increasing the level of NAD+. On the contrary, in a high-energy state such as high-fat diet and obesity, SIRT1 activity would decrease with a decreased level of NAD+ [16-19]. In liver hepatocytes, impaired SIRT1 signaling contributes to the pathogenesis of both alcoholic and non-alcoholic fatty liver disease [20-22].
Nicotinamide is the amide form of nicotinic acid (vitamin B3, Niacin) and an endogenous precursor for intracellular NAD+ biogenesis. The biosynthesis of NAD+ in mammalian cells occurs through two major pathways, salvage and de novo synthesis (the Preiss-Handler pathway) [23-25]. Both pathways involve nicotinamide as a precursor. As nicotinamide is also the general product of SIRT1-catalyzed deacetylation reactions, high concentrations of nicotinamide has been generally used as a SIRT1 inhibitor [26-28]. However, several recent studies, including ours, have documented that exogenous nicotinamide exposure was indeed associated with elevated NAD+ content, causing activated SIRT1 in a variety of cell types [29-31]. Moreover, SIRT1 is involved in the regulation of autophagy, as autophagy related genes such as Atg3, Atg7 and Atg8 (LC3) are all substrates that could be de-acetylated by SIRT1 [32].
Our group previously reported that nicotinamide activated SIRT1 in hepatocytes, whereby ameliorating palmitate-induced ER stress [30]. In this study, we extended our investigations and hypothesized that nicotinamide may be protective against lipotoxicity in hepatocytes. Our results demonstrated that nicotinamide supplementation protects against palmitate-induced hepatotoxicity via a mechanism involving SIRT1-dependent autophagy induction.
2. Methods and materials
2.1. Chemicals
Chemicals including palmitate (P0500), bovine serum albumin (BSA, A7030), myriocin (M1177), bay11-7082 (B5556), STF-083010 (SML0409), and DMSO (D2650) were purchased from Sigma-Aldrich. Other chemicals used in this study were purchased as follows: CLI-095 (InvivoGen, 14I24-MM), TLR4 neutralizing antibody (PAb hTLR4, InvivoGen, pab-hstlr4). Palmitate-BSA conjugates were prepared step by step as follows: powdered palmitate was dissolved in ethanol and saponified with sodium hydroxide. The sodium salt was dried, re-suspended in saline and heated at 80°C until completely dissolved. While the solution was still warm, isovolumetric 20% (w/v) BSA was added and the mixture was stirred at 50°C for 4 hours to allow palmitate to bind to BSA. The palmitate-BSA complex (3 mmol/L fatty acid: 1.5 mmol/L BSA; molar ratio, 2:1) was then sterilized by filtering using 0.2 mm diameter filter, and aliquoted for future use. In all the experiments except specific explanations, the control group was exposed to an equal amount of solvent (e.g. BSA, ethanol, DMSO).
2.2. Cell Culture
HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA), and cultured using Dulbecco's Modified Eagle Medium (DMEM from Sigma-Aldrich, D5648) containing 10% (v/v) fetal bovine serum (PAA Laboratories, A15-701), 2 mmol/L glutamine (Sigma-Aldrich, G3126), 100 U/mL penicillin, and 100 μg/mL streptomycin (Life technologies, 15140-122) at 37°C in a humidified O2/CO2 (95:5) atmosphere. Alpha mouse liver (AML)-12 hepatocyte culture was established from a mouse transgenic for human transforming growth factor α, and was obtained from the American Type Culture Collection (ATCC, CRL-2254), and was cultured in Dulbecco's Modified Eagle Medium/Ham's Nutrient Mixture F-12, 1:1 (DMEM/F-12, Sigma-Aldrich, 051M8322) containing 10% (v/v) fetal bovine serum (PAA Laboratories, A15-701), 5 mg/ml insulin (Sigma-Aldrich, I9278), 5 μg/ml transferrin (Sigma-Aldrich, T8158), 5 ng/ml selenium (Sigma-Aldrich, 229865), 40 ng/ml dexamethasone (Sigma-Aldrich, D4902), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life technologies, 15140-122) at 37°C in a humidified O2/CO2 (95:5) atmosphere.
