

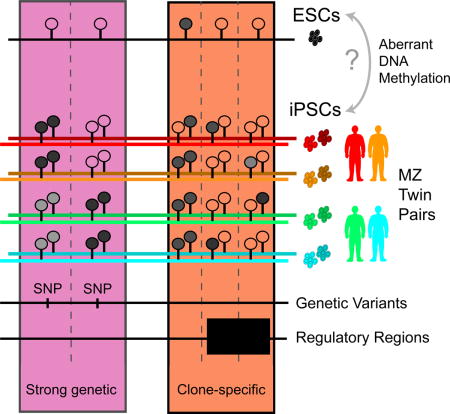
SUMMARY
iPSCs show variable methylation patterns between lines, some of which reflect aberrant differences relative to ESCs. To examine whether this aberrant methylation results from genetic variation or non-genetic mechanisms, we generated human iPSCs from monozygotic twins to investigate how genetic background, clone, and passage number contribute. We found that aberrantly methylated CpGs are enriched in regulatory regions associated with MYC protein motifs and affect gene expression. We classified differentially methylated CpGs as being associated with genetic and/or non-genetic factors (clone, passage) and found that aberrant methylation preferentially occurs at CpGs associated with clone-specific effects. We further found that clone-specific effects play a strong role in recurrent aberrant methylation at specific CpG sites across different studies. Our results argue that a non-genetic biological mechanism underlies aberrant methylation in iPSCs, and that it is likely based on a probabilistic process involving MYC that takes place during or shortly after reprogramming.
Graphical abstract
INTRODUCTION
Induced pluripotent stem cells (iPSCs) and their derived cell types are a powerful tool to model and potentially treat human disease. However, the epigenetic differences between iPSCs and embryonic stem cells (ESCs) present after reprogramming are still not well understood. During the past decade, studies have observed that CpG sites differ in methylation status between individual iPSC lines (methylation variation), as well as CpG sites that differ in methylation status between iPSCs and ESCs (methylation aberrancy) (Deng et al., 2009; Doi et al., 2009; Lister et al., 2011; Nazor et al., 2012; Ruiz et al., 2012). Aberrantly methylated CpG sites are further classified relative to their methylation status in the parental tissue of origin, and iPSCs can show loss of methylation, gain of methylation, or retain patterns similar to the tissue of origin (somatic memory). Some studies have identified aberrant CpG sites that are clone-associated or disappear with extended passage (Hussein et al., 2014; Kim et al., 2010; Nazor et al., 2012; Ohi et al., 2011). Other studies have suggested that genetic variation is an important regulator of gene expression and DNA methylation, as well as aberrant methylation in iPSCs (Burrows et al., 2016; Kyttala et al., 2016; Rouhani et al., 2014). However, previous studies aimed at identifying factors contributing to aberrant methylation have had limitations in part because they did not simultaneously examine the relative contributions of genetic (DNA variation) and non-genetic (i.e. clonality and passage) factors, or take into account the background rates of these factors. Thus, the mechanisms driving methylation aberrancy in iPSC lines are unclear and the relative importance of genetic versus non-genetic factors in this phenomenon has not yet been determined.
Genetic and non-genetic factors can potentially affect the methylation status of a large proportion of CpGs in iPSCs; therefore, aberrantly methylated CpGs can be associated with these factors by chance if background rate is not taken into consideration. Thus, it is necessary to first estimate how methylation levels at CpG sites across the genome associates with these factors. A study design using multiple pairs of monozygotic twins could be used to partition differentially methylated CpG sites across the genome based on association with genetic background, clone, or passage. Methylation variation associated with genetic variants will tend to look similar in all clones derived from either individual in a pair of monozygotic twins, but will vary between individuals in different twin pairs. Clone-specific variation that arises in a random or probabilistic manner, however, will show consistency within a specific clone at different passages, or between multiple clones across individuals from different genetic backgrounds, but won’t tend to cluster consistently by genetic background. Passage-associated variation will be consistently different in multiple clones between early and late passages, but won’t show biases for specific clones or genetic backgrounds. To establish that these associations are biologically meaningful, enrichments of the CpG sites for functional annotations such as the proximity of genetic variants, overlap with epigenetic regulatory regions and transcription factor motifs, and gene ontologies can be conducted, while correlated gene expression changes can be used to identify potential functional consequences of methylation. Thus, across the genome, differentially methylated CpGs can be classified according to their associations with genetic background, clone, and passage, as well as functional annotations.
Here, we generated 22 iPSC clonal lines from six individuals (3 pairs of older monozygotic twins). We profiled the 22 iPSC lines at early (passages 5 (p5) and 9 (p9)) and late (passage 20 (p20)) passages as well as fibroblasts (tissue of origin) using genome-wide methylation arrays and RNA-seq data. We estimate aberrant methylation of the iPSCs relative to ESCs and show that aberrant methylation affects gene expression and is enriched for CpGs associated with MYC and MYC-related protein motifs. We then identify genome-wide associations between CpG methylation variation and genetic background, clone, and passage and show that these associations likely result from relevant biological processes. We examine whether aberrant CpGs are enriched for CpGs associated with genetic and non-genetic effects and show that aberrant methylation preferentially occurs at CpGs showing clone-associated effects and is less enriched at sites associated with genetic background. Our study shows that non-genetic regulatory mechanisms associated with clone-specific effects most strongly underlie iPSC aberrancy.
RESULTS
Methylation and RNA-seq profiling of fibroblasts and iPSCs from identical twins
We generated 22 iPSC lines from fibroblasts obtained from 3 pairs of monozygotic twins (all female, Caucasian, >50 years of age) to examine and distinguish the roles of genetic background, clonal lines, and passage on iPSC methylation. iPSC lines from each twin set group were derived and cultured under parallel conditions, and all iPSC lines were characterized using standard criteria (Figure S1A–E). Samples were collected for analysis at p5, p9, and p20 (for a total of 51 iPSC samples) (Figure 1A). We successfully determined genome-wide patterns of DNA methylation of fibroblast parental populations and respective derived iPSC lines using the Illumina 450K HumanMethylome BeadChip for 49 of these samples. To examine if observed methylation changes were associated with altered gene expression, we also generated RNA-seq data for the 44 samples at passages 9 and 20 (Figure 1A). Finally, to examine whether DNA methylation patterns that were similar within twins were associated with genetic variants, we performed whole genome sequencing (WGS) of blood samples from the six participants (Figure 1A). To determine if fibroblast samples from the same genetic background showed similar DNA methylation profiles, we examined the methylation patterns in the fibroblast parental populations of each individual twin, and compared these to previously established methylation profiles of 62 fibroblasts from unrelated individuals (Wagner et al., 2014). Hierarchical methylation clustering showed the individual twin fibroblast lines interspersed randomly between the 62 unrelated individuals (Figure 1B), in agreement with previous studies demonstrating epigenetic divergence in aging twins (Fraga et al., 2005; Wong et al., 2010). Of note, the fibroblasts did cluster by twin set when analyzing 65 SNPs that are present on the methylation array, confirming they are correctly genetically matched (Figure 1C). These results show that fibroblast cells from individuals with the same genetic background show divergent DNA methylation, likely as a result of changes during the lifetime.
Figure 1. DNA methylation in iPSCs is associated with genetic background, clone and passage (see also Figure S1).
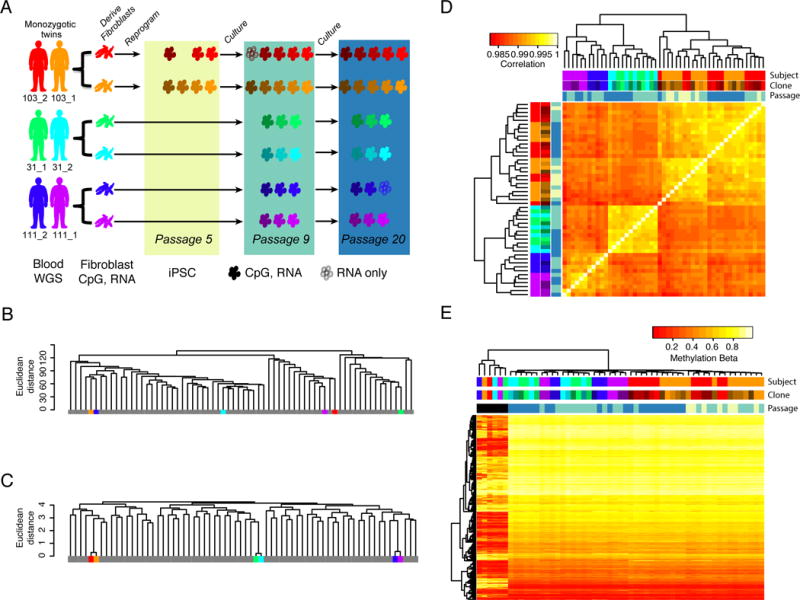
A) Study design indicating the fibroblast and iPSC samples derived from each subject in the three twin sets (103_2,103_1; 31_1,31_2; 111_2,111_1). iPSC clones are colored by shades of the subject’s color code and the colored rectangles depict indicated passages. Color codes are consistent throughout the paper. Blood samples were used for whole genome sequencing (WGS), while fibroblast and iPSC samples were used for DNA methylation (CpG) and RNA-seq (RNA) analyses. iPSCs indicated by filled cells have both methylation and RNA-seq data, while those indicated by outlines only have RNA-seq data. B) Dendrogram showing clustering of genome-wide methylation data of fibroblast samples from this study (color-coded) with data from 62 previously published fibroblast samples (grey) showing that fibroblasts do not cluster by genetic background. C) Dendrogram showing clustering at 65 SNPs present on the methylation arrays showing that twins cluster together based on genetic information. D) Hierarchical clustering and heat map of correlation of genome-wide methylation patterns of iPSC samples showing clustering by subject (genetic background), clone and passage (colored based on rectangle shades in 1A). E) Hierarchical clustering and heat map of methylation levels at 3,270 CpGs that have been shown to distinguish pluripotent and somatic cell types and also passed QC in our analysis. The fibroblasts (labeled black in passage annotation, six left most columns) randomly cluster whereas iPSC cluster by genetic background, clone and passage.
