

Abstract
Objective:
We report a case series of 10 patients with γ-aminobutyric acid (GABA)–transaminase deficiency including a novel therapeutic trial and an expanded phenotype.
Methods:
Case ascertainment, literature review, comprehensive evaluations, and long-term treatment with flumazenil.
Results:
All patients presented with neonatal or early infantile-onset encephalopathy; other features were hypotonia, hypersomnolence, epilepsy, choreoathetosis, and accelerated linear growth. EEGs showed burst-suppression, modified hypsarrhythmia, multifocal spikes, and generalized spike-wave. Five of the 10 patients are currently alive with age at last follow-up between 18 months and 9.5 years. Treatment with continuous flumazenil was implemented in 2 patients. One patient, with a milder phenotype, began treatment at age 21 months and has continued for 20 months with improved alertness and less excessive adventitious movements. The second patient had a more severe phenotype and was 7 years of age at initiation of flumazenil, which was not continued.
Conclusions:
GABA-transaminase deficiency presents with neonatal or infantile-onset encephalopathy including hypersomnolence and choreoathetosis. A widened phenotypic spectrum is reported as opposed to lethality by 2 years of age. The GABA-A benzodiazepine receptor antagonist flumazenil may represent a therapeutic strategy.
γ-Aminobutyric acid (GABA) was reported in 1950 as a ubiquitous compound arising from α-decarboxylation of glutamic acid,1 and subsequently recognized as the major inhibitory neurotransmitter of the CNS.2,3 GABA is metabolized by a combination of GABA transaminase (GABA-T or ABAT) and succinic semialdehyde dehydrogenase (SSADH). When GABA-T is absent, GABA is unable to convert to succinic semialdehyde resulting in a buildup of GABA and β-alanine, homocarnosine, and the GABA ketone, 2-pyrrolidinone.4,5
GABA-T deficiency is caused by biallelic, recessive mutations in the ABAT gene (MIM 137150). Since the first report of GABA-T deficiency in 1984,6 7 additional cases have been reported.7–11 The syndrome was previously characterized as early-onset epileptic encephalopathy with mortality within the first 2 years of life. We present a case series of 10 patients, including a previously unreported case and 2 with follow-up subsequent to initial publication, with an expanded phenotype and a novel therapeutic intervention.
METHODS
Following identification of our index case, a literature review was completed to identify all previously reported cases. Authors and care providers of living patients were contacted for follow-up. Patients were seen clinically and a comprehensive review of their medical history, EEG, imaging, and laboratory studies were conducted. With the exception of the brother of the initial proband, who was diagnosed posthumously, the remaining 9 patients have documented ABAT mutations. This study was approved by the Boston Children's Hospital Institutional Review Board.
Standard protocol approvals, registrations, and patient consents.
Standard consent was received from the parents of the patient shown in the video at Neurology.org to print and post the video online and they have been given the opportunity to review the manuscript.
RESULTS
Ten patients were identified with GABA-T deficiency; 5 are currently alive with age at last follow-up 18 months to 9.5 years (mean 6 years) (table). All patients presented with neonatal/infantile onset encephalopathy, hypotonia, and hypersomnolence. Median age at onset was 3 months (range 0–7 months) with 4 having neonatal presentations.