2.3. RNA interference
Cultured cells were transfected with human Sirt1 siRNA (Santa Cruz Biotechnology, sc40986) using Lipofectamine 2000 according to the manufacturer's instructions. In the control group, cells were transfected with scrambled siRNA (Santa Cruz Biotechnology, sc37007).
2.4. LDH release Assay
Cells were seeded at 2×105/mL in 24 well plates and cultured overnight. After the indicated treatments, culture medium was collected and LDH was detected using an LDH assay kit (Thermo Scientific Inc., NC9674653) according to the manufacturer's instructions. The absorption at OD510 nm was measured using a SPECTRAmax 340PC microplate reader.
2.5. Fluorescence microscopy
HepG2 cells were seeded at a density of 1×104 cells/cm2 onto a sterilized cover slip on 6-well plates for overnight incubation. After each treatment for the indicated length of time, the cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature. The fixed cells were then incubated with propidium iodide (PI, Thermo Fisher Scientific, P3566) for 30 minutes, followed by three washes with ice-cold PBS. Images were captured using an Olympus fluorescence microscope.
2.6. Cell lysates and Western Blotting
Cells were seeded at 2×105/mL in 24 well plates and cultured overnight. After the indicated treatments, cells were lysed in lysis buffer consisting of the following: 20 mM Tris/HCl (pH 7.4), 150 mM NaCl, 10% glycerol, 2% NP-40, 1mM ethylene diamine tetra-acetic acid (pH 8.0), 20 mM sodium fluoride, 30 mM sodium pyrophosphate, 0.2% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1 mM dithiothreitol, 1 mM sodium vanadate, 50 μM leupeptin, and 5 μM aprotinin. Samples were held on ice with frequent vortexing for 15 minutes and centrifuged for 10 minutes at 12500 g. The protein content of each supernatant was quantified using the protein assay reagent from Bio-Rad Laboratories (Hercules, CA) in accordance with the manufacturer's instructions.
Fifteen micrograms of protein were separated via electrophoresis through a 12% polyacrylamide gel (15% for LC3I and LC3II) and transferred to 0.45 μm Immobilin-P polyvinylidene difluoride membrane (PerkinElmer Life Sciences). After transfer, membranes were blocked in 5% (wt/vol) BSA in phosphate-buffered saline (PBS) with 0.1% Tween 20 and probed with the antibodies specified, followed by secondary antibodies incubation. Specific proteins were detected with an enhanced chemiluminescence kit (PerkinElmer Life Science).
2.7. Statistical analysis
All data were expressed as means ± SD of at least three independent experiments. Statistical analyses were carried out using SPSS 11.0 software (Chicago, IL, USA). The differences between treatments were performed using one-way ANOVA and analyzed by post-hoc test with Fisher's least significant difference (LSD). Differences between treatments were considered to be statistically significant at p < 0.05.
3. Results
3.1. Nicotinamide protects against palmitate-induced cell death in hepatocytes
The effect of nicotinamide on palmitate-elicited hepatotoxicity was determined by pretreating both HepG2 cells (a human hepatoma cell line) and AML-12 cells (a non-transformed mouse hepatocyte cell line) with various doses of nicotinamide for 1 hour, followed by 0.5 mM palmitate exposure for overnight. LDH release was measured. As shown in Fig. 1, nicotinamide pretreatment alleviated palmitate-induced cell death in a dose-dependent manner in both HepG2 (Fig. 1A) and AML-12 cells (Fig. 1B), evidenced by a dose-dependent decline of LDH release at the presence of nicotinamide pretreatment. This protective effect of nicotinamide on palmitate-induced lipotoxicity was further confirmed by propidium iodide (PI) staining. PI is a popular red-fluorescent nuclear and chromosome counterstain, which only can permeant to dead cells and bind to DNA, resulting in an enhanced fluorescence. HepG2 cells were incubated with/without 1 mM nicotinamide for 1 hour, followed by an 8-hour exposure to 0.5 mM palmitate. The cells were subsequently fixed and stained with PI. As shown in Fig. 1C, palmitate at 0.5 mM caused a significant increase in positively stained cells, which showed a red fluorescence. Nicotinamide pretreatment significantly reduced the number of PI-positive cells when compared with cells treated with palmitate alone.