To examine methylation changes associated with reprogramming, we performed hierarchical clustering of the genome-wide methylation profiles of twin iPSCs. Unlike their parental somatic sources, the iPSCs clustered by twin set, and even showed interspersing of individual twins within the clustering of twin set groups (Figure 1D), demonstrating a strong role for genetic background in regulating their overall methylation status. Within these twin sets, there was also clustering based on clones, as well as by time in culture, indicated by passage number. When we included the fibroblast samples and focused on sites that were previously shown to distinguish pluripotent stem cells from somatic cell types (Nazor et al., 2012), we observed that while there was very little variation among iPSCs, and the majority of methylation changes between iPSCs and the fibroblasts had occurred by p5, we still observed clustering according to genetic background, as well as clone and passage (Figure 1E). Analysis of the RNA-seq data (hierarchical clustering of the 500 most variable genes) showed a similar pattern to the methylation data: iPSC lines clustered together based on genetic background as well as passage (Figure S1F). These combined results confirm previous findings that genetic background plays a large role in regulating overall CpG methylation (Burrows et al., 2016) (Kyttala et al., 2016) and gene expression differences between iPSC lines during reprogramming (Rouhani et al., 2014), and support previous studies showing that non-genetic factors including clone and passage also contribute to these differences (Nazor et al., 2012; Ruiz et al., 2012).
Aberrant CpG methylation is recurrent and varies by regulatory region
We next characterized aberrant methylation at CpG sites in each of the 49 iPSC samples in our study by whether they differed from ESCs. Sites classified as aberrant required a CpG to be different from a panel of 15 ESCs (Nazor et al., 2012) by an absolute methylation value of 0.2 as well as by a Z-score that corresponded to a 1% FDR across a single iPSC sample (see STAR Methods). In addition, because genetic variants near the single base extension (SBE) of the probe can produce assay artifacts (Chen et al., 2013) (see below), we removed 6,093 sites associated with these variants. This resulted in an average of 1,300 CpGs being called aberrant in each sample (Figure 2A). We also classified aberrant sites into three aberrant CpG classes according to how the iPSC methylation level related to the paired fibroblast sample: 1) within 0.2 of the paired fibroblast methylation level - somatic memory; 2) more than 0.2 higher - iPSC gain; and 3) more than 0.2 lower - iPSC loss. We identified on average 533 somatic memory, 168 iPSC loss and 436 iPSC gain sites per sample. In total, we identified 9,310 sites that were aberrant in one or more iPSC sample, of which 7,372 sites (79%) were aberrant in either one sample or had consistent classification across multiple samples, and 1,938 sites (21%) that were aberrant in multiple samples but with different classifications (Figure 2B, Table S1A). A total of 466,154 sites were not considered aberrant in any iPSC sample. In general, aberrant sites were present in one (3,794; 41%) or a few (2–5, 2926; 31%) samples, but in some cases, sites were aberrant in 10 or more samples (1749; 19%). Thus, while only 0.3% of CpGs are aberrantly methylated in a given sample, the same sites are recurrently aberrant across multiple samples, suggesting that the process underlying methylation aberrancy is not random.
Figure 2. Aberrant DNA methylation in iPSCs (see also Tables S1A, S1B, S2, S3A, and Figure S2).
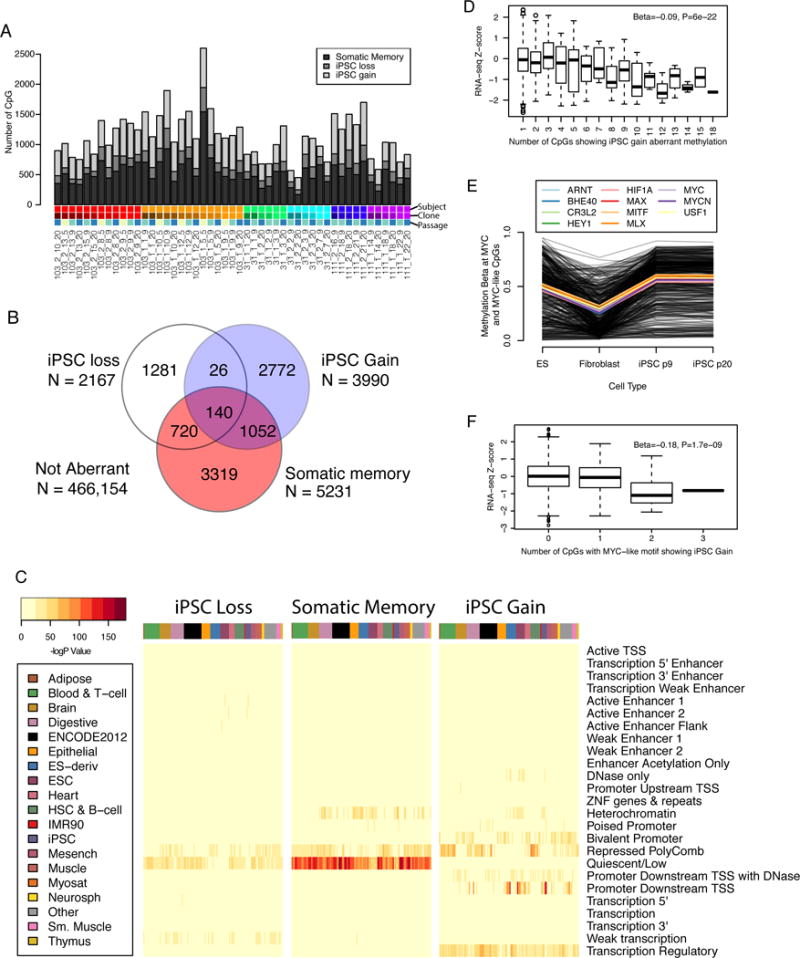
A) Barplot showing the number of aberrant methylation sites in each of the 49 samples, broken down into aberrant types. iPSC lines are color coded by subject, clone, and passage (Figure 1A). B) Venn diagram showing the classification of CpG sites aberrant in one or more samples. C) Heat maps showing enrichment −log P-value for hypergeometric association between ROADMAP regulatory regions (25 states) in 127 reference epigenomes and CpG sites associated with each aberrant classification. D) Boxplot showing the RNA-seq normalized expression values according to the number of aberrantly methylated iPSC gain CpGs annotated to the gene. Each data point that goes into the box plot corresponds to the expression value of a single gene in a single sample considering the number of neighboring aberrant CpGs. Beta and P-values derive from linear regression of raw data after including sample name as a covariate. E) Average methylation Beta values for CpGs that carry MYC or MYC-like motifs (identified as enriched in iPSC gain sites by CentriMo) and show iPSC gain. Each black line indicates an individual CpG and the colored lines indicate the average expression value for all CpGs associated with each motif type. F) Boxplot as in D, but restricted to the CpGs carrying the MYC and MYC-like motifs that showed at least 1 iPSC gain in 1 individual.
We investigated the distribution of aberrant sites across the genome to determine if they preferentially occurred in functional elements or were associated with gene annotations. We tested if the three different classes of aberrant sites were enriched in any of 25 chromatin states based on imputed data in 127 reference epigenomes (Ernst and Kellis, 2015) using a hypergeometric test. We observed that iPSC loss sites were associated with repressed regions bound by polycomb, quiescent regions (not bound by protein), and weak transcription regions in the majority of tissue-types, but were also associated with active enhancers and active enhancer flanking regions in a few of the ESC and iPSC samples (Figure 2C). Somatic memory sites showed strong enrichment for quiescent regions, as well as being enriched for repressed polycomb regions and heterochromatin across a variety of tissue-types. iPSC gain sites were associated with bivalent promoters, repressed polycomb regions, promoters downstream of transcription start sites (TSS), and regulatory transcription regions across a variety of tissue and cell-types. In ESC and iPSC cell-types, the iPSC gain sites were also enriched in DNaseI sensitive sites, heterochromatin, and promoters downstream of TSS, but not in repressed polycomb regions. To examine enrichment of functional and regional gene annotations, we first identified genes that were enriched for specific subtypes of aberrant methylation relative to overall rates (Table S1B). We performed gene set enrichment on these gene lists using GOseq with Gene Ontology (GO) and the Molecular Signatures Database, which also includes chromosome region information, and although no functional gene annotations were observed, genes associated with either iPSC gain or any type of aberrant CpG showed enrichment at a telomeric region of chromosome 3 (chr3p26) (Table S2). These findings demonstrate that dependent on their classification (memory, gain or loss) aberrantly methylated sites in iPSCs are enriched in functionally distinct chromatin states and can show regional association in the genome.
iPSC gain aberrant CpG sites affect expression of associated genes
We next investigated whether aberrant methylation affected the expression levels of associated genes. For each of the 42 iPSC samples (with methylation arrays at p9 and p20) (Figure 1A), we matched the DNA methylation data with normalized RNA-seq expression values for each gene and categorized the genes (none, iPSC loss, Somatic Memory, iPSC gain, multiple) according to the number and types of aberrant sites annotated to that gene in that sample. For each category, we pooled all gene expression levels (42 expression levels per gene) and after adjusting for sample, tested whether the categories showed overall differences in gene expression using multiple linear regression. We observed systematically lower gene expression values for iPSC gain aberrant methylation that correlated with the number of aberrant CpGs per gene (i.e. genes with more aberrant CpGs had lower expression) (Figure 2D), but did not see gene expression changes for other types of aberrant methylation (Figure S2A). These results show that aberrant methylation, and specifically iPSC gain aberrant methylation, is associated with gene expression changes.
To investigate altered transcription factor binding as a potential mechanism underlying the association between aberrant methylation and gene expression changes, we conducted a motif enrichment analysis. We observed that all three classes of aberrant methylation were enriched for short AT-rich motifs that did not match known transcription factor motifs (Table S3A). In addition, iPSC gain CpGs were enriched for transcription factor motifs including CXXC1 and MYC, as well as 10 other motifs that were similar to the MYC motif and shared at least four of the six central base pairs (CACGTG), which we defined as MYC-like motifs (Table S3A). The CpGs that contained MYC and MYC-like motifs tended to be intermediately methylated (Beta values between 0.2 and 0.8) and were on average higher in the iPSC lines compared to ESC lines (Figure 2E). Furthermore, genes near aberrantly methylated CpG probes overlapping MYC and MYC-like motifs had significantly lower expression levels compared with genes near non-aberrantly methylated probes (Figure 2F). Genes associated with two or three iPSC gain CpGs overlapping MYC and MYC-like sites showed on average 0.97 Z-score units lower expression than genes in iPSC samples with non-aberrantly methylated MYC and MYC-like CpGs. These data show that the iPSC gain aberrant methylation of CpGs consistently lowers the expression of associated genes potentially through the binding of regulatory proteins including MYC.