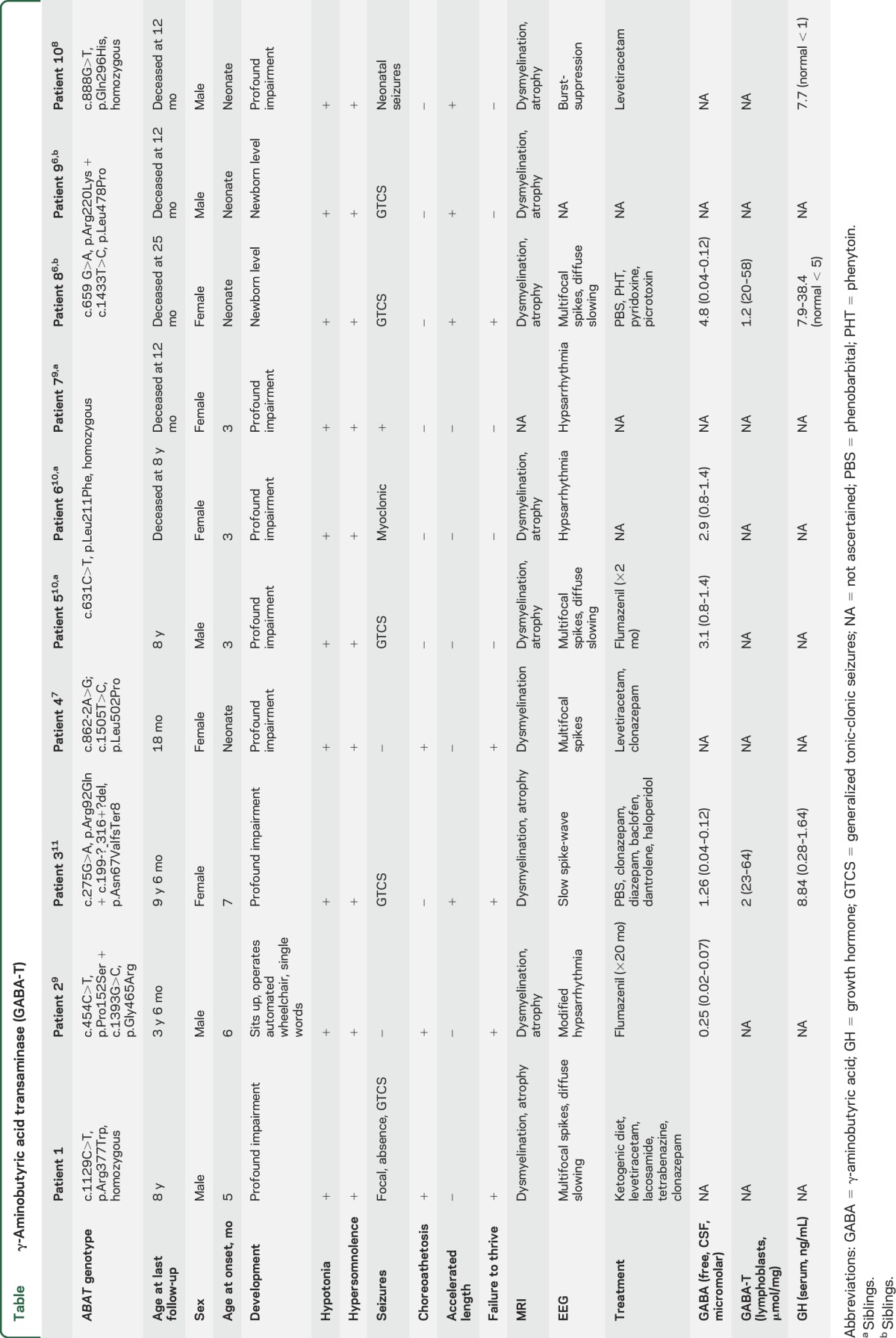
Table.
γ-Aminobutyric acid transaminase (GABA-T)
Patient 1, age 8 years, was born to first cousin parents of Mexican descent and presented at 5 months of age with decreased developmental progress, hypotonia, hypersomnolence, focal seizures, and virtually continuous choreoathetosis. Generalized tonic-clonic seizures occurred at 2 years, and absence seizures developed at age 6 years. Generalized convulsive status epilepticus occurred at age 6 years. EEGs have shown multifocal spikes and diffuse background slowing. MRI demonstrated progressive atrophy, particularly involving the sylvian fissures and pontomesencephalic cisterns (figure 1). Genetic studies were obtained starting with chromosomal microarray with single nucleotide polymorphism genotyping that demonstrated extensive regions of homozygosity, consistent with known parental consanguinity. These regions of homozygosity were specifically analyzed on exome sequencing. Clinical whole exome sequencing revealed a novel homozygous variant ABAT NM_000663.4 c.1129C>T; p.R377W. Sanger sequencing confirmed each parent as a heterozygous carrier of the mutation and provided orthogonal validation of the presence of the variant in the proband. The variant has not been previously reported in a patient with ABAT deficiency, but is predicted to be pathogenic by CADD, PolyPhen-2, SIFT, and MetaSVM, and is not present in the ExAC database of 121,000 chromosomes sequenced at this position in the genome.