Fig. 1. Nicotinamide protects against palmitate-induced cell death in hepatocytes.
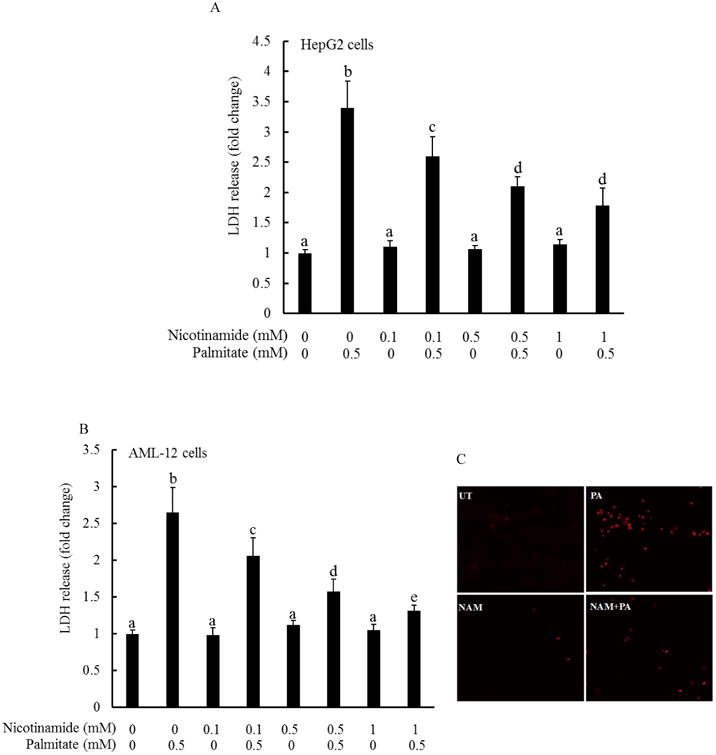
HepG2 cells (A) and AML-12 cells (B) were treated with palmitate at 0.5 mM at the presence/absence of various doses of nicotinamide for 16 hours. Cell viability was determined by LDH release measurement. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly (p < 0.05). C. Fluorescence microscopic examination of PI staining. HepG2 cells were treated with palmitate at 0.5 mM at the presence/absence of 1 mM nicotinamide for 8 hours. NAM, nicotinamide.
3.2. Nicotinamide is an autophagy inducer
Nicotinamide was reported to induce autophagy in fibroblasts [34] and neurons [35]. To determine the effect of nicotinamide on autophagy induction in hepatocytes, HepG2 cells were supplemented with nicotinamide for two hours. Total RNA and proteins were isolated. Autophagy is regulated by a group of proteins named autophagy-related protein (ATGs). We first measured the effect of nicotinamide on expression of microtubule-associated protein 1A/1B-light chain 3 (LC3) and Beclin-1 (Atg6), two ATGs. As shown in Fig. 2A, a two-hour exposure of nicotinamide led to a significant increases of both ATGs in HepG2 cells. LC3 is a soluble protein with a molecular mass of approximately 17 kDa and is distributed ubiquitously in mammalian tissues and cultured cells. During autophagy, the LC3 (LC3-I) originated in the cytosol is conjugated to phosphatidylethanolamine, forming the LC3-phosphatidylethanolamine conjugate (LC3-II). Then LC3-II is translocated to autophagosome membrane. When the fusion of lysosomes and autophagosomes take place, LC3-II is degraded together with other components in autophagosome. Thus, LC3-II acts as a marker which reflects autophagic activity [33]. Moreover, during protein degradation in autophagy, the recognition of ubiquitinated protein is facilitated by ubiquitin receptors. As p62 is the adaptor mediating the degradation of the substrate, its level is inversely correlated with autophagy. Upon nicotinamide exposure, LC3-II was markedly increased, while p62 protein abundance was significantly decreased (Fig. 2B). These results altogether suggest nicotinamide exposure induces autophagy in hepatocytes.