Genome-wide association of CpG methylation levels with genetic background, clone, and passage
We next identified differentially methylated CpGs associated with genetic and/or non-genetic (clone, passage) factors in order to be able to test whether aberrant methylation occurs preferentially at CpGs associated with these effects above and beyond background rates. We first characterized how methylation variation at genome-wide CpGs (481,557 sites on the 450K BeadChip) was associated with these factors across the 49 iPSC samples. We performed two separate ANOVA analyses that varied according to: 1) genetic background, 2) clone, and 3) passage. Statistical tests were performed on the 42 samples from p9 and p20 to ensure that groups of samples were balanced across individuals, although we show examples including data from all passages. We then classified each CpG according to its association with genetic background, clone, passage, or a combination of these three factors (hereafter referred to as the seven CpG predictor classes, Figure 3A, Table S1A). We expected strong genetic effects to be associated with both genetic background and clone because the multiple clones from each twin pair have the same genetic background. On the other hand, weak genetic effects were expected to show higher variability between clones from the same twin pair and thus only be associated with genetic background and not clone. We therefore named the CpGs associated with both genetic background and clone “strong genetic” and those associated with only genetic background “weak genetic”. Most sites associated with both genetic background and clone showed a proportional relationship between the −logP values of these two factors, consistent with a genetic effect (Figure S3A); but there was also a group of CpGs that showed higher clone association, which are likely clone-specific effects that were falsely associated with genetic background because the multiple samples (p9 and p20) have the same genetic background. Therefore, we applied an additional criterion to correctly classify the CpG sites that were more strongly associated with clone effects, specifically the CpGs with a stronger clone P-value were grouped with the other CpGs showing “clone-specific” effects. This resulted in 12,063 “strong genetic” and 2,903 “strong genetic + passage” CpGs moving respectively into the “clone-specific” and “clone-specific + passage” classes (see STAR Methods). In total, 154,724 (32.1%) of the CpG sites tested showed methylation variation associated with genetic background, clone, or passage across the iPSC lines (Figure 3A). Strong and weak genetic effects, with or without passage, were associated with 113,758 CpGs (73.5% of associated sites), while non-genetic effects were associated with 40,966 CpGs (26.5%), including those showing clone-specific (N=33,754), passage-specific (N=3,351), or both clone and passage (N=3,861) associations. We use these background rates of association between differentially methylated CpGs and genetic background, clone, and passage below when we examine factors influencing aberrant methylation; however, we first set out to confirm that the CpG predictor classes are biologically relevant and have functional consequences.
Figure 3. Methylation variation predictor classification has functional significance (see also Tables S1B, S2, S3B, S4, and Figure S3).
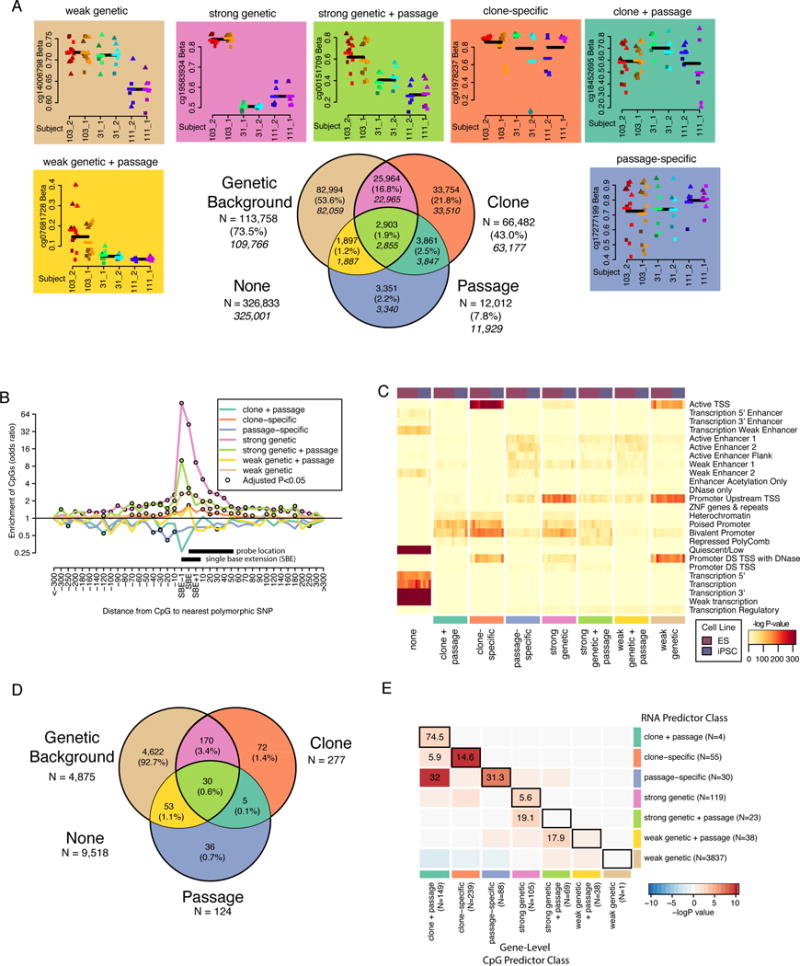
A) Venn diagram showing the overlap of CpG sites associated with genetic background, clone, and passage by ANOVA (FDR < 0.05). Percentages are of the total number of CpGs associated with one or more factor and the italicized numbers indicate the number of CpGs in each group after removal of SBE sites. Plots above and next to the Venn diagram show examples of CpGs that fall into each of the seven categories of the Venn diagram and are colored according to their classification. Within each plot, points are colored according to clone and shapes indicate passage (circle = P5, square = P9, triangle = P20). Black lines indicate the mean of all samples with the same genetic background and colored lines indicate mean of all samples from the same individual. B) Line plot showing odds ratios (OR) of the relationship between a CpG being associated with genetic background and harboring a polymorphic genetic variant at a given distance from the probe for each CpG predictor class from Figure 2A. CpGs are grouped according to distance from SBE site (e.g. −10 includes −2 to −10 and 10 includes +2 to +10). Open black circles indicate that the association was significant at FDR < 0.05. X-axis indicates distance from SBE site. Y-axis is on a log scale. Black bars indicate the position of the assay probe or bases considered to be SBE variants. C) Heat maps showing enrichment –log P-value for hypergeometric association between ROADMAP regulatory regions (25 states) in 8 ES and 5 iPSC reference epigenomes and CpG sites associated with each predictor classification. D) Venn diagram showing the number of genes associated with each RNA predictor class by ANOVA (FDR < 0.01). E) Heatmap showing OR’s for the overlap between gene-level CpG predictor class (columns) and RNA predictor class (rows). Black boxes surround comparisons where the same predictor classification group was compared between methylation and gene expression. Cells are colored according to −logP-value. Inf corresponds to “infinite” and reflects a positive association when an OR cannot be calculated due to a missing cell value.
Specific genetic variants near and distal to CpG sites explain association with genetic background
To validate that the association of CpG methylation levels with genetic background was due to genetic variants and not to experimental artifacts such as batch effects, we examined the contribution of specific genetic variants. Using WGS (see STAR Methods, Table S4), we identified the nearest polymorphic variant to each CpG probe and compared whether CpG probes that were closer to a polymorphic variant were more likely to be associated with each of the seven CpG predictor classes (Figure 3B). We observed association between the distance of the polymorphic variant and the odds of being associated with genetic background predictors that peaked at the probe and extended up to ~200bp on either side. CpGs with a genetic variant at or near the position of the 3′ SBE were highly likely to be associated with strong genetic, strong genetic + passage, and weak genetic effects, consistent with genetic variants at the SBE disrupting the methylation assay. Because these are likely artifacts of the assay, we removed 6,093 sites associated with SBE variants (positions −1, 0, and 1) from further analyses (see italicized numbers in Figure 3A). The elevated enrichment up to ~200bp upstream and downstream from the probe (which has a length of 50 bp), however, suggests that nearby variants influence methylation at CpG sites. CpGs near monomorphic variants did not show this effect (Figure S3B). Previously identified methylation quantitative trait loci (meQTL) were also enriched in strong and weak genetic classes (Figures S3C and S3D). Thus, CpG sites in genetic background predictor classes are more likely to be in close proximity and associated with functional genetic variants, while CpGs in clone and passage predictor classes tend to not be near genetic variants. This supports the use of the seven CpG predictor classes as a tool to differentiate genetic vs. clone and passage effects on aberrant methylation.
CpGs predictor classes are differentially enriched in regulatory regions and for transcription factors
To examine whether CpGs in different CpG predictor classes reflect different regulatory processes, we examined how CpGs in each of the seven CpG predictor classes associated with regulatory regions. First, we examined enrichment in regulatory elements, as defined by chromatin states in ESC and iPSC lines (Figure 3C). The 325,001 CpGs not associated with any of the CpG predictor classes (indicated as none in Figure 3C) were more likely to be in regions not targeted by regulatory proteins (protein-free quiescent regions and transcribed regions of genes), while associated CpGs tended to lie in transcription start sites, promoters and enhancers. Clone-specific CpGs were most associated with active TSS, but not associated with enhancers, while strong and weak genetic CpGs were more associated with promoters upstream of the TSS. We then examined transcription factor motifs and observed that genetic (both strong and weak) and clone-specific CpG predictor classifications were enriched for CG-rich motifs that did not match known transcription factors (Table S3B). However, CpGs in the passage-specific predictor class, as well as those in the combined genetic + passage classes (weak genetic + passage and strong genetic + passage), were enriched for a motif similar to the JUN/FOS motif. The CpGs associated with this motif tended to show decreased methylation between p9 and p20 (average decrease 0.054, Wilcox P=1×10−19: Figure S3E), which may be biologically important given that c-Jun suppresses the expression of pluripotent genes (Liu et al., 2015). Thus, changes at JUN/FOS targets are associated with changes during passaging, in a manner that may be modified by genetics. These results show that CpGs associated with different CpG predictor classifications occur in different regulatory regions and thereby suggest that genetic and non-genetic factors affect different regulatory processes.
Non-genetic factors are associated with methylation and expression of genes relevant for stem cell function
We next examined if genes associated with different CpG predictor classes showed gene expression changes consistent with that class. We analyzed the RNA-seq data (44 iPSC samples at p9 and p20) (Figure 1A) using the same ANOVA approach that we used for the methylation data and classified gene expression patterns according to seven RNA-seq predictor classes, resulting in 4,988 associated genes (Figure 3D, Table S1B). To enable gene-level comparisons with the methylation data, we generated gene-level CpG predictor classifications by identifying genes enriched for CpGs from each CpG predictor class using a hypergeometric test, identifying 1,147 genes enriched for any CpG predictor class (Table S1B, Figure S3F). Using a Fisher’s exact test, we then estimated the overlap between gene-level CpG predictor class and RNA predictor class (Figure 3E). For most predictor classes (i.e. “clone-specific”), genes associated with the gene-level CpG class were enriched for genes in the respective RNA predictor class. When we observed cross-category enrichment, this was generally in similar categories (e.g. clone+passage gene-level predictor class vs. passage-specific RNA predictor class). Strong genetic gene-level CpG effects showed the largest overlap with strong genetic + passage RNA effects, possibly indicating a delayed effect of methylation changes on RNA expression. Weak genetic gene-level CpG effects were not supported in the RNA data, but this could be due to low number of genes enriched for weak genetic CpG effects. Because clone-specific, clone + passage, and passage effects showed strong overlaps, we analyzed the list of genes that showed overlap between the gene-level CpG and RNA predictor classes of these three classes (Figure S3G). Interestingly, this list contains genes that have been previously shown to be differentially methylated in iPSCs (e.g. TMEM132D, DPP6, and FAM19A5) (Lister et al., 2011; Ruiz et al., 2012). The long non-coding RNA gene MEG3 from the DLK1-D103 imprinting locus, which has been shown to affect pluripotent stem cell function (Benetatos et al., 2014; Carey et al., 2011; Mo et al., 2015; Stadtfeld et al., 2010) was also differentially expressed. These results show that the patterns of the CpG and RNA predictor classes are similar, and that non-genetic factors (e.g. clonality, passage), by affecting methylation variation, may alter expression of important genes relevant for stem cell function.