Figure 1. MRI in γ-aminobutyric acid transaminase deficiency.
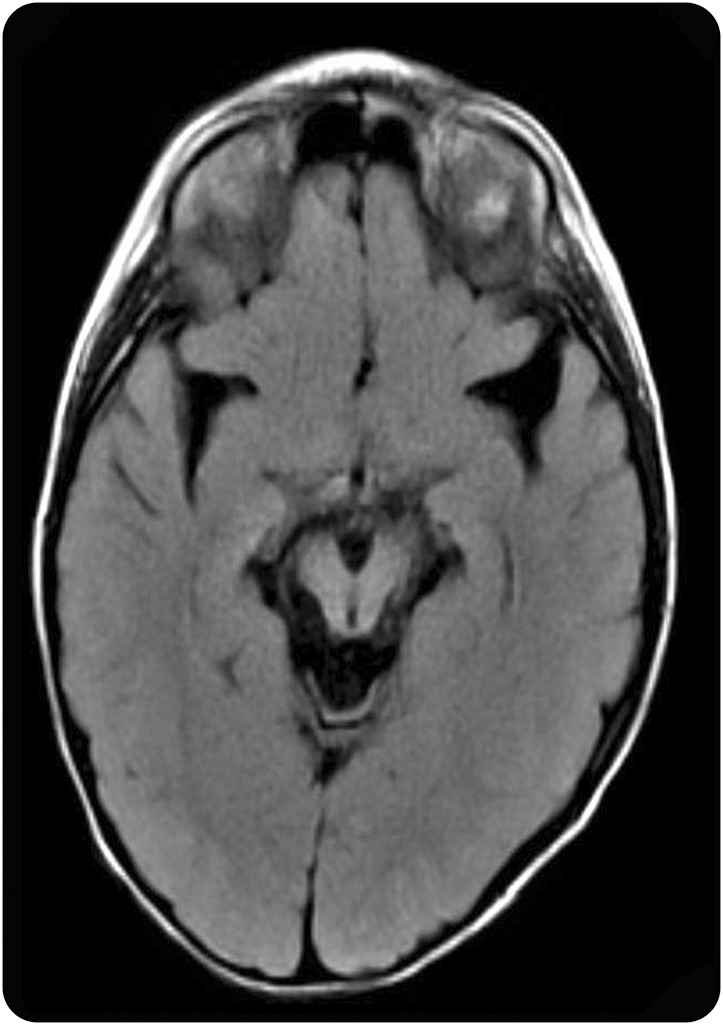
Axial MRI, fluid-attenuated inversion recovery sequence (patient 1, age 4 years), with prominent enlargement of sylvian fissures and pontomesencephalic cisterns.
Patient 2 presented at 6 months of age with feeding problems, developmental impairment, failure to thrive, hypersomnolence, decreased visual fixation, oculomotor apraxia, profound hypotonia, and hyperreflexia. Laboratory testing for routine chemistries as well as plasma ammonia, lactate, amino acids, creatine kinase, and urine organic acids was normal. Comparative genomic hybridization microarray was normal, and CSF amino acids and neurotransmitters were normal. A plasma metabolomics panel showed an increase in the GABA lactone 2-pyrrolidinone. Subsequent clinical whole exome sequencing showed compound heterozygosity for the ABAT variants c.454C>T; p.P152S and c.1393G>C; p.G465R. Sanger sequencing confirmed parental segregation and both variants were predicted to be pathogenic by SIFT, PolyPhen-2, LRT, and Mutation Taster. This patient's genotype was recently reported along with utilization of cellular models demonstrating impaired enzymatic activity and mtDNA copy number.9 CSF was then measured for GABA with elevations in the free (247 nmol/L, reference range 17–67) and total (33.4 μmol/L, reference range 4.2–13.4) levels. EEG at age 21 months showed high-voltage polymorphic delta and multifocal and generalized spike-wave consistent with modified hypsarrhythmia (figure 2A). The patient demonstrated virtually continuous choreoathetosis and myoclonus (video). The patient was initiated on a flumazenil infusion utilizing initial IV boluses of 0.01 mg/kg and reaching an infusion of 0.05 mg/kg/h, maintained on a regimen of 1.7 mg/kg/d of flumazenil. The EEG background improved after the first infusion and a follow-up EEG showed continued improvement in organization and background, although with some persistent generalized epileptiform activity (figure 2B). Clinical improvement was noted with improved alertness and interactions and less excessive adventitious movements, which has been sustained over 11 months. In addition, the patient continues to progress with an increase in purposeful movements, consistent response to verbal cues and commands, as well as ability to control a motorized wheelchair.