Fig. 2. Nicotinamide induces autophagy activation in hepatocytes.
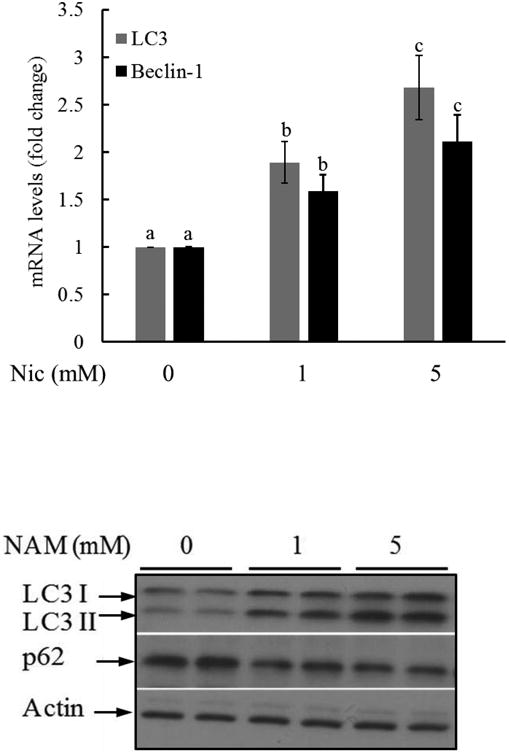
HepG2 cells were supplemented with nicotinamide (at 1 and 5 mM) for two hours. Total RNA and proteins were isolated and subjected to real time PCR for LC3 and Beclin-1 expression and Western blot for the detection of LC3-I/II and p62 protein abundance. A. mRNA levels of LC3 and Beclin-1. Bars with different characters differ significantly (p < 0.05). B. Western blot for LC3-I/II and p62 protein abundance.
3.3. Autophagy induction protects against palmitate-induced hepatotoxicity
The effect of autophagy suppression/induction on palmitate-elicited cell death was subsequently investigated. HepG2 cells were pretreated with or without bafilomycin A1 (Baf, 100 nM), chloroquine (CQ, 20 μM), or rapamycin (100 nM) for 1 hour, followed by 0.5 mM palmitate exposure for overnight. LDH release was measured after 16 hours. CQ and Baf are two widely used autophagy inhibitors. Bafilomycin A1 prevents the maturation of autophagic vesicles by inhibiting the fusion between lysosomes and autolysosomes. Chloroquine disrupted late-stage autophagy by inhibition of lysosomal function, leading to an accumulation of autophagic vesicles unable to undergo lysosomal digestion. Rapamycin is an autophagy inducer via inhibiting mTOR activity. As shown in Fig. 3A, both Baf and CQ pretreatment exacerbated palmitate-induced cell death, evidenced by robustly increased LDH release from these cells in comparison to cells exposed to palmitate alone. In contrast, rapamycin pretreatment almost completely prevented LDH release in HepG2 cells after a 16-hour palmitate exposure (Fig. 3B). To determine the role of autophagy in nicotinamide's protection against palmitate-induced lipotoxicity in hepatocytes, HepG2 cells were pretreated with Baf or CQ for 1 hour, followed by nicotinamide and palmitate exposure overnight. LDH release was measured 16 hours later. As shown in Fig. 4, both Baf and CQ abolished the protective effect of nicotinamide on palmitate-induced cytotoxicity.
Fig. 3. Autophagy induction protects against palmitate-induced hepatotoxicity.
