

Most cells in the human body possess a primary cilium, which is singularly expressed at the cell surface.1 This tiny organelle serves as a cellular “antenna” for the perception of its surrounding extracellular environment. The signaling pathways that transmit through the cilium play indispensable roles in proper development and homeostatic maintenance of the human body. Primary cilium and mitotic spindle share a common structural origin where they arise from, the centriole. However, their expression is incompatible in that the cilium is generally assembled in interphase cells, while the spindles are formed during mitosis. Therefore, the cilium, or the lack thereof has close correlations with cell cycle progression. Defects associated with the cilium lead to a wide spectrum of genetic diseases, collectively recognized as ciliopathies. Primary cilium is also implicated in cancer development, since loss of cilia is commonly observed in a large number of tumor types. Thus far, it is not clear whether the loss of primary cilia plays a direct role in cancer progression or a mere consequence of transformation and remains a subject of keen debate. Nevertheless, the biological events, such as cell cycle progression, Hedgehog (Hh), canonical and non-canonical Wnt, and Notch signaling to name a few that are regulated by or processed at the primary cilium make it plausible to consider that the integrity of the cilium is associated with tumor formation.
Pancreatic ductal adenocarcinoma (PDAC), which accounts for 90% of pancreatic tumor cases, is one of the most lethal type of cancer, and activating Kras mutation is nearly universal (> 90%) in PDAC. The exocrine pancreas, which produces digestive enzymes, is anatomically composed of duct, centroacinar, and acinar cells. PDAC has long been thought to originate from duct cells, although recent evidences show that PDAC mainly arise from acinar cells.2 The acinar cells gain metaplastic ductal characteristics (acinar-to-ductal metaplasia: ADM), followed by conversion of ADM to pancreatic intraepithelial neoplasia (PanIN) that finally progress to PDAC. As is the case with many cancers, primary cilia are absent from the majority of human PanIN and PDAC lesions.3 Despite the lack of primary cilia in acinar cells, ADM cells assemble the organelle, indicating that PanIN and following PDAC developments are accompanied with loss of primary cilia.
It has been previously shown that blocking the oncogenic Kras signaling by administration of PI3K- or MEK-inhibitor restores primary cilia in pancreatic cancer cells3, suggesting that activated Kras signaling directly contributes to prevent ciliogenesis in PDAC cells. Moreover, in a recent study, HDAC2, which belongs to the histone deacetylase family, was identified as a suppressor of primary cilia formation in PDAC cells.4 Compromising HDAC2 function by pharmacological intervention or siRNA-mediated gene silencing results in the “re-”ciliation of several pancreatic cancer cells. Of note, HDAC1, which is an isoform of HDAC2 with similarities in both structure and function, was found irrelevant to cilia formation in these cells. Since HDAC2 is unable to associate with centrioles and primary cilia owing to its localization at the nucleus, HDAC2 was hypothesized to control the expression of gene(s) that promote(s) deciliation. Indeed, HDAC2 positively regulates the expression of Aurora A kinase (AURKA), which facilitates the disassembly of primary cilia in that the expression and subsequent kinase activity of AURKA was compromised by ablation of HDAC2. Although Kras enhances the transcription of AURKA probably through the Kras-MAPK pathway-dependent activation of the ETS2 transcription factor, combined depletion of HDAC2 and Kras leads to a higher increase in ciliation than singularly ablating the genes. This implies that HDAC2 exerts its effect on AURKA expression, independent of Kras. Furthermore, silencing of HDAC2 or Kras increases cells in quiescence due to ciliation, suggesting that HDAC2 and Kras could promote proliferation by suppression of primary cilia formation. Taken together, these findings allow us to propose that HDAC2 causes loss of primary cilia via controlling AURKA transcription in a Kras-independent manner, thus leading to proliferation in PDAC cells (Fig. 1).
Figure 1.
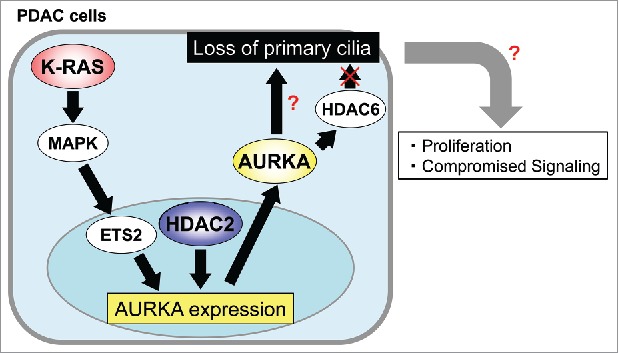
HDAC2 and Kras positively control AURKA transcription. AURKA induces loss of primary cilia through unidentified downstream protein(s) in PDAC cells. Deciliation could promote proliferation and/or compromised signaling in PDAC cells.
AURKA phosphorylates HDAC6, which allows it to deacetylate microtubules of ciliary axoneme, resulting in the disassembly of primary cilia in non-transformed cells.5 However, inhibition or ablation of HDAC6 was inefficacious against ciliation in pancreatic cancer cells.4 Given that AURKA is overexpressed in multiple cancers including PDAC, aberrant activation of AURKA might phosphorylate unexpected targets to execute deciliation, and it will be interesting to identify the downstream components of AURKA in PDAC cells.
As loss of primary cilia in normal pancreatic duct cells up-regulates Hh signaling and excess Hh signaling is a hallmark of pancreatic ductal adenocarcinoma6, it is tempting to speculate that deciliation contributes to tumor progression by enhancing Hh signaling. However, recent evidences suggest that activation of the Hh signaling in adjacent stroma cells, but not PDAC cells, is required for tumor progression; that is, Hh secreted from PDAC cells exerts paracrine effect on the stroma to activate the Hh signaling pathway.7 Therefore, impact of deciliation in PDAC cells is unclear at present. Future works will be needed to elucidate whether and how loss of primary cilia in PDAC cells affects primary cilium-mediated signaling and/or cell cycle progression, thereby practically accelerating the onset and growth of PDAC in vivo.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol 2011; 193:435-44; PMID:21536747; http://dx.doi.org/ 10.1083/jcb.201101005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rooman I, Real FX. Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development?. Gut 2012; 61:449-58; PMID:21730103; http://dx.doi.org/ 10.1136/gut.2010.235804 [DOI] [PubMed] [Google Scholar]
- [3].Seeley ES, Carrière C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res 2009; 69:422-30; PMID:19147554; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kobayashi T, Nakazono K, Tokuda M, Mashima Y, Dynlacht BD, Itoh H. HDAC2 promotes loss of primary cilia in pancreatic ductal adenocarcinoma. EMBO Rep 2017; 18:334-43; PMID:28028031; http://dx.doi.org/ 10.15252/embr.201541922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007; 129:1351-63; PMID:17604723; http://dx.doi.org/ 10.1016/j.cell.2007.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cervantes S, Lau J, Cano DA, Borromeo-Austin C, Hebrok M. Primary cilia regulate Gli/Hedgehog activation in pancreas. Proc Natl Acad Sci U S A 2010; 107:10109-14; PMID:20479231; http://dx.doi.org/ 10.1073/pnas.0909900107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, et al.. A paracrine requirement for hedgehog signalling in cancer. Nature 2008; 455:406-10; PMID:18754008; http://dx.doi.org/ 10.1038/nature07275 [DOI] [PubMed] [Google Scholar]