Figure 2. Pre and post-flumazenil EEG.

EEG of patient 2 at (A) 21 months of age, with high-voltage polymorphic delta and multifocal and generalized spike-wave; and (B) 26 months, with improved background organization but some persistence of generalized epileptiform activity (settings in all figures: HFF 70 Hz, LFF 1 Hz, sensitivity 10 μV/mm).
Patient 3 was previously reported at age 28 months.11 Follow-up of this patient, now age 9 years and 6 months, indicates she is nonverbal, nonambulatory with spastic quadriparesis and dystonia, and dependent on gastrostomy tube feedings, but is relatively stable with rare convulsive seizures. EEG background is slow spike-wave and her medications are phenobarbital, clonazepam, diazepam, dantrolene, baclofen, and haloperidol.
Patient 4 is a recently published case from an unrelated family from the South Indian state of Kerala.7 This patient presented similarly with choreoathetosis and hypersomnolence. Initial EEG at 5 months was normal, and follow-up at 18 months revealed multifocal epileptiform discharges.
Patients 5–7 are siblings. The sole surviving affected child, patient 5, is 8 years of age with little to no developmental progress following onset at 3 months of age with epileptic encephalopathy. The oldest sibling, patient 6, presented similarly and died at 8 years of age, and the youngest affected sibling, patient 7, died at 12 months of age.9,10 Based on the therapeutic trial in patient 2, patient 5 underwent a trial of flumazenil 1.7 mg/kg/d at the age of 7 years. After 2 months, this was discontinued due to increased wakefulness with agitation and no observed clinical benefit.
Patients 8 and 9 are the index sibpair associated with the original report of the disorder with mortality at 12 and 25 months of age, respectively.6 Patient 10 was published as a postmortem diagnosis using whole exome sequencing with fatality at 12 months of age.8
DISCUSSION
We describe a series of 10 cases of GABA-T deficiency and a therapeutic flumazenil trial in 2 patients. All patients presented with profound neurodevelopmental impairment and were unable to consistently visually track. A prominent movement disorder characterized by virtually continuous choreoathetosis while awake was observed in patients 1, 2, and 4. CSF values demonstrated a 2- to 40-fold increase in free GABA concentration. Serum growth hormone levels were elevated in the patients in whom it was measured (table), consistent with GABAergic stimulation of growth hormone secretion,12 and correlated with accelerated linear growth in those patients. EEG patterns were burst-suppression, modified hypsarrhythmia, multifocal spikes, slow spike-wave, and diffuse background slowing. MRI showed dysmyelination and cerebral atrophy.