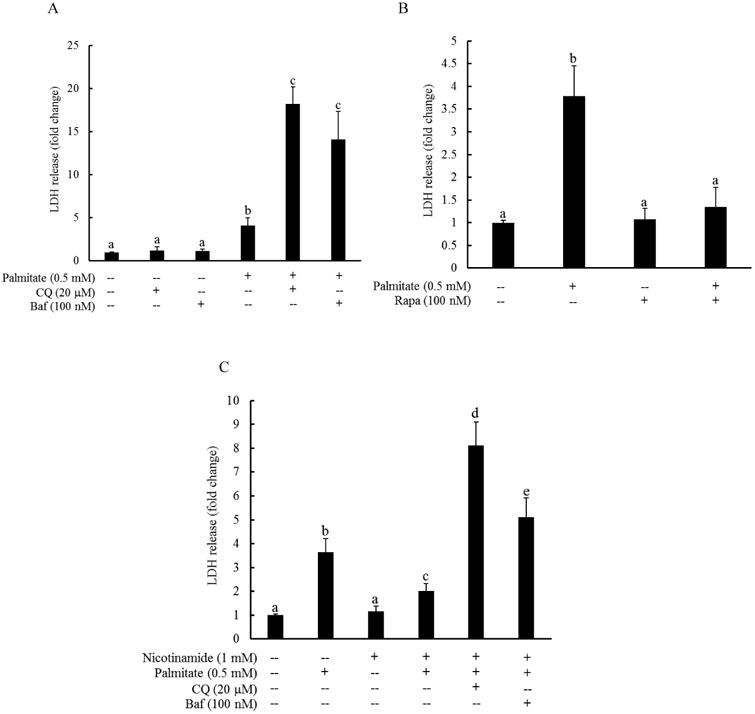
HepG2 cells were pretreated with or without bafilomycin A1 (Baf, 100 nM), chloroquine (CQ, 20 μM) (A), or rapamycin (Rapa, 100 nM) (B) for 1 hour, followed by 0.5 mM palmitate exposure for overnight. LDH release was measured after 16 hours. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly (p < 0.05). (C) HepG2 cells were pretreated with bafilomycin A1 (Baf, 100 nM) or chloroquine (CQ, 20 μM) for 1 hour, followed by nicotinamide and palmitate exposure for overnight. LDH release was measured 16 hours later. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly (p < 0.05).
Fig. 4. Nicotinamide-induced autophagy activation is SIRT1-dependent.
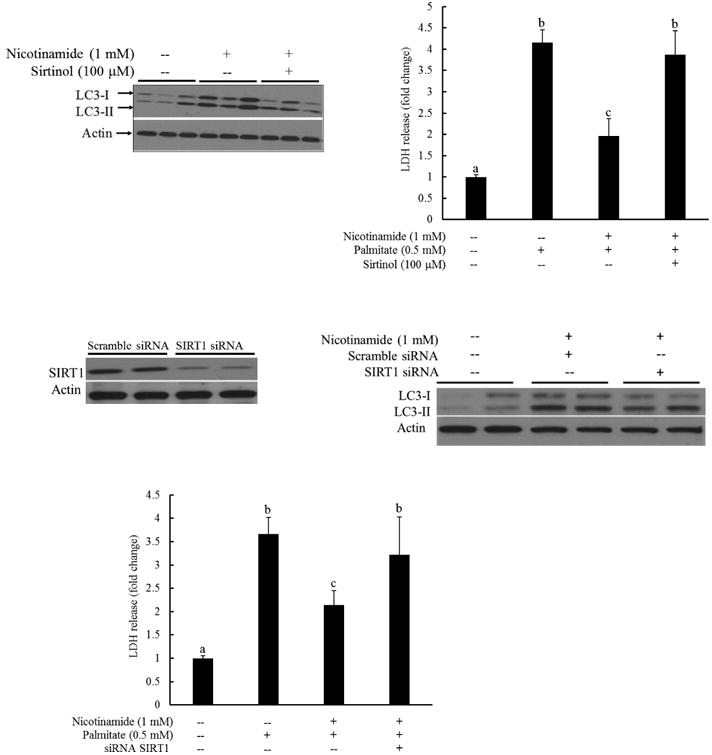
A. HepG2 cells were pretreated with sirtinol (100 μM) for 1 hour, followed by nicotinamide (1 mM) for another 2 hours. LC3-I and LC3-II levels were determined by western blotting using whole cell lysates. B. HepG2 cells were pretreated with/without sirtinol (100 μM) for 1 hour, followed by nicotinamide (1 mM) supplementation. Palmitate (0.5 mM) exposure was initiated 1 hour later and LDH release was measured after 16 hours to determine cell viability. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly (p < 0.05). C. HepG2 cells were transfected with SIRT1 siRNA for overnight. SIRT1 protein abundance was detected by Western blot. D. HepG2 cells were transfected with siRNA for SIRT1 for overnight, followed by nicotinamide (1 mM) for another 2 hours. LC3-I and LC3-II levels were determined by western blotting using whole cell lysates. E. HepG2 cells were transfected with siRNA for SIRT1 for overnight and treated with nicotinamide (1 mM) for one hour, followed by palmitate exposure. LDH release was measured 16 hours later. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly (p < 0.05).
