

Cyclin-dependent kinase, or CDKs, are known1 to orchestrate DNA repair (stitching) and cell cycle control (timing). In G1 and early S phase, double-stranded DNA breaks (DSBs) are predominantly repaired by non-homologous end-joining (NHEJ). This occurs due to the favored recruitment of 53BP1 and associated proteins, which excludes the scaffolding protein BRCA1. In late S phase, homologous recombination (HR) is the favored method of DSB repair. Mechanistically, HR occurs due to high CDK activity resulting in the phosphorylation of multiple proteins, including NBS1 and CtIP, which in turn stimulate the endonuclease activity of the MRN complex, and allows the binding of BRCA1 to CtIP and BRCA1s recruitment to the DSB. Recruitment of BRCA1 to the break, blocks 53BP1 binding and favors generation of single-stranded DNA promoting resection and repair by HR.
A slowly growing body of work demonstrates that non-cell cycle CDKs may also play a role in monitoring DNA integrity. For example, CDK9 (best known as the catalytic subunit of transcription factor P-TEFb) was found to be essential for recovery from replication stress.2 It was further demonstrated that CDK9 accumulated on chromatin in response to replication stress and that CDK9 and cyclin K interact with ataxia telangiectasia and Rad3-related protein and other checkpoint signaling proteins. Subsequent work3 has demonstrated that CDK9's role in the replication stress response may be activated by deacetylation by SIRT2 at lysine 48.
In the current volume of Cell Cycle,4 Nepomuceno and colleagues demonstrate CDK9 also plays a novel role in DSB repair. Specifically, the Nepomuceno team follows up on their previous demonstration5 of a potential interaction between BRCA1 (known to be a key regulator in DSB repair) and CDK9 via BRCA1s tBRCT domain. Now they demonstrate that this interaction is functionally relevant through a series of well-controlled experiments. First, they use co-immunoprecipitation assays to demonstrate that endogenous BRCA1 (and BARD1) stably interacts with endogenous CDK9. Next, they deplete MCF7 cells of endogenous CDK9 with shRNAi and exposed them to ionizing radiation (IR). This experiment revealed reduced formation of γ-H2AX foci following IR, as well as reduced HR repair, in cells depleted of CDK9. NHEJ repair was unaffected by CDK9 depletion. Next, the authors used immunofluorescence (IF) to demonstrate that CDK9 was physically co-localized with BRCA1-positive foci at DNA damage sites following IR. Again, using shRNAi to deplete cells of CDK9 and IF to measure the recruitment of repair protein to sites of DSB repair, the authors demonstrate significantly reduced recruitment of BRCA1 and RAD51 (which would temporally follow BRCA1 recruitment in HR repair), without measurable effect on the recruitment of 53BP1 to site of DNA damage. Finally, the authors demonstrate that depletion of CDK9 results in significantly reduced ability to survive IR long-term based on clonogenicity assays.
Figure 1 highlights an interpretation of the data presented. DSBs are recognized by the MRN complex (Mre11, Rad50 and Nbs1) and ATM, as highlighted in Fig. 1. It is yet unclear whether CDK9 enters the complex before or after MRN and ATM. For this review, I assume that CDK9 enters after ATM (Fig. 1). CDK9 may enter alone, or more likely in preformed complex with BRCA1. Once CDK9 has entered the complex, γ-H2A phosphorylation accumulates to high levels and BRCA1 blocks access of 53BP1, favoring DNA end resection and HR.
Figure 1.
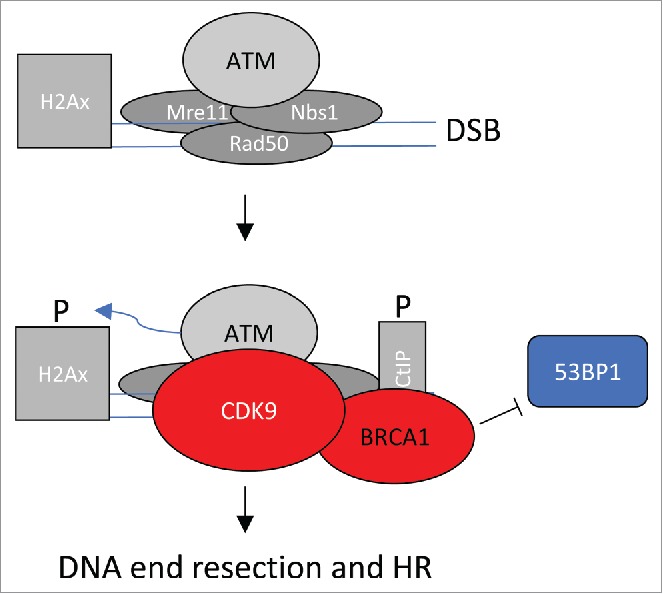
CDK9-dependent steps in DSB repair. DSBs are recognized by the MRN complex (Mre11, Rad50 and Nbs1) and ATM. CDK9, likely in complex with BRCA1, enters the complex as γ-H2A phosphorylation events accumulate. BRCA1 blocks access of 53BP1 favoring DNA end resection and HR.
It is tempting to speculate that CDK9 synergizes with the cell cycle-regulating CDKs to favor repair by HR as mediated by BRCA1 in cells late in S phase and to suppress NHEJ as mediated by 53BP1. Of course, many questions remain. The order of recruitment of other factors, such as CtIP, is unexplored in the current work. Does CDK9 require either cyclin T or K in this process? Does CDK9 even require its catalytic activity or is it serving merely a scaffolding role? If so, is BRCA1 a substrate or are other proteins in the complex or downstream complexes. Does CDK9 acetylation at lysine 48 play a role, as it does in the replication stress response? In any case, the data presented in this manuscript open a novel area of exploration and it will be exciting to see how CDK9 integrates DNA repair and cell cycle events.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol 2016; 19:1-9; PMID:28008184; http://dx.doi.org/ 10.1038/ncb3452 [DOI] [PubMed] [Google Scholar]
- [2].Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, Shyr Y, Cortez D. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep 2010; 11:876-82; PMID:20930849; http://dx.doi.org/ 10.1038/embor.2010.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang H, Park SH, Pantazides BG, Karpiuk O, Warren MD, Hardy CW, Duong DM, Park SJ, Kim HS, Vassilopoulos A, Seyfried NT, Johnsen SA, Gius D, Yu DS.. SIRT2 directs the replication stress response through CDK9 deacetylation. Proc Natl Acad Sci U S A 2013; 110:13546-51; PMID:23898190; http://dx.doi.org/ 10.1073/pnas.1301463110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nepomuceno TC, Fernandes VC, Gomes TT, Carvalho RS, Montiero AN, Carvalho MA. BRCA1 recruitment to damaged DNA sites is dependent on CDK9. Cell Cycle 2017; 16(7):665-672; PMID:28278048; http://dx.doi.org/ 10.1080/15384101.2017.1295177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Woods NT, Mesquita RD, Sweet M, Carvalho MA, Li X, Liu Y, Nguyen H, Thomas CE, Iversen ES Jr., Marsillac S, Karchin R, Koomen J, Monteiro ANA.. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci Signal 2012; 5(242):rs6; PMID:22990118; http://dx.doi.org/ 10.1126/scisignal.2002255 [DOI] [PMC free article] [PubMed] [Google Scholar]