The differential diagnosis of GABA-T deficiency includes the early-onset epileptic encephalopathies presenting in the neonatal and infantile periods. Demonstration of elevated CSF GABA levels, GABA peak on magnetic resonance spectroscopy, or presence of 2-pyrrolidinone on metabolomic screening would suggest the presence of this disorder, which can be confirmed by gene sequencing. SSADH deficiency is more common and the other known inherited disorder of GABA catabolism. While the clinical onset is usually later and less severe than GABA-T deficiency, acute infantile encephalopathy with choreoathetosis and myoclonus has been reported in a minority of patients.13 In the latter, evidence of excessive 4-OH-butyric acid is expected in physiologic fluids. While sleep disorders are prevalent in SSADH deficiency, characterized by polysomnographic demonstration of reduced stage REM and prolonged REM latency,14 the hypersomnolence of GABA-T deficiency is a distinguishing feature. This may be related to higher CSF GABA levels in GABA-T deficiency compared to the 1- to 8-fold increase reported in SSADH deficiency.15
Flumazenil intervention was implemented as targeted therapy in an attempt to decrease GABAergic innervation as the antagonist of the benzodiazepine binding site of the GABA(A) receptor. The intervention appears to be associated with clinical and electrophysiologic improvement in patient 2, who has been maintained on this for 20 months without adverse effects. The treatment did not show efficacy in patient 5, initiated at age 7 years. A milder phenotype and earlier intervention may be associated with enhanced treatment efficacy. The explanation for a broadening phenotypic spectrum may be less severe effects from certain mutations, although genotype–phenotype correlation is unclear at this time. Intrauterine and early developmental effects of deficiency of GABA degradative enzymes are likely based on the ontogeny of GABA concentrations, as well as glutamate and glutamine, including regional effects of distribution.16,17 Developmental characteristics of GABA are particularly dynamic, with prenatal/early postnatal depolarizing effects that transition to inhibitory responses in the postnatal period. The GABAergic system continues to evolve during childhood and even adolescence, with evidence of increasingly precise and rapid synaptic actions via alteration of interneuron properties, synchronization of GABA release, and selected receptor subunits.18 Inhibitory GABAergic systems control the size and propagation of neuronal assemblies within neural networks.19 In addition, GABA-mediated depolarization induces the differentiation of dendrites, development of excitatory synaptic mechanisms, and patterning of oscillatory activities.20
We present an updated series of 10 patients with GABA-T deficiency including a previously unreported case and a targeted therapeutic trial in 2 cases. This includes 4 patients surviving past age 7 years—the oldest at 9.5 years—in contrast to mortality previously reported in the first 2 years of life. More diagnosed cases are anticipated with the increasing utilization of comprehensive gene panels that include ABAT and whole exome sequencing for evaluation of children with unexplained early-onset epileptic encephalopathies and movement disorders.
Supplementary Material
GLOSSARY
- GABA
γ-aminobutyric acid
- GABA-T
γ-aminobutyric acid transaminase
- SSADH
succinic semialdehyde dehydrogenase
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Drs. Koenig, Riviello, Chung, Bain, Chiriboga, Ichikawa, Osaka, and Tsuji: acquisition of data, analysis and interpretation. R. Hodgeman: study concept and design, analysis and interpretation, preparation of manuscript. Drs. Gibson and Bonnen: study concept and design, critical revision of the manuscript for important intellectual content. Dr. Pearl: study concept and design, acquisition of data, analysis and interpretation, preparation of manuscript, study supervision.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
M.K. Koenig has received research funding from People Against Leigh Syndrome, Reata Pharmaceuticals Corporation, Ultragenyx Pharmaceuticals Corporation, Novartis Pharmaceuticals Corporation, EryDel S.P.A., and the Memorial Hermann Foundation; serves on the scientific advisory board for the Tuberous Sclerosis Alliance and editorial board for Journal of Child Neurology; and consults for Novartis Pharmaceuticals Corporation and Lundbeck Pharmaceuticals Services LLC. R. Hodgeman reports no disclosures relevant to the manuscript. J. Riviello reports his spouse is medical editor for Up To Date. W. Chung and J. Bain report no disclosures relevant to the manuscript. C. Chiriboga served on the scientific advisory board for Avexys and Roche Pharmaceuticals, is a practicing child neurologist as part of a Faculty Practice Organization (30% effort), is funded by Ionis Pharmaceutical and by the Department of Defense, Award 10259004, and received research support from the SMA Foundation. K. Ichikawa, H. Osaka, and M. Tsuji report no disclosures relevant to the manuscript. K. Gibson receives research support from the NIH (R01NS082286). P. Bonnen is a paid consultant to Retrophin and is supported by National Institute of Neurological Disorders and Stroke R01NS083726. P. Pearl receives research support from the NIH (R01NS082286) and the SSADH Foundation and receives royalties from Demos Medical Publishers for Inherited Metabolic Epilepsies and Neuro-Logic: A Primer on Localization. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Roberts E, Frankel S. gamma-Aminobutyric acid in brain: its formation from glutamic acid. J Biol Chem 1950;187:55–63. [PubMed] [Google Scholar]
- 2.Krnjević K, Schwartz S. The action of gamma-aminobutyric acid on cortical neurones. Exp Brain Res 1967;3:320–336. [DOI] [PubMed] [Google Scholar]
- 3.Hayashi TaKN. Action of c-amino acids on the motor cortex of higher animals, especially gamma-amino-b-oxy-butyric acid as the real inhibitory principle in brain. Presented at the 20th International Physiological Congress; July 30, 1956; Bruxelles, France; 410.
