

Abstract
Purpose
Little is known about the signaling mechanisms controlling meibomian gland (MG) homeostasis and the pathogenic processes leading to MG atrophy and dysfunction in dry eye disease (DED). We investigated the role of fibroblast growth factor receptor 2 (FGFR2) in the MG homeostasis of adult mice.
Methods
A triple transgenic mouse strain (Krt14-rtTA; tetO-Cre; Fgfr2flox/flox), referred to as Fgfr2CKO mice, was generated in which the Fgfr2 gene is ablated by Cre recombinase in keratin 14 (Krt14)-expressing epithelial cells on doxycycline (Dox) induction. FGFR2 expression in normal human and mouse MGs was evaluated by immunohistochemistry. Pathologic MG changes in transgenic mice with conditional deletion of FGFR2 were examined by lipid staining, histology, and immunostaining.
Results
FGFR2 was highly expressed in normal human MGs and adult mouse MGs. Two-month-old Fgfr2CKO mice fed Dox-containing chow for 2 weeks developed severe MG atrophy. MG acinar atrophy in the Fgfr2CKO mice was associated with reduced lipid (meibum) production and the development of clinical findings similar to those in humans with evaporative DED related to MG dysfunction (MGD). Immunohistochemical analyses showed that FGFR2 deletion severely affected proliferation and differentiation of MG acinar cells but affected MG ductal cells to a lesser extent.
Conclusions
FGFR2 deletion results in significant MG acinar atrophy and clinical manifestations of MGD in Fgfr2CKO mice, suggesting that MG homeostasis is FGFR2 dependent. The Fgfr2CKO mice with inducible MG atrophy can serve as a valuable animal model for investigating the pathogenesis of MGD and developing novel therapeutic strategies for MGD-related DED.
Keywords: meibomian gland dysfunction, FGFR2, dry eye disease, meibomian gland atrophy, knockout animals
Dry eye disease (DED) is a multifactorial disease characterized by ocular surface desiccation and related ocular irritation. The prevalence of DED is estimated to be 7% to 34% of the US population, and the number of affected persons is increasing with the aging of our population.1–3 The hallmark of DED is tear film instability, caused by a reduction of aqueous tear secretion, and an increase in tear evaporation, either alone or in combination.1,4 The most common cause of evaporative DED is meibomian gland dysfunction (MGD), which is associated with a decrease in the quantity and/or quality of lipids in tear film.5,6 Meibomian glands (MGs), which are embedded in the tarsal plates of the eyelids, synthesize and secrete a mixture of lipids, termed “meibum,” which is spread over the eye surface during blinking.7 Meibum constitutes the outer layer of tear film and overlies the aqueous and mucin gels produced by the lacrimal glands and conjunctival goblet cells, respectively. As such, meibum plays a critical role in slowing the evaporation of tear fluids, protecting the ocular surface from desiccating stress, and enhancing tear film stability by lowering the surface tension of tears.7,8 Despite the high prevalence of MGD among patients with DED, our understanding of the pathogenesis of MGD remains extremely limited, which undoubtedly has hampered the development of effective therapeutics to ameliorate MGD or restore the various MG functions. Current MGD therapies are mostly palliative and unsatisfactory, as they often aim primarily at symptomatic relief of DED and not directly at remediating the underlying pathogenesis of MGD.9,10 To facilitate the development of therapeutic approaches that target the pathogenic mechanisms of MGD and produce a long-lasting improvement in MG functions, it is of paramount importance to understand the molecular mechanisms that modulate MG homeostasis and help maintain tear film stability.
MGs are considered large and modified sebaceous glands (SGs).11 MGs and SGs share common glandular characteristics, including their holocrine secretion of lipids, by which the terminally differentiated acinar epithelial cells disintegrate themselves to release the intracellular lipids.12 Prior studies of immortalized sebocytes (acinar epithelial cells of the sebaceous glands) have shown that several types of cell surface receptors, including those for peptides, sex hormones, neurotransmitters, and growth factors, are expressed in sebocytes, suggesting that SG homeostasis is likely to be regulated by some of these receptors.13
It is well known that fibroblast growth factor receptors (FGFRs) and their ligands participate in embryonic and postnatal development and regulate cell proliferation, survival, migration, and differentiation in adult mice.14–16 In mammals, there are 22 FGF ligands that can signal via one or more of the four FGFRs. Among the four Fgfr genes (Fgfr1–4), Fgfr1–3 genes contain alternative splicing sites at the C-terminal IgIII domain, resulting in two isoforms known as Fgfrb and Fgfrc. An in vivo mouse genetic study showed that conditional gene deletion of one of the isoforms of FGFR2, namely Fgfr2b, in epidermal cells, causes striking abnormalities in hairs and SGs without affecting animal survival.14 These findings suggest that FGFR2b signaling activity is required for the normal development and homeostasis of SGs.