Functional characterization of genes enriched for CpG predictor classes
To further examine the functional implications of the CpG predictor classes, we conducted a gene set enrichment using GOseq for each of the seven gene-level CpG predictor classes (Table S1A, Figure S3F). We observed modest functional enrichment, with strong-genetic genes enriched for MHC protein complex and antigen presentation, clone-specific genes enriched for cell-cell adhesion and genes on the X-chromosome, and passage-related genes (passage-specific, clone + passage, and strong genetic + passage) associated with genes on the X-chromosome (Table S2). These results suggest that genes enriched for differentially methylated CpGs in specific predictor classes have similar functions, with genetic classes being associated with highly genetically variable loci, and clone and passage classes reflecting mechanisms relevant to reprogramming, such as X-chromosome reactivation. Taken together, these results suggest that the CpG predictor classes reflect physiological differences that are consistent with the class type (i.e. genetic, clone, or passage) and have downstream effects on gene expression.
iPSC gain aberrant CpG sites are associated strongly with clone-specific effects
To explore the relative contributions of genetic and non-genetic factors to aberrant methylation, we first performed hierarchical clustering of aberrant calls and examined how the samples clustered by these factors (Figure 4A). We observed that clones showed strong similarity to each other (only a few sets of iPSC samples did not cluster by clone) and that genetic background and passage showed some, but not complete, clustering (Figure 4A). Of note, sites with genetic variants near the SBE, when included, incorrectly drove clustering by genetic background because SBE sites appeared as strong somatic memory calls (see STAR Methods and Figure S4A–C). These results suggest that although SBE variation can cause a specific type of aberrant methylation artifact, when these sites are removed, aberrantly methylated CpGs are more similar between samples of the same clone than between samples with the same genetic background.
Figure 4. Association of aberrant methylation with clone-specific effects (see also Tables S1A, S1B, and S3A, and Figure S4).
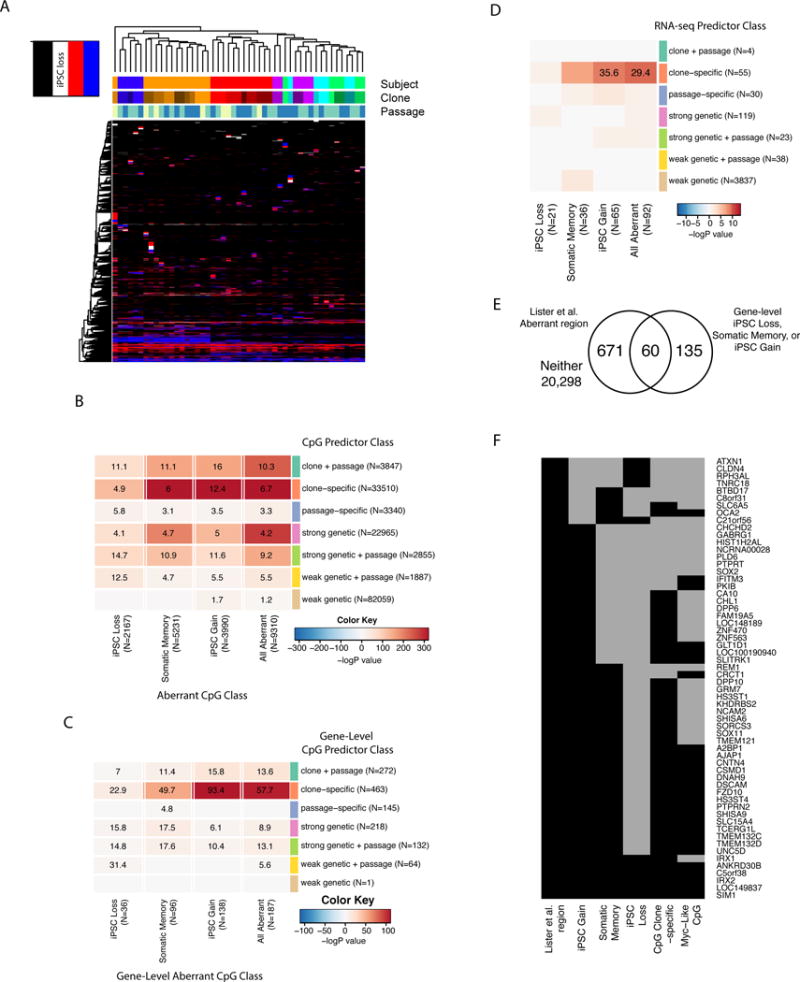
A) Heat map showing hierarchical clustering of 9,310 aberrant CpGs, cells are colored according to whether they are not aberrant (None), iPSC loss, somatic memory, or iPSC gain in each of the 49 samples. B) Heatmap showing odds ratios (ORs) from Fisher’s exact test of overlap between CpGs in aberrant CpG classes (columns) and those in CpG predictor classes. For CpG predictor classes, the reference is sites not associated with any category (the “None” category). For aberrant CpG classes, the reference is sites showing no aberrant methylation in any sample (the “Not Aberrant” category). Cells are colored according to −logP-value with positive values indicating over-enrichment and negative indicating under-enrichment. Cells with non-significant results (FDR > 0.05) do not have an OR reported. C) Heatmap showing ORs for the overlap between gene-level aberrant CpG classes and gene-level CpG predictor classes. Cells are colored according to −logP-value. D) OR’s for the overlap of genes enriched for aberrant classification (columns) and genes where the expression values from RNA-seq were associated with each predictor classification (rows). Cells are colored according to −logP-value. (E) Venn diagram showing the intersection of genes in aberrant regions in Lister et al. and genes aberrantly methylated in this study. F) List of 60 genes that show overlap in aberrant methylation between Lister et al. and this study. Genes are annotated by whether they showed gene-level aberrant methylation for iPSC gain, somatic memory, iPSC loss, or gene-level clone-specific enrichment. MYC-like CpG indicates the gene carried one or more iPSC gain CpGs associated with a MYC or MYC-like binding site. Cells are black if the variable is present (overlaps a Lister et al region; shows gene-level enrichment for iPSC gain, somatic memory, or iPSC loss; shows gene-level enrichment for the clone-specific CpG predictor class, or carries at least one CpG with a predicted Myc bindings site), and grey if absent.
Clustering patterns based on aberrant CpGs do not take into account the background rates of differentially methylated CpGs associated with genetic background, clone, and passage; therefore, we integrated the aberrant results with the CpG and RNA predictor classes and examined whether aberrant methylation was significantly enriched in these classes above background rates. We first calculated the overlap between the CpGs in the three aberrant CpG classes and those in the seven CpG predictor classes described in Figure 3A. For each classification of CpG predictor class and aberrant CpG class, we calculated whether the two groups overlapped relative to the “None” CpG predictor class and “Not Aberrant” aberrant class reference groups using a Fisher’s exact test (Figure 4B). We observed significant positive enrichment for almost all comparisons showing that aberrant methylation tends to be associated with genetic background, clone, and passage. The most strongly associated CpG predictor classes were clone + passage, clone-specific, strong genetic, and strong genetic + passage. We next examined overlap at the gene level. As described above for CpG predictor classes, we identified genes that were enriched for specific subtypes of aberrant methylation relative to overall rates (Table S1B). Using the genes that were enriched for aberrant CpGs or CpG predictor classes, we compared the overlap of genes across classes using a Fisher’s exact test (Figure 4C). Consistent with the single CpG site observations, we observed the highest overlap between aberrant genes and clone-specific, strong genetic, and strong genetic + passage genes. However, genes associated with iPSC gain showed particularly high enrichment for clone-specific genes (OR=93.4, P=6.9×10−111). Consistent with these observations, when we compared the gene-level aberrant CpG classes to the RNA predictor classes (from Figure 3D), we observed strong enrichment for clone-specific effects only (Figure 4D). Our findings suggest that the aberrant methylation is strongly associated with clone-specific effects, particularly for iPSC gain, and modestly associated with genetic effects.
Comparison of aberrant genes across studies supports non-genetic mechanisms
Because subsets of aberrantly methylated genes are reproduced across studies, we compared aberrant regions identified in this study to those in Lister et al. (Lister et al., 2011), and examined their associations according to genetic background, clone, and passage. Lister et al. reports “aberrant regions”, which we converted to gene-level aberrant annotations by labeling genes as aberrant if they contained a CpG on the methylation array that overlapped an aberrant region. We then compared genes in the Lister el al. aberrant regions to the combined gene-level aberrant CpG classes and calculated overlap using a Fisher’s exact test. We found a significant overlap of 60 genes (OR=13.4, P=2.8×10−40, Figure 4E), supporting reproducibility of aberrancy across studies. We partitioned the genes into CpG predictor classes and observed the strongest reproducibility in clone-specific genes (Figure S4D). We further examined the 60 genes linked to aberrancy that were common to both studies, and observed that 51 were enriched for iPSC gain sites (Figure 4F). A majority of those genes were also enriched for somatic memory sites and clone-specific effects. Since we had found that iPSC gain aberrant methylation of CpGs affects the expression of associated genes, possibly through the binding of regulatory proteins including MYC (Figures 2E and 2F, Table S3A), we next examined whether the 60 overlapping genes carried CpGs containing MYC and MYC-like binding sites. Approximately 50% of the 51 genes associated with iPSC-gain contained MYC and MYC-like sites, including genes that have been previously linked to aberrancy in other studies (e.g. A2BP1/RBFOX1, CSMD1, TMEM132C, TMEM132D, IRX2) (Choi et al., 2015; Ruiz et al., 2012). These findings suggest that genes identified as aberrantly methylated across multiple studies are enriched for CpGs that show clone-specific and iPSC gain methylation, and overlap binding sites of transcription factors associated with reprogramming.