3.4. Nicotinamide-induced autophagy activation is SIRT1-dependent
SIRT1 activation is positively associated with autophagy induction in a variety of cell types [36]. We previously reported that nicotinamide exposure activated SIRT1 in HepG2 cells [30]. To determine the involvement of SIRT1 activation in nicotinamide's protection against palmitate hepatotoxicity, sirtinol, a selective SIRT1 inhibitor, was employed to suppress SIRT1 activity. To determine the effect of SIRT1 on nicotinamide-instigated autophagy induction, HepG2 cells were first pretreated with sirtinol (100 μM) for 1 hour, followed by nicotinamide (1 mM) for 2 more hours. LC3-I and LC3-II levels were determined by Western blotting using whole cell lysates. Nicotinamide exposure increased LC3-II level, which was blunted by the pretreatment of sirtinol (Fig. 4A). The role of SIRT1 activation in the anti-lipotoxic effect of nicotinamide in hepatocytes was subsequently examined via pretreating HepG2 cells with/without sirtinol (100 μM) for 1 hour, followed by nicotinamide (1 mM) supplementation. Palmitate (0.5 mM) exposure was initiated 1 hour later and LDH release was measured after 16 hours to determine cell viability. As shown in Fig. 4B, sirtinol pretreatment almost completely abrogated the protective effect of nicotinamide on palmitate-induced lipotoxicity. The mechanistic involvement of SIRT1 in nicotinamide-elicited autophagy induction and hepatoprotective effect was further investigated via SIRT1 siRNA transfection experiments. As shown in Fig. 4C and D, SIRT1 knockdown by siRNA transfection alleviated LC3-II increases in response to nicotinamide supplementation. Accordingly, the hepatoprotective effect of nicotinamide in palmitate-treated HepG2 cells was offset (Fig. 4E).
4. Discussion
Nicotinamide as a dietary supplementation has been a source of vitamin B3 for decades. It is believed to have beneficial effects on energy metabolism due to its role as a predominant NAD+ precursor. In the present study, we used a cell culture model to investigate the anti-lipotoxic effect of nicotinamide and potential mechanism. Our data support our hypothesis that nicotinamide protected against palmitate-induced lipotoxicity in hepatocytes. Subsequent mechanistic investigations revealed that nicotinamide-instigated autophagy induction contributed to its anti-lipotoxic effect. Our data further unveiled that SIRT1 upregulation plays an important role in nicotinamide-triggered autophagy induction and the protection against palmitate-elicited lipotoxicity.