Very little is known about the signaling pathways that regulate MG homeostasis.13,14,17 Specifically, the role of FGFR2 and its related ligands in MG development and homeostasis has not been explored. To determine whether FGFR2 signaling is crucial for MG homeostasis in mice, we used an inducible conditional gene knockout strategy to delete Fgfr2 in keratin 14 (Krt14)-expressing epithelial cells and generated a mouse model of MG atrophy inducible with doxycline (Dox) (Krt14-rtTA; tetO-Cre; Fgfr2flox/flox, referred as Fgfr2CKO) (Fig. 1). Herein, we describe the histopathologic features of MG atrophy in this triple-transgenic mouse strain and demonstrate for the first time that the induced loss of FGFR2 (both b and c isoforms) in 2-month old mice leads to severe MG atrophy, substantiating that FGFR2 plays a pivotal role in MG homeostasis and tissue maintenance. To the best of our knowledge, this mouse model constitutes the first animal model of inducible MG atrophy, phenotypically resembling MG atrophy in human MGD-related DED. Our findings also suggest that the Fgfr2CKO mouse model of Dox-induced MG atrophy can serve as a valuable tool for delineating the pathogenic mechanisms of MGD and for evaluating potential therapeutic interventions for MGD-related DED.
Figure 1.

Schematic of Dox-induced conditional deletion of Fgfr2 gene. Double transgenic mice (K14rtTA-tetOCre) were created to express Cre recombinase, on Dox induction, in epithelial cells that express Krt14 promoters. The double transgenic mice were then bred to Fgfr2flox/flox mice to generate triple transgenic mice (K14rtTA-tetOCre-Fgfr2 fl/fl). Ablation of Fgfr2 in K14-expressing cells is induced by feeding the triple transgenic mice Dox chow for various lengths of time.
Materials and Methods
Acquisition of Donor Eyelid Tissues
Tarsal plates removed from fresh cadaver's upper and lower eyelids of a healthy Caucasian female donor (36 years of age) were obtained from Mid-America Transplant (St. Louis, MO, USA). The use of human tissue in research conformed to the provisions of the Declaration of Helsinki and was exempted by the Washington University Human Subjects Protection Office. Prior to removal of eye tissues, medical and ocular histories of the donor had been deidentified and reviewed to ensure no evident ocular or systemic diseases. The removal of donor eyelid tissues and corneas was performed under a standard protocol for procurement of ocular tissues.
Mouse Strains and Genotyping
All transgenic mice were bred at the Animal Science Research Center of the University of Missouri (Columbia, MO, USA). Animal experiments were conducted in accordance with the institutional guidelines on the Care, Welfare and Treatment of Laboratory Animals, and the protocols were approved by the University of Missouri Institutional Animal Care and Use Committee. The experiments conformed to the standards in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Transgene alleles were screened by PCR using tail DNA, following the conditions and primer pairs recommended for JAX mice (Krt14-rtTA stock#008099 and tetO-Cre stock#006224) and previously reported for Fgfr2-flox mice.18,19
Generation of Conditional Knockout Mice on Dox Administration
Compound transgenic mice of K14rtTA-tetOCre-Fgfr2fl/fl were generated via natural mating following the scheme illustrated in Figure 1. In this deletion system, Cre expression in Krt14- (or K14)-expressing cells can be initially induced in mice of any age by systemic administration of Dox, and consequently the floxed Fgfr2 is ablated by Cre recombinase (Fig. 1). In our study, conditional knockout of Fgfr2 was induced in 2-month-old adult mice by ad libitum Dox chow feeding, for 4 days to 2 weeks, at the dosage of 1 g Dox/kg chow (Dox diet #AD3008; Custom Animal Diets, Bangor, PA, USA). Littermates fed with regular chow without Dox served as control animals.