DISCUSSION
Our study, which used multiple iPSC clones from three monozygotic twin pairs measured at multiple time points in culture, allowed us to characterize how aberrant DNA methylation proportionally varied according to genetic background, clone, and passage and provided important insights into the underlying mechanisms behind aberrant methylation in iPSCs. Although previous studies showed that genetic factors play a major role in regulating the pluripotent cell epigenome (Kyttala et al., 2016; Rouhani et al., 2014), we show here that aberrant methylation is most strongly enriched for non-genetic factors, specifically clone-specific effects, and only modestly enriched for genetic effects. Furthermore, we demonstrate that aberrant methylation is enriched at functionally relevant sequences, including regulatory regions and transcription factor motifs, is likely to have functional effects on gene expression, and occurs reproducibly at specific CpG sites across studies. Our findings provide insight into the regulatory mechanisms that may be controlling iPSC methylation and highlight an important role for clone-specific effects in aberrant methylation.
The overall functional consequences of aberrant iPSC methylation on cellular fate and function remain unclear. We examined CpGs aberrancies and, in agreement with other studies, found that only 2% of iPSC methylation sites were aberrant (Figure 2). We divided the aberrant sites into three classes (somatic memory, iPSC gain, iPSC loss) and observed differential functional associations among them. Different classes of aberrancies are present in functionally distinct chromatin states, with iPSC gain sites showing enrichments in bivalent promoters, repressed polycomb regions, promoters downstream of TSS, and transcription regulatory sequences, suggesting that these aberrancies occur in important regulatory elements. Somatic memory and iPSC gains were also associated with repressed polycomb regions, but were also enriched for quiescent regions that may not have functional effects. Additionally, motif enrichment analysis showed that iPSC gain CpGs were enriched for motifs important in transcriptional regulation and metabolism. For example, we observed an enrichment of the MYC motif and MYC-like motifs, such as HIF1A. c-Myc is thought to act early in the process of reprogramming, and to promote an active chromatin state prior to pluripotency regulators being activated (Sridharan et al., 2009). HIF1A can positively affect reprogramming by promoting the glycolytic shift that occurs during somatic cell reprogramming (Prigione et al., 2014), which may be an early event in facilitating the epigenetic changes required for reprogramming (Folmes and Terzic, 2016). We further observed that gene expression in samples containing iPSC gain methylation at MYC and MYC-like motifs was decreased, consistent with functional consequences of these changes. Other classes of aberrant methylation (somatic memory and iPSC loss) had less clear functional implications as they were enriched in quiescent regions and were not associated with gene expression changes. Our analyses suggest that while aberrancies in all three classes may have functional consequences, iPSC gain aberrancies are most likely to contribute to changes in gene expression and thus could positively affect acquisition and/or maintenance of a pluripotent state.
We show that while genetic background is often associated with CpG methylation levels, it does not play a primary role in aberrant methylation. We observed that monozygotic twin-derived iPSCs show highly similar methylation and approximately 73.5% of the 154,724 associated CpGs being associated with genetic background either uniquely (53.6%) or in association with clone (16.8%), passage (1.2%) or both clone and passage (1.9%) (Figure 3). The presence of specific genetic variants near the probe and association with previously reported meQTL loci supports the model these CpGs are driven by genetic variants and not systematic variation such as batch effects. When we examined whether aberrant CpGs were preferentially enriched for CpGs associated with genetic background above genome-wide rates, however, we observed only modest enrichment. The association between aberrant methylation and genetic background further weakened when we examined gene regions or gene expression, suggesting that genetic variation does not play a primary role in aberrant methylation. Importantly, we did observe an important exception to this pattern when genetic variants disrupted the methylation array single base extension assay. For these SBE sites, we observed strong association with genetic background as well as somatic memory aberrant methylation. These variants, if not removed, cause aberrant methylation to appear similar across iPSCs from the same genetic background and may result in false interpretations of the relative importance of genetic variation on aberrant methylation. Thus, while genetic variation ubiquitously affects methylation levels, including aberrant methylation, it is not strongly preferentially enriched at CpGs showing aberrant methylation and is therefore not a primary determinant.
Although only a quarter of the differentially methylated CpGs were associated with clone and passage effects, aberrant methylation was highly enriched at CpGs with clone-specific effects. Genes containing clone and passage-associated CpGs were enriched for functional annotations relevant to iPSCs, including cell-cell adhesion and the X-chromosome, suggesting that they reflect underlying biological process physiologically relevant to reprogramming including the X-chromosome activation state in human iPSCs (Pasque and Plath, 2015). Additionally, passage-associated CpGs were enriched for JUN/FOS binding sites, which consistently showed decreased methylation between passage 9 and 20. It is known that Jun inhibits somatic cell reprogramming and is important for differentiation (Liu et al., 2015), and has been shown to regulate cell adhesion genes as ESCs exit a pluripotent ground state (Veluscek et al., 2016). When we examined the overlap between CpGs associated with non-genetic factors and aberrant methylation, we observed high enrichment for clone-specific effects above that expected from genome-wide rates, particularly for iPSC gain sites. This enrichment strengthened when we examined gene-level methylation enrichment as well as gene expression patterns. Further, genes that were identified in this study as well as Lister et al. showed high levels of iPSC gain and clone-specific effects, suggesting that this phenomenon is replicable across studies, samples, and methylation assay platforms. Thus, non-genetic factors moderately contribute to CpG methylation variation, but clone-specific effects are particularly enriched in and are therefore likely to be an important factor in mediating aberrant methylation.
Overall, we show that CpG methylation in iPSCs is clearly influenced by genetic variation, and that in some cases this variation can explain aberrantly methylated sites. However, the fact that aberrant methylation is enriched in functionally distinct regions, is correlated with gene expression changes, most often occurs independently of genetic background in a clone-specific manner, and shows enrichment for MYC and MYC-like protein binding sites suggests that there are non-genetic physiological mechanisms underlying aberrant methylation. Our work provides a foundation for future studies aimed at elucidating the specific molecular mechanisms underlying aberrancy as well as the physiological consequences of these epigenetic modifications.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests should be directed to the corresponding author Kelly Frazer (kafrazer@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Sample collection
Individuals were recruited from the Twin Sibling Pedigree cohort (TSP; a population based twin registry spanning counties in Southern California) (Pasha et al., 2013), and informed consent was received from all individuals. These collections were approved by the Institutional Review Boards of the University of California at San Diego and of The Salk Institute. Fibroblasts were generated from skin biopsies of three pairs of Caucasian female monozygotic twins in the iPSCORE collection (Panopoulos et al., in Press): iPSCORE _31_1 and iPSCORE _31_2 were age 63 at collection, iPSCORE _111_1 and iPSCORE _111_2 were age 55, and iPSCORE _103_1 and iPSCORE _103_2 were age 70.
iPSC derivation and culture
To make retroviruses for reprogramming, moloney-based retroviral vectors (pMX-OCT4, pMX-SOX2, pMX-KLF-4, pMX-c-MYC) were obtained from Addgene (see Key Resources Table). Packaging plasmids (pCMV-gag-pol-PA and pCMV-VSVg) were kindly provided by Dr. Gerald Pao (The Salk Institute, La Jolla, CA). Retroviruses were collected 24 hours after 293T cells were transfected with plasmids using Lipofectamine (Invitrogen) according to manufacturer’s recommendations. To derive iPSCs, twin fibroblast lines were infected with retroviruses encoding OCT4, SOX2, KLF-4 and c-MYC by spinfection at 800×g for 1 hour at room temperature in the presence of polybrene (8 μg/ml), and replated onto MEFs (Millipore) before switching to ESC medium for iPSC colony formation, as described (Lutz et al., 2008; Panopoulos et al., 2012). Individual colonies were expanded in mTeSR-1 media (STEMCELL Technologies) on Matrigel-coated plates. iPSC are named according to iPSCORE collection name (i.e. 31_1), followed by the clone and passage (e.g. 31_1_1_20 corresponds to family ID 31_individual 1_clone1_passage 20).
iPSC characterization
iPSC lines were evaluated for pluripotency by flow cytometry and gene expression analysis. Flow cytometry analysis was performed using fluorescently conjugated antibodies to the pluripotent cell surface markers TRA-1-60 and SSEA-4 (BD Biosciences). For gene expression analysis, Total RNA was isolated using Trizol Reagent (Invitrogen) and was reverse transcribed using the SuperScript II Reverse Transcriptase kit (Invitrogen). Real-time PCR was performed using SYBR-Green PCR Master mix (Applied Biosystems). The expression levels were normalized to GAPDH and the individual pluripotent genes (OCT4, SOX2, NANOG, and CRIPTO) were compared to the fibroblast marker COL6A2. Karyotype analysis was performed by Wicell Cytogenetics.
METHOD DETAILS
Methylation arrays
DNA was isolated (DNeasy kit, Qiagen) from iPSCs at passages 5, 9, and 20 and from fibroblasts at passage 7 and 500 ng was bisulfite converted (EZ DNA Methylation Kit, Zymo Research), of which 250 ng was hybridized to Infinium HumanMethylation450 BeadChip arrays (Illumina). To prevent batch effects samples from each individual were arrayed across different BeadChips. Samples were processed (whole genome amplification, bead hybridization, immunostaining, scanned) as described (Smith et al., 2015). Methylation levels were processed using the minifi package in R (Fortin et al., 2014) and initially normalized using background subtraction (preprocessIllumina). Then, probes with missing data (detection P < 0.01 in any sample, N = 3,955) or associated with cross-reactivity (Chen et al., 2013) (N = 29,233) were removed. The data was normalized using SWAN (Maksimovic et al., 2012) (preprocessSWAN) and beta values obtained (getBeta, type = “Illumina”).
HumanCoreExome arrays
Genomic DNA from fibroblasts from all six individuals (passage 7) and one iPSC clone from each individual (passage 20) was extracted (AllPrep DNA/RNA Mini Kit, Qiagen), normalized to 200 ng, hybridized to HumanCoreExome chips (Illumina) and stained and scanned per standard protocol. We observed an average call rate of 99.2% across the 12 arrays (dbGaP phs000924).
RNA-seq
Total RNA was extracted from the fibroblast and iPSC lines using RNeasy mini kits (Qiagen) following the manufacturer’s protocol. RNA quality was assessed based on RNA integrity number (RIN) using an Agilent Bioanalyzer: all samples had a RIN value >= 8.4. Libraries were prepared using the Illumina TruSeq stranded mRNA kits and sequenced using an Illumina HiSeq4000 to an average of ~31 million read pairs. 2×100 bp RNA-seq reads were aligned with STAR (2.5.0a) to the hg19 reference (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit) using Gencode v19 splice junctions with default alignment parameters except –outFilterMultimapNmax 20, –outFilterMismatchNmax 999, –alignIntronMin 20, –alignIntronMax 1000000, –alignMatesGapMax 1000000 (Dobin et al., 2013; Harrow et al., 2012). Bam files were coordinate sorted using Sambamba (0.5.9) (Tarasov et al., 2015) and duplicate reads were marked using biobambam2 (2.0.21) bammarkduplicates (Tischler and Leonard, 2014). The minimum uniquely mapped read percentage was 85% and the median was 92%. We estimated transcript and gene expression using the STAR transcriptome bam file and RSEM (1.2.20) rsem-calculate-expression (–seed 3272015 –estimate-rspd –forward-prob 0) (Li and Dewey, 2011). We filtered RSEM gene TPM values by removing any genes whose expression was not greater than 2 TPM in 10 or more iPSC samples and adjusted for duplicate rate using linear regression. We then transformed the residual expression values for each of the 14,506 genes passing these filters to match a standard normal distribution.