Although much progress has been made over the last several decades, the exact mechanism underlying palmitate-induced lipotoxicity remains to be clear and generally considered to be multifactorial. Nevertheless, evidence has been accumulating to support that autophagy induction protects hepatocytes against palmitate-induced cell death, although the effect of palmitate exposure on autophagy induction remains controversial [3, 4, 10-12]. We previously reported that tBHQ, a potent Nrf2 inducer, protected hepatocytes against palmitate-induced cell death via inducing autophagy [4]. Nicotinamide has been reported to induce mitophagy (mitochondria-specific autophagy) via elevating cellular NAD+/NADH ratio and SIRT1 protein activation [34]. These observations prompted us to examine if nicotinamide is capable of conferring its anti-lipotoxic benefit via inducing autophagy activation. This hypothesis was indeed supported by the data obtained in this study. We found that nicotinamide protected palmitate-induced cell death in a dose-dependent manner. Nicotinamide increased LC3-II levels, an indicator of autophagy induction, whereas the abundance of p62 protein (a specific autophagy substrate) was decreased, indicating that nicotinamide is an autophagy inducer. Furthermore, our data demonstrated that autophagy inhibitors efficiently blunted nicotinamide's protection against palmitate-induced lipotoxicity. However, rapamycin, a well-studied autophagy inducer, showed protection against palmitate-induced lipotoxicity. These data taken together indicate that nicotinamide protects against lipotoxicity in hepatocytes via autophagy induction.
Autophagy is a well-regulated intracellular event which refers to self-eating. Although the concept is simple, the development and regulation of the process is highly complicated [5, 6]. SIRT1, the founding member of sirtuin family, catalyzes the deacetylation of protein targets using NAD+ as an acetyl group acceptor [13-15]. Hepatic SIRT1 signaling impairment contributes to the pathogenesis of a variety of liver diseases, including both alcoholic and non-alcoholic fatty liver [20-22]. Accumulating evidence supports that SIRT1 positively regulates autophagy activation in a variety of cell types [36]. We previously reported that nicotinamide supplementation conferred hepatocytes resistance to palmitate-induced ER stress, wherein we demonstrated that the upregulation/activation of SIRT1 by nicotinamide contributed to its ER stress-alleviating effect [30]. These observations inspired us to examine the potential involvement of SIRT1 activation in nicotinamide-induced autophagy activation. Our data clearly demonstrated that SIRT1 inhibition prior to nicotinamide supplementation abolished nicotinamide-induced LC3-II elevation, confirming that nicotinamide-provoked autophagy induction is SIRT1-dependent. Moreover, SIRT1 inhibition by sirtinol also abolished the protection of nicotinamide on palmitate-induced hepatotoxicity, further confirming the involvement of SIRT1-depedent autophagy induction upon nicotinamide exposure.
In summary, our results demonstrated that nicotinamide supplementation protects hepatocytes against palmitate-induced cell death in hepatocytes and autophagy induction contributes to the anti-lipotoxic property of nicotinamide. Nicotinamide supplementation results in SIRT1 activation in hepatocytes, which plays a mechanistic role in nicotinamide-elicited autophagy induction and protection against lipotoxicity in hepatocytes. Although both HepG2 and AML-12 cell lines were used, it is obvious that our conclusion will be further strengthened with primary hepatocytes in the future studies. Moreover, in vivo investigations are definitely warranted to test the therapeutic potential of nicotinamide in NAFLD.
Highlights.
Nicotinamide protects hepatocytes against palmitate-induced cell death (lipotoxicity).
Nicotinamide is an autophagy inducer.
Nicotinamide protects hepatocytes against lipotoxicity via inducing autophagy activation.
Nicotinamide-induced autophagy induction is SIRT1-dependent.
Acknowledgments
This research was supported by grants from the National Institutes of Health NIAAA grants R01 AA017442 (Z Song) and Nature Science Foundation of China (81102111).