Oil-Red-O Staining for Lipids
Fresh mouse eyelids were collected from transgenic and control mice, immediately fixed with 4% paraformaldehyde overnight, and rinsed with PBS. For Oil-Red-O (ORO) staining of the eyelid whole mount, eyelids were placed in 60% 2-propanol for 15 minutes, stained with ORO solution for 30 minutes, and then destained with 60% 2-propanol for 15 to 30 minutes to achieve optimal lipid staining over the background color. For ORO staining on cryosections, eyelid tissues were embedded in OCT and sectioned at 10 μm of thickness. Frozen sections were placed in 60% 2-propanol for 1 minute, stained with ORO solution for 15 minutes, rinsed with PBS, and counterstained with hematoxylin.
Histology
Mouse eyes with eyelids were dissected, fixed overnight in 4% paraformaldehyde (PFA) in PBS, and then processed for embedding in paraffin. Tissue sections (5 μm) were deparaffinized with xylene, rehydrated with ethanol, and stained with either hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) reagent per the manufacturer's instructions.
5-Bromo-2′-Deoxyuridine Incorporation Assay
5-Bromo-2′-deoxyuridine (BrdU), 0.1 mg/g of body weight, was injected intraperitoneally and labeled for 2 hours before mice were euthanized. BrdU immunostaining was performed following the conditions previously described by Fromm et al.20
Immunofluorescence and Immunohistochemistry
Paraffin sections were deparaffinized in xylene, rehydrated in a decreased ethanol series, and subjected to antigen retrieval in 10 mM sodium citrate buffer by boiling for 10 minutes.21,22 Sections were treated with 3% hydrogen peroxide in PBS for 20 minutes to block endogenous peroxidase activity. Cyrosections (7 μm) were dipped in PBS to remove OCT, fixed with 4% PFA for 10 minutes, and then rinsed with PBS three times. Tissue sections were blocked with 3% horse serum in PBST (PBS plus 0.1% Tween-20 for paraffin sections) or PBS plus 0.5% Triton X-100 (for cryosections) for 1 hour at room temperature and then incubated overnight at 4°C, with primary antibodies diluted in the same buffer. Slides were washed with PBS, incubated at room temperature for 1 hour with either fluorophore-conjugated or biotinylated secondary antibodies, and then washed with PBS. For immunofluorescence, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and slides were mounted with Mowiol (Sanofi-Aventis, Bridgewater, NJ, USA) for viewing under a Leica microscope equipped with a charge-coupled device (CCD) camera for photography. For immunohistochemistry, sections were incubated with Vectastain Elite ABC Reagent (PK-6100; Vector Laboratories, Burlingame, CA, USA), and color was developed by using 3,3′-diaminobenzidine as a substrate (D4293; Sigma-Aldrich Corp., St Louis, MO, USA). Sections were counterstained with hematoxylin for cell nuclei.
Primary antibodies were purchased from the following sources: anti-FGFR2 (ab10648; Abcam, Cambridge, MA, USA); anti-keratin 10 and anti-keratin 14 (PRB-159P and PRB-155P, respectively; Covance, Emeryville, CA, USA); anti-keratin 16 (NBP2-45538; Novusbio, Littleton, CO, USA); anti-p63 (GTX102425, GeneTex, Irvine, CA, USA), anti-PPARγ (#2435; Cell Signaling, Danvers, MA, USA); anti-PCNA (ab29; Abcam); and anti-BrdU (M-0744; Dako, Carpinteria, CA, USA). Fluorophore-conjugated secondary antibodies were obtained from ThermoFisher Scientific (Rockford, IL, USA) and biotinylated secondary antibodies were from Vector Laboratories.
Statistical Analysis
For quantitative analysis of BrdU index in MG ductal epithelial cells, two eyelids of different mice were excised from either control or Fgfr2CKO fed with Dox for 6 days. A minimal of four cross sections along a MG central duct and a minimal of six ducts on each section were included in analyses. Data were expressed as mean ± SEM, and P values were calculated using paired Student's t-tests, with P < 0.05 being significant.