QUANTIFICATION AND STATISTICAL ANALYSIS
Copy Number Variation Determination
Raw scan data were processed by Genome Studio (Illumina, Inc) using the supplied cluster files for SNP calling on the HumanCoreExome arrays (average call rate 0.99, GenCall threshold 0.15). The process array data was subjected to both manual and computerized analysis to detect CNVs. Both approaches were used, as it is known that automated CNV calling can fragment segments due to non-normal distributions of probes on the arrays; hence a single contiguous CNV can be erroneously broken up into several smaller regions. For systematic manual inspection of the data, chromosome plots were created to visualize the B-allele frequencies (proportion of A and B alleles at each genotype) and log R ratios (ratio of observed to expected intensities) for each chromosome. The plots were scanned independently by two operators, comparing iPSCs and matched germline samples for signatures of abnormalities in the iPSCs.
For computerized analysis genotype data were exported to Nexus CN where CNV calling was carried out with the hg19/GRCh37 reference version of the human genome. The X and Y chromosomes were removed due to the complexity of reliably determining copy number in these copy variable and highly repetitive chromosomes. A descriptor sheet was supplied with the six sample pairings for germline to corresponding iPSC results files. The Nexus files and settings used were: Systematic Correction File: Catlg_ILM_HumanCoreExome-12v1-1_B_20140311.bed_hg19_ilum_correction.txt (as supplied by Biodiscovery Inc). Manual analysis did not identify any CNVs. Nexus CN initially detected 13 autosomal CNVs, however due to issues with borderline thresholding for low confidence CNVs, the list of 13 was manually curated and low confidence CNV calls were removed. The quality controlled and condensed list contained 10 unique and verified CNVs. The combined set of variants are shown in Supplement Figure 1D.
Fibroblast clustering
Raw methylation arrays for 62 fibroblast samples that were previously published (Wagner et al., 2014) were downloaded from GEO (accession: GSE52025). Sites with missing data were removed and the arrays were normalized as described for the samples in this study using background subtraction and SWAN. We performed batch correction between the 6 fibroblast samples generated in this study and the 62 from Wagner et al. using sva (Leek et al., 2012) package in R (R Core Team, 2015) (ComBat function). Clustering was performed in R, using Euclidean distance and complete clustering on all sites that passed QC in both datasets. Because the SNP beta values are excluded during normalization, the SNP beta values were obtained from the raw data and calculated as B signal/(A Signal + B Signal + 100) and compared to the 6 samples from this study where the signal was calculated in a similar way.
Identification of aberrant CpG sites
To identify aberrant sites methylation levels at individual CpG sites were compared with 15 methylation profiles of ES cells previously published (Nazor et al., 2012). Raw methylation levels of the ES cells were downloaded from GEO (accession GSE31848) and a single ES line per subject was chosen (i.e. GSM867941, GSM867939, GSM867940, GSM867947, GSM867948, GSM867949, GSM867938, GSM867950, GSM867945, GSM867946, GSM867943, GSM867944, GSM867942, GSM867952, GSM867936). The averages and standard deviations of the beta values (B signal/(A Signal + B Signal + 100) were calculated for each CpG. Sites were excluded if a probe failed in any of the 15 ES lines. SBE CpGs that contained a genetic variant at the 1,0, or −1 position of the CpG probe as identified by whole genome sequencing were removed. For each iPSC sample, a Z-score for each CpG was calculated by comparing the iPSC beta value to the mean and standard deviation of the ES cells. The Z-score distribution for each iPSC sample was then compared to a normal distribution. A cutoff was chosen such that the number of expected Z-scores (absolute value) from the normal distribution was 1% or less of the number of observed Z-scores (absolute value). The average cutoff was 3.42 (range 3.2–3.8) and the average number of sites associated was 18,780 (range 4,597–40,110). Sites that exceeded this Z-score cutoff (positive or negative) were considered aberrant in the iPSC line if they were also at least 0.2 Beta away from the mean of the ESC lines (average 1300; range 599–2,799). Sites were further classified into iPSC loss, somatic memory, and iPSC gain by comparing the sample to their respective fibroblast sample. If the iPSC was within +/− 0.2 of the fibroblast, it was considered somatic memory; if the iPSC was more than 0.2 less than the fibroblast, it was considered iPSC loss; and if the iPSC was more than 0.2 more than the fibroblast, it was considered iPSC gain.
ROADMAP analysis
The 25-state chromHMM state predictions based on imputed data in 127 reference epigenomes were downloaded (http://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/imputed12marks/jointModel/final/*segments.bed.gz; 6/30/2015). For each CpG we determined the state of the closet segment in each of the 127 epigenomes using bedtools (closestBed) (Quinlan and Hall, 2010). Enrichment of CpGs sites based on their classification (CpG predictor class or aberrant CpG class) in the 25 different states across of either the iPSCs and ESCs or all 127 epigenomes was calculated using the hypergeometric distribution (phyper in R).
Aberrant methylation and gene expression
For all RNA samples for which there was methylation data (N=42), gene expression values from each sample were paired with methylation status at CpGs annotated to their gene (the first gene listed in the Illumina annotation file) in the same sample. Each gene was classified by the presence and type of aberrant methylation (none, iPSC gain, somatic memory, iPSC loss, or multiple different types). Expression values were adjusted for each iPSC sample using linear regression and the category of aberrant type was tested as a predictor for the residual gene expression as compared to the “none” category using multiple linear regression (Figures 3D and S3). For CpGs with Myc or Myc-like binding sites, the same approach was used except only CpGs that showed aberrant iPSC gain in any sample and any of the Myc-like binding sites (Table S5) were examined (Figure 2F).
Whole Genome Sequencing/Variant Calling
DNA was isolated from blood samples (DNeasy kit, Qiagen) quantified, normalized and sheared with a Covaris LE220 instrument. DNA libraries were prepared (TruSeq Nano DNA HT kit, Illumina), characterized in regards to size (LabChip DX Touch, Perkin Elmer) and concentration (Quant-iT, Life Technologies), normalized to 2–3.5nM, combined into 6-sample pools, clustered and sequenced on the HiSeqX (150 base paired-end). Base call (BCL) files were used to map reads to the hg19 reference sequence (which had the pseudoautosomal region of chrY masked) using ISIS Analysis Software (v. 2.5.26.13; Illumina) (Raczy et al., 2013). The ISIS Isaac Aligner (v. 1.14.02.06) identified and marked duplicate reads, and these were removed from downstream analysis. Bam files were characterized using Picard (v. 1.113–1.131), and input to the ISIS IsaacVariant Caller (v. 2.0.17) (default settings), which yielded genomic VCF files. For computation of accuracy, single nucleotide variants with a “PASS” flag were compared to GIAB (v. 2.19; ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/NISTv2.19).
Resulting VCF files were filtered according to HighDPFRatio, fraction of basecalls filtered at a site is greater than 0.4; HighDepth: locus depth is greater than 3× the mean chromosome depth; HighSNVSB: SNV strand bias value (SNVSB) exceeds 10; IndelConflict: locus is in region with conflicting indel calls; LowGQX: locus GQX (minimum genotype quality assuming variant position and the genotype quality assuming non-variant position) is less than 30 or not present; SiteConflict: site genotype conflicts with proximal indel call. Resulting filtered files were combined using GATK and variants called in one or more samples were assumed to be homozygous reference in all samples in which they were not called. Non-reference concordance was calculated for each sample based on the proportion of non-reference calls that had the exact (same allele call) genotype in the appropriate twin. We obtained an average of 3,603,710 SNPs and 518,796 indel variants per individual with an average TiTv ratio of 2.09 at biallelic SNPs (Table S6). We observed high non-reference SNP concordance between twin samples (average 98%).
ANOVA
Analysis of variance (ANOVA) was performed in R using the anova function on a linear (lm) model. We conducted two analyses for both the 42 DNA methylation and 44 RNA-seq data from iPSC samples at passages 9 and 20 to maintain approximately balanced groups. The first analysis estimates the effect of genetic background after adjusting for passage: we performed an ANOVA including a variable indicating the twin pair (3 sets) with passage as a covariate. The second analysis estimates the effect of clone and also the effect of passage: we performed an ANOVA including clone and passage as variables. For each predictor (genetic background, clone, passage), we adjusted the resulting P-values for multiple testing using the Benjamini-Hochberg method for false discovery rate (FDR) (Benjamini and Hochberg, 1995) and considered associated sites significant at a 5% FDR for DNA methylation and associated genes at 1% FDR for RNA-seq (Table S2). A more stringent threshold was used for RNA-seq because of the high density of borderline significant results.
We expected that in general, for a genetic effect, we would see a proportional, but less significant clone association than genetic background association because there are numerically fewer twin pairs (3) then clones (22), and the test statistic for genetic background has fewer degrees of freedom compared with clone. On the other hand, clone effects associated with reprogramming would not be expected to track with genetic background and would likely occur in only one or a subset of clones within a genetic background, resulting in the P-value for clone being more significant.
To classify CpGs and genes into predictor classes (Tables S1, S2), we grouped them according to the combination of predictors that were significant. Additionally, in order to be considered “strong genetic” or “strong genetic + passage”, we imposed an additional requirement that the P-value associated with genetic background needed to be smaller than the P-value associated with clone, consistent with expectations for a genetic effect. Those that did not meet this criterion were grouped “clone-specific” or “clone-specific + passage”. This step ensures that strongly clone-specific sites do not drive false association with genetic background because the multiple samples (p9 and p20) have the same background.
Genetic variants near the CpG probe
To determine if CpG sites associated with genetic background were enriched for the presence of proximal genetic variants, we first identified the closest non-reference variant across all 6 WGS samples to each CpG using bedtools (closestBed). For each CpG, the distance from the 0-position to the variant was calculated as the distance reported from closestBed (negative for upstream) multiplied by −1 if the probe was reported on the reverse (R) strand. This resulted in a distance where the positions 0–49 corresponded to the bases of the probe and the −1 position to the site where SBE occurs. We excluded all CpGs where the genotypes between twins did not agree (N = 38,137), although the results were similar whether they were included or not. We then split the CpGs into those near a polymorphic variant in which twin pairs had different genotypes from each other, and those that were monomorphic (i.e. all reference alternate homozygotes or all heterozygotes). Within each group, CpGs were grouped into distance bins and the odds ratio and 95% confidence interval of the CpG being associated with genetic background was calculated using the combined >300bp or <−300bp categories as a null group using the Fisher’s exact test (fisher.test in R).