Abbreviation
- NAFLD
non-alcoholic fatty liver disease
- SIRT1
sirtuin 1
- SFA
saturated fatty acid
- NAD
nicotinamide adenine dinucleotide
- Baf
bafilomycin-A1
- CQ
chloroquine
- ER
endoplasmic reticulum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schaffer JE. Lipotoxicity: when tissues overeat. Current opinion in lipidology. 2003;14:281–7. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P. Nonalcoholic fatty liver disease. The New England journal of medicine. 2002;346:1221–31. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 3.Wu J, Wu JJ, Yang LJ, Wei LX, Zou DJ. Rosiglitazone protects against palmitate-induced pancreatic beta-cell death by activation of autophagy via 5'-AMP-activated protein kinase modulation. Endocrine. 2013;44:87–98. doi: 10.1007/s12020-012-9826-5. [DOI] [PubMed] [Google Scholar]
- 4.Li S, Li J, Shen C, Zhang X, Sun S, Cho M, Sun C, Song Z. tert-Butylhydroquinone (tBHQ) protects hepatocytes against lipotoxicity via inducing autophagy independently of Nrf2 activation. Biochim Biophys Acta. 2014;1841:22–33. doi: 10.1016/j.bbalip.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–76. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madrigal-Matute J, Cuervo AM. Regulation of Liver Metabolism by Autophagy. Gastroenterology. 2016;150:328–39. doi: 10.1053/j.gastro.2015.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukuo Y, Yamashina S, Sonoue H, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res. 2014;44:1026–1036. doi: 10.1111/hepr.12282. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Rodriguez A, Mayoral R, Agra N, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179. doi: 10.1038/cddis.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–1908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Chang F, Li F, Fu H, Wang J, Zhang S, Zhao J, Yin D. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun. 2015;463:262–7. doi: 10.1016/j.bbrc.2015.05.042. [DOI] [PubMed] [Google Scholar]
- 11.Park M, Sabetski A, Kwan Chan Y, Turdi S, Sweeney G. Palmitate induces ER stress and autophagy in H9c2 cells: implications for apoptosis and adiponectin resistance. J Cell Physiol. 2015;230:630–9. doi: 10.1002/jcp.24781. [DOI] [PubMed] [Google Scholar]
- 12.Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–98. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirschey MD. Old enzymes, new tricks: sirtuins are NAD(+)-dependent de-acylases. Cell Metab. 2011;14:718–9. doi: 10.1016/j.cmet.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong L, Mostoslavsky R. Fine tuning our cellular factories: sirtuins in mitochondrial biology. Cell Metab. 2011;13:621–6. doi: 10.1016/j.cmet.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Ann Med. 2011;43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshino J, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HJ, et al. Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J Proteome Res. 2011;10(2):722–31. doi: 10.1021/pr100892r. [DOI] [PubMed] [Google Scholar]
- 18.Tao R, et al. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem. 2011;286:14681–90. doi: 10.1074/jbc.M110.201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canto C, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–9. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Min HK, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15:665–74. doi: 10.1016/j.cmet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You M, et al. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am J Physiol Gastrointest Liver Physiol. 2008;294:G892–8. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- 22.Yin H, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–11. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkinson A, Day J, Bowater R. Bacterial DNA ligases. Mol Microbiol. 2001;40:1241–8. doi: 10.1046/j.1365-2958.2001.02479.x. [DOI] [PubMed] [Google Scholar]
- 24.Rutter J, et al. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–4. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- 25.Burkle A. Physiology and pathophysiology of poly (ADP-ribosyl) ation. Bioessays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 26.Bitterman KJ, et al. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 27.Hou X, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–26. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magni G, et al. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang SY, Kang HT, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem. 2012;287:19304–14. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, et al. Nicotinamide ameliorates palmitate-induced ER stress in hepatocytes via cAMP/PKA/CREB pathway-dependent Sirt1 upregulation. Biochim Biophys Acta. 2015;1853:2929–36. doi: 10.1016/j.bbamcr.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D, et al. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee IH, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374–9. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanida I, Ueno T, Kominami E. In: LC3 and Autophagy, in Autophagosome and Phagosome. Deretic V, editor. Humana Press; Totowa, NJ: 2008. pp. 77–88. [Google Scholar]
- 34.Jang SY, Kang HT, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem. 2012;287:19304–14. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu D, et al. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging. 2013;34:1564–80. doi: 10.1016/j.neurobiolaging.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salminen A, Kaarniranta K. SIRT1: regulation of longevity via autophagy. Cell Signal. 2009;21:1356–60. doi: 10.1016/j.cellsig.2009.02.014. [DOI] [PubMed] [Google Scholar]