- 4.Parviz M, Vogel K, Gibson KM, Pearl PL. Disorders of GABA metabolism: SSADH and GABA-transaminase deficiencies. J Pediatr Epilepsy 2014;3:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaeken J, Casaer P, Haegele KD, Schechter PJ. Normal and abnormal central nervous system GABA metabolism in childhood. J Inherit Metab Disord 1990;13:793–801. [DOI] [PubMed] [Google Scholar]
- 6.Jaeken JCP, de Cock P, Corbeel L, Eeckels R, Eggermont E. Gamma-aminobutyric acid-transaminase deficiency: a newly recognized inborn error of neurotransmitter metabolism. Neuropediatrics 1984;15:165–169. [DOI] [PubMed] [Google Scholar]
- 7.Nagappa M, Bindu PS, Chiplunkar S, et al. Hypersomnolence-hyperkinetic movement disorder in a child with compound heterozygous mutation in 4-aminobutyrate aminotransferase (ABAT) gene. Brain Dev 2016;39:161–165. [DOI] [PubMed] [Google Scholar]
- 8.Louro P, Ramos L, Robalo C, et al. Phenotyping GABA transaminase deficiency: a case description and literature review. J Inherit Metab Dis 2016;39:743–747. [DOI] [PubMed] [Google Scholar]
- 9.Besse A, Petersen AK, Hunter JV, Appadurai V, Lalani SR, Bonnen PE. Personalized medicine approach confirms a milder case of ABAT deficiency. Mol Brain 2016;9:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besse A, Wu P, Bruni F, et al. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab 2015;21:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuji MAN, Obata T, Tomiyasu M, et al. A new case of GABA transaminase deficiency facilitated by proton MR spectroscopy. J Inherit Metab Disord 2009;33:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Racagni G, Apud JA, Cocchi D, Locatelli V, Muller EE. GABAergic control of anterior pituitary hormone secretion. Life Sci 1982;31:823–838. [DOI] [PubMed] [Google Scholar]
- 13.Zeiger WA, Sun LR, Bosemani T, Pearl PL, Stafstrom CE. Acute infantile encephalopathy as presentation of succinic semialdehyde dehydrogenase deficiency. Pediatr Neurol 2016;58:113–115. [DOI] [PubMed] [Google Scholar]
- 14.Pearl PL, Shamim S, Theodore WH, et al. Polysomnographic abnormalities in succinic semialdehyde dehydrogenase (SSADH) deficiency. Sleep 2009;32:1645–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria). Biol Psychiatry 2003;54:763–768. [DOI] [PubMed] [Google Scholar]
- 16.Das SK, Ray PK. Ontogeny of GABA pathway in human fetal brains. Biochem Biophys Res Commun 1996;228:544–548. [DOI] [PubMed] [Google Scholar]
- 17.Jansen EE, Struys E, Jakobs C, Hager E, Snead OC, Gibson KM. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol 2008;8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilb W. Development of the GABAergic system from birth to adolescence. Neuroscientist 2012;18:613–630. [DOI] [PubMed] [Google Scholar]
- 19.Swadlow HA. Fast-spike interneurons and feedforward inhibition in awake sensory cortex. Cereb Cortex 2003;13:25–32. [DOI] [PubMed] [Google Scholar]
- 20.Wang DD, Kriegstein AR. Defining the role of GABA in cortical development. J Physiol 2008;587:1873–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.