Association with previously identified meQTL
CpGs associated with meQTL were previously identified (Lemire et al., 2015). When testing CpGs associated with genetic background in our study for enrichment with CpGs associated with meQTLs, only CpGs that met the criteria for inclusion in the Lemire et al. paper were used: SBE sites (at position 1,0, or −1) with a European population variant (frequency > 0) and probes spanning a European population variant (frequency > 0.05) as defined in (Chen et al., 2013) were excluded. Enrichment was calculated using a Fisher’s exact test.
Gene-level CpG predictor classification
CpGs were assigned to genes based on the UCSC_RefGene_Name field in the Illumina HumanMethylation450 annotation file (HumanMethylation450_15017482_v.1.1.csv). When multiple genes were listed, the CpG was assigned once to both. For each gene, enrichment was assessed by a hypergeometric test in R using phyper. The number of CpGs that were associated with each CpG predictor class or aberrant CpG class out of the total number of CpGs associated with that gene was compared to the overall number of CpGs associated to all genes. The P-values were corrected for multiple testing using p.adjust (method = “fdr”) in R and considered significant if the adjusted P-value was less than 0.05. Because each CpG predictor class and aberrant CpG class was tested independently, a gene can be enriched for multiple CpG predictor or aberrant CpG classes.
Motif enrichment
Regions flanking CpG sites (122bp with 60 bp on either side of the CpG) were obtained from Illumina 450K Methylation BeadArray annotation (file). Sites showing association with genetic background, clone, or passage were compared to those sites not found to be associated with any of the CpG predictor classes, while aberrant classifications were compared to sites showing no aberrant methylation in any sample. Regions were compared using MEME-ChIP (v4.11.2) (Machanick and Bailey, 2011), which incorporates Dreme (Bailey, 2011), Tomtom (Gupta et al., 2007), and CentriMo (Bailey and Machanick, 2012) to the human and mouse HOCOMOCOv10 databases. Only human associations that showed an E-value less than 0.01 are reported. Because many of the motifs identified showed similarity to the MYC motifs, MYC-like motifs were defined as those that shared at least four of the six central base pairs of the MYC motif (CACGTG). Because the ENOA_HUMAN.H10MO.A motif was retracted from the HOCOMOCO database (personal communication Ivan Kulakovskiy), we removed this motif from the results.
GOseq gene set enrichment
Genes were tested for enrichment using GOseq (Young et al., 2010) adjusting for the number of methylation probes associated with each gene (Geeleher et al., 2013). Sites were annotated to one or more genes based on the Illumina HumanMethylation450 BeadChip manifest file. Gene categories were downloaded from the Molecular Signatures Database v5.1 (http://www.broadinstitute.org/gsea/msigdb/index.jsp, msigdb.v5.1.symbols.gmt) or accessed using the “org.Hs.eg.db” library (Carlson) in R. Associations reaching a false discovery rate of less than 0.05 are reported.
Overlap of aberrant CpGs with Lister et al
To identify CpG sites that overlap the regions previously reported as aberrantly methylated in Lister et al we used bedtools (intersectBed). Because the Lister et al. regions were reported in hg18, the hg18 positions from the Illumina HumanMethylation450 annotation file were used. For genes, a gene was considered to overlap if any CpG annotated to the gene overlapped one of the Lister et al. regions. The strength of the overlap between aberrantly methylated regions in Lister et al. and aberrantly methylated CpGs in this study were estimated by Fisher’s exact test across all sites or stratified by CpG predictor or aberrant CpG class.
SBE variation filtering
While we used WGS to identify SBE sites and subsequently filtered them, most studies do not have access to this data. Because many SBE sites are polymorphic in populations, we examined whether filtering CpGs based on the European allele frequency of SNPs at the SBE positions could effectively identify SBE sites discovered by WGS. We performed a receiver operating characteristic (ROC) analysis for one subject in each twin pair using the ROC command in R package Epi (Bendix Carstensen, 2016)(URL http://CRAN.R-project.org/package=Epi) (Figure S4C). For each subject, a CpG was positive for an SBE variant if that subject’s WGS data contained a non-reference variant at positions −1,0, or 1. The frequency of SBE variants in the European population at the position was obtained as previously reported (Chen et al., 2013). Across the three samples, we observed that filtering at an average allele frequency cutoff of 0.003 removed 77% percent of the SBE sites identified by whole genome sequencing with 97% specificity (see Figure S4C for results from a single sample), but that rare variants and individual-specific variants were missed.
DATA AND SOFTWARE AVAILABILITY
Methylation, array genotype, RNA-seq expression values, and whole genome sequence genotype data is available through dbGaP (phs000924 and phs001325).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TRA-1-60-FITC | BD Biosciences | 560380 |
| SSEA-4-PE | BD Biosciences | 560128 |
| Bacterial and Virus Strains | ||
| pMXs-hOCT3/4 | Addgene | 17217 |
| pMXs-hSOX2 | Addgene | 17218 |
| pMXs-hKLF4 | Addgene | 17219 |
| pMXs-hc-MYC | Addgene | 17220 |
| pCMV-gag-pol-PA | gift from Dr. Gerald Pao; The Salk Institute, La Jolla, CA, USA | N/A |
| pCMV-VSVg | gift from Dr. Gerald Pao; The Salk Institute, La Jolla, CA, USA | N/A |
| Critical Commercial Assays | ||
| AllPrep RNasy Blood & Tissue Kit | Qiagen | Cat no: 80204 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat no: 69506 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat no: RS-122-2103 |
| EZ DNA Methylation Kit | Zymo Research | Cat no: D5001 |
| Infinium HumanMethylation450 BeadChip | Illumina | Cat no: WG-314-1003 |
| Infinium HumanCoreExome BeadChip | Illumina | Cat no: WG-331-1101 |
| Deposited Data | ||
| Whole genome sequencing data | (DeBoever et al., in Press) | dbGaP phs000924 |
| RNA-seq | This paper | dbGaP phs000924 |
| DNA Methylation | This paper | dbGaP phs000924 |
| Embryonic stem cell DNA methylation | (Nazor et al., 2012) | GEO GSE31848 |
| ROADMAP chromHMM | (Ernst and Kellis, 2015) | http://egg2.wustl.edu/roadmap/data/ |
| Experimental Models: Cell Lines | ||
| Human: Twin-derived iPSCs and fibroblasts | This paper | N/A |
| Human: BJ-Fibroblasts | Salk Stem Cell Core | N/A |
| Human: H1 (WA01) ESCs | Salk Stem Cell Core | N/A |
| Human: H9 (WA09) ESCs | Salk Stem Cell Core | N/A |
| Mouse Embryonic Fibroblasts | Millipore | PMEF-CF |
| Oligonucleotides | ||
| Endogenous OCT4 F: GGGTTTTTGGGATTAAGTTCTTCA R: GCCCCCACCCTTTGTGTT |
Panopoulos et al, 2012 | N/A |
| Endogenous SOX2 F: CAAAAATGGCCATGCAGGTT R: AGTTGGGATCGAACAAAAGCTATT |
Panopoulos et al, 2012 | N/A |
| NANOG F: ACAACTGGCCGAAGAATAGCA R: GGTTCCCAGTCGGGTTCAC |
Panopoulos et al, 2012 | N/A |
| CRIPTO F: CACGATGTGCGCAAAGAGA R: TGACCGTGCCAGCATTTACA |
Panopoulos et al, 2012 | N/A |
| GAPDH F: GGACTCATGACCACAGTCCATGCC R: TCAGGGATGACCTTGCCCACAG |
Panopoulos et al, 2012 | N/A |
| Software and Algorithms | ||
| R | (R Core Team, 2015) | https://cran.r-project.org/ |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Nexus CN | Biodiscovery | http://www.biodiscovery.com/nexus-copy-number/ |
| minifi | (Fortin et al., 2014) | http://bioconductor.org/packages/release/bioc/html/minfi.html |
| SWAN | (Maksimovic et al., 2012) | http://bioconductor.org/packages/release/bioc/html/minfi.html |
| Sambamba | (Tarasov et al., 2015) | http://lomereiter.github.io/sambamba/ |
| bammarkduplicates | (Tischler and Leonard, 2014) | https://github.com/gt1/biobambam/blob/master/src/programs/bammarkduplicates. 1 |
| RSEM | (Li and Dewey, 2011) | https://deweylab.github.io/RSEM/ |
| Genome Studio | Illumina, Inc | https://www.illumina.com/techniques/microarrays/array-data-analysis-experimental-design/genomestudio.html |
| sva | (Leek et al., 2012) | https://bioconductor.org/packages/release/bioc/html/sva.html |
| bedtools | (Quinlan and Hall, 2010) | http://bedtools.readthedocs.io/en/latest/ |
| ISIS Analysis Software | Illumina | https://support.illumina.com/sequencing/sequencing_software.html |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| MEME-ChIP | (Machanick and Bailey, 2011) | http://meme-suite.org/tools/meme-chip |
| Dreme | (Bailey, 2011) | http://meme-suite.org/tools/meme-chip |
| Tomtom | (Gupta et al., 2007) | http://meme-suite.org/tools/meme-chip |
| CentriMo | (Bailey and Machanick, 2012) | http://meme-suite.org/tools/meme-chip |
Supplementary Material
S1A: List of 158,466 CpGs that were associated with a CpG predictor class and/or an aberrant CpG class. For each CpG the chromosome, position, classification by CpG predictor class, the number of samples that showed aberrant methylation in each of the three aberrant CpG classes, and whether or not the CpG is an SBE site is shown. S1B: Gene-based CpG analyses. Genes enriched for CpGs associated with each CpG predictor class, each aberrant CpG class, and each gene’s RNA-seq predictor class annotation are given. NA in CpG class categories indicates that the gene did not have CpGs annotated to it on the 450K BeadChip, while NA in RNA predictor class indicates that the gene was not expressed in the iPSCs and therefore not analyzed.
List of gene sets showing significant enrichment (FDR < 0.05) for genes in one of the seven gene-level CpG predictor classes or in gene-level aberrant CpG classes. For aberrant genes, a group including all genes showing significant enrichment of any aberrant CpG was also used (“Any Aberrant”). Analyses were performed using GOseq adjusting for the number of CpGs annotated to each gene, and the Gene Ontology (GO) and Molecular Signatures Databases were used.
S3A: List of enriched motifs for CpGs associated with aberrant CpG classes. Results from Dreme and CentriMo analyses are shown. Only human motifs are reported. All Dreme motif results with E-values < 0.01 are listed. When a significant motif was identified with Dreme, TomTom was used to try to identify similar known motifs – none were observed. When CentriMo was used to identify a motif, the Fisher E-value, central enrichment P-value, and central enrichment distribution are shown. MYC and MYC-like motifs (defined here as sharing at least four bases out of the six central CACGTG bases of the MYC motif) are indicated for the CentriMo analysis of iPSC gain sites. S3B: List of enriched motifs for CpGs associated with CpG predictor classifications. Results from Dreme and CentriMo analyses are shown. Only human motifs are reported. All Dreme motif results with E-values < 0.01 are listed. When a significant motif was identified with Dreme, TomTom was used to identify similar known motifs. When multiple “similar known motifs” were identified by TomTom for the same “significant motif”, all results are listed with the same Motif Number. A “similar motif” (i.e. FOSB) may be listed with multiple Motif Numbers if it was identified by TomTom in multiple analyses. When CentriMo was used to identify a motif, the Fisher E-value, central enrichment P-value, and central enrichment distribution are shown.
Acknowledgments
This work was supported in part by a CIRM grant GC1R-06673 (to KAF) and NIH grants HG008118-01 (to KAF), HL107442-05 (to KAF and JCIB), DK105541 (to KAF), DK112155 (to KAF), and EY021237 (to KAF). Work in the laboratory of JCIB was supported by grants from The Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED002), Universidad Catolica San Antonio de Murcia (UCAM) and the G. Harold and Leila Y. Mathers Charitable Foundation. We thank the Gallagher Family for their generous gift to the University of Notre Dame to support stem cell research. Methylation array data were generated at the UCSD IGM Genomics Center of with support from NIH grant P30CA023100. We thank Dr. Roy Williams for CNV calling with Nexus software and Hiroko Matsui for processing of array data. We would also like to thank Fangwen Rao and Dr. Daniel T. O’Conner for providing us with twin samples from the University of California San Diego Twins cohort. We dedicate this work to the memory of our dear colleague Dr. O’Conner.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, A.D.P., E.N.S., W.T.B., J.C.I.B., and K.A.F.; Validation, A.D.P. and E.N.S.; Formal Analysis, E.N.S. and P.J.S.; Investigation, A.D.P., A.D.A., Y.H., V.M., K.E.D., C.C., W.B., E.S., and A.D.C.; Data Curation, E.N.S.; Writing – Original Draft, A.D.P., E.N.S., and K.A.F.; Writing – Review & Editing, A.D.P., E.N.S., and K.A.F.; Visualization, A.D.P., E.N.S., P.J.S., and K.A.F.; Supervision, J.C.I.B., K.A.F., and W.T.B.; Project Administration, A.D.P., J.C.I.B., and K.A.F.; Funding Acquisition, A.D.P., J.C.I.B., and K.A.F.
References
- Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40:e128. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendix Carstensen MP, Laara Esa, Hills Michael. Epi: A Package for Statistical Analysis in Epidemiology. R package version 20 2016 [Google Scholar]
- Benetatos L, Vartholomatos G, Hatzimichael E. DLK1-DIO3 imprinted cluster in induced pluripotency: landscape in the mist. Cell Mol Life Sci. 2014;71:4421–4430. doi: 10.1007/s00018-014-1698-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Burrows CK, Banovich NE, Pavlovic BJ, Patterson K, Gallego Romero I, Pritchard JK, Gilad Y. Genetic Variation, Not Cell Type of Origin, Underlies the Majority of Identifiable Regulatory Differences in iPSCs. PLoS Genet. 2016;12:e1005793. doi: 10.1371/journal.pgen.1005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Hanna JH, Faddah DA, Buganim Y, Kim J, Ganz K, Steine EJ, Cassady JP, Creyghton MP, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588–598. doi: 10.1016/j.stem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Carlson M. org.Hs.eg.db: Genome wide annotation for Human [Google Scholar]
- Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics: official journal of the DNA Methylation Society. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Lee S, Mallard W, Clement K, Tagliazucchi GM, Lim H, Choi IY, Ferrari F, Tsankov AM, Pop R, et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol. 2015;33:364–376. doi: 10.1038/nbt.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Terzic A. Energy metabolism in the acquisition and maintenance of stemness. Semin Cell Dev Biol. 2016;52:68–75. doi: 10.1016/j.semcdb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics. 2013;29:1851–1857. doi: 10.1093/bioinformatics/btt311. [DOI] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Puri MC, Tonge PD, Benevento M, Corso AJ, Clancy JL, Mosbergen R, Li M, Lee DS, Cloonan N, et al. Genome-wide characterization of the routes to pluripotency. Nature. 2014;516:198–206. doi: 10.1038/nature14046. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttala A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK, Nakanishi M, Nishimura K, Ohtaka M, Weltner J, et al. Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Reports. 2016;6:200–212. doi: 10.1016/j.stemcr.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire M, Zaidi SH, Ban M, Ge B, Aissi D, Germain M, Kassam I, Wang M, Zanke BW, Gagnon F, et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat Commun. 2015;6:6326. doi: 10.1038/ncomms7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Han Q, Peng T, Peng M, Wei B, Li D, Wang X, Yu S, Yang J, Cao S, et al. The oncogene c-Jun impedes somatic cell reprogramming. Nat Cell Biol. 2015;17:856–867. doi: 10.1038/ncb3193. [DOI] [PubMed] [Google Scholar]
- Lutz M, Modesto V, Panopoulos A. Protocol for making retroviral reprogramming factors. StemBook (Cambridge (MA)) 2008 [PubMed] [Google Scholar]
- Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo CF, Wu FC, Tai KY, Chang WC, Chang KW, Kuo HC, Ho HN, Chen HF, Lin SP. Loss of non-coding RNA expression from the DLK1-DIO3 imprinted locus correlates with reduced neural differentiation potential in human embryonic stem cell lines. Stem Cell Res Ther. 2015;6:1. doi: 10.1186/scrt535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Muller FJ, Wang YC, Boscolo FS, et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 2011;13:541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos AD, D’Antonio M, Benaglio P, Williams R, Hashem SI, Schuldt BM, DeBoever C, Arias AD, Garcia M, Nelson B, et al. iPSCORE: A systematically derived resource of iPSC lines from 222 individuals for use in examining how genetic variation affects molecular and physiological traits across a variety of cell types. Stem Cell Reports. doi: 10.1016/j.stemcr.2017.03.012. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerias A, Batchelder EM, Plongthongkum N, Lutz M, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168–177. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V, Plath K. X chromosome reactivation in reprogramming and in development. Curr Opin Cell Biol. 2015;37:75–83. doi: 10.1016/j.ceb.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, Blumlein K, Wanker EE, Ralser M, Cramer T, et al. HIF1alpha modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells. 2014;32:364–376. doi: 10.1002/stem.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. [Google Scholar]
- Raczy C, Petrovski R, Saunders CT, Chorny I, Kruglyak S, Margulies EH, Chuang HY, Kallberg M, Kumar SA, Liao A, et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics. 2013;29:2041–2043. doi: 10.1093/bioinformatics/btt314. [DOI] [PubMed] [Google Scholar]
- Rouhani F, Kumasaka N, de Brito MC, Bradley A, Vallier L, Gaffney D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014;10:e1004432. doi: 10.1371/journal.pgen.1004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Diep D, Gore A, Panopoulos AD, Montserrat N, Plongthongkum N, Kumar S, Fung HL, Giorgetti A, Bilic J, et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16196–16201. doi: 10.1073/pnas.1202352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EN, Ghia EM, DeBoever CM, Rassenti LZ, Jepsen K, Yoon KA, Matsui H, Rozenzhak S, Alakus H, Shepard PJ, et al. Genetic and epigenetic profiling of CLL disease progression reveals limited somatic evolution and suggests a relationship to memory-cell development. Blood Cancer J. 2015;5:e303. doi: 10.1038/bcj.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, Zhou Q, Plath K. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler G, Leonard S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code for Biology and Medicine. 2014;9:13. [Google Scholar]
- Veluscek G, Li Y, Yang SH, Sharrocks AD. Jun-Mediated Changes in Cell Adhesion Contribute to Mouse Embryonic Stem Cell Exit from Ground State Pluripotency. Stem Cells. 2016;34:1213–1224. doi: 10.1002/stem.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, Moffitt TE, Mill J. A longitudinal study of epigenetic variation in twins. Epigenetics: official journal of the DNA Methylation Society. 2010;5:516–526. doi: 10.4161/epi.5.6.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1A: List of 158,466 CpGs that were associated with a CpG predictor class and/or an aberrant CpG class. For each CpG the chromosome, position, classification by CpG predictor class, the number of samples that showed aberrant methylation in each of the three aberrant CpG classes, and whether or not the CpG is an SBE site is shown. S1B: Gene-based CpG analyses. Genes enriched for CpGs associated with each CpG predictor class, each aberrant CpG class, and each gene’s RNA-seq predictor class annotation are given. NA in CpG class categories indicates that the gene did not have CpGs annotated to it on the 450K BeadChip, while NA in RNA predictor class indicates that the gene was not expressed in the iPSCs and therefore not analyzed.
List of gene sets showing significant enrichment (FDR < 0.05) for genes in one of the seven gene-level CpG predictor classes or in gene-level aberrant CpG classes. For aberrant genes, a group including all genes showing significant enrichment of any aberrant CpG was also used (“Any Aberrant”). Analyses were performed using GOseq adjusting for the number of CpGs annotated to each gene, and the Gene Ontology (GO) and Molecular Signatures Databases were used.
S3A: List of enriched motifs for CpGs associated with aberrant CpG classes. Results from Dreme and CentriMo analyses are shown. Only human motifs are reported. All Dreme motif results with E-values < 0.01 are listed. When a significant motif was identified with Dreme, TomTom was used to try to identify similar known motifs – none were observed. When CentriMo was used to identify a motif, the Fisher E-value, central enrichment P-value, and central enrichment distribution are shown. MYC and MYC-like motifs (defined here as sharing at least four bases out of the six central CACGTG bases of the MYC motif) are indicated for the CentriMo analysis of iPSC gain sites. S3B: List of enriched motifs for CpGs associated with CpG predictor classifications. Results from Dreme and CentriMo analyses are shown. Only human motifs are reported. All Dreme motif results with E-values < 0.01 are listed. When a significant motif was identified with Dreme, TomTom was used to identify similar known motifs. When multiple “similar known motifs” were identified by TomTom for the same “significant motif”, all results are listed with the same Motif Number. A “similar motif” (i.e. FOSB) may be listed with multiple Motif Numbers if it was identified by TomTom in multiple analyses. When CentriMo was used to identify a motif, the Fisher E-value, central enrichment P-value, and central enrichment distribution are shown.