Results
FGFR2 Expression in Human and Mouse MGs
To investigate the importance of FGFR2 in MG homeostasis, we first examined FGFR2 expression patterns in the tarsal plate of human and mouse eyelids. We noted high FGFR2 levels were expressed in both acinar and ductal epithelial cells of human and mouse MGs (Fig. 2). The finding of mouse MGs having a similar FGFR2 expression pattern to that of human MGs suggests that FGFR2 signaling is required for MG homeostasis in both species, similar to what has been reported in mouse SGs.14
Figure 2.
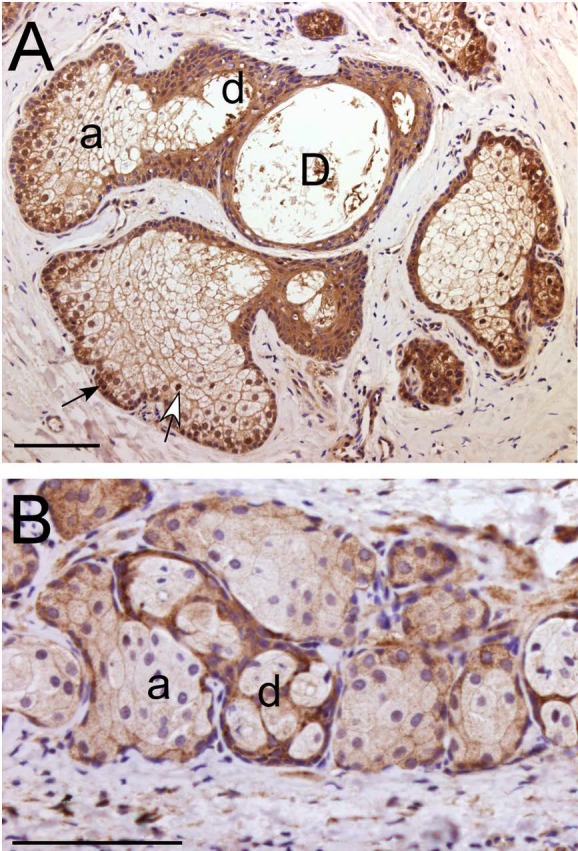
FGFR2 expression in normal human and mouse MGs detected by immunohistochemistry. (A) In human MG, FGFR2 is expressed in the acini (a), ductules (d), and central duct (D). In the peripheral MG acinus, FGFR2 immunoreactivity was detected in both the cell membrane and nuclei of basal epithelial cells (black arrow) and in newly differentiated meibocytes (white arrow). (B) In mouse MG, the FGFR2 expression pattern was similar to that seen in human MG (A). Scale bars denote 100 μm.
MG Atrophy as a Result of Fgfr2 Conditional Deletion on Dox Induction
Conditional deletion mediated by keratin 5 (Krt5) promoter has shown that FGFR2b is required for SG homeostasis in the skin of adult mice.14 However, to our knowledge, the role of FGFR2 in MG homeostasis has never been investigated in that mouse model. Keratin 14 (Krt14) is a keratin pair of Krt5 and is known to be expressed in various ocular surface epithelial tissues, including MGs.21 To investigate whether FGFR2 plays a role in MG homeostasis, we generated a triple transgenic mouse model (Krt14TA-tetOCre-Fgfr2fl/fl), as depicted in Figure 1, in which conditional deletion of Fgfr2 only occurs via activation of Krt14 promoter on Dox feeding. At 2 months of age, the mice were fed either regular chow (control mice) or Dox chow (Fgfr2 conditional knockout or Fgfr2CKO mice). The triple transgenic mice fed with regular chow display no evident MG pathologies but develop MG atrophy on feeding with Dox-containing chow. After 7 to 10 days of Dox chow feeding, Fgfr2CKO mice were reluctant to keep their eyes open wide and showed symptoms of ocular irritation. With extended Dox feeding (14 days), more advanced symptoms were noted, with macerated periorbital hairs and eyelids, suggesting excessive eye rubbing due to ocular irritation (Fig. 3). Control mice did not exhibit such symptoms. Lipid (meibum) production by MGs in the upper eyelids was assessed by eyelid whole mounts stained with ORO In control mice, meibum was seen in the central duct, but in Fgfr2CKO mice, a reduction of meibum staining in the central duct and a reduction in MG acinar size were visible (compare Fig. 3A with 3B). These findings suggest that FGFR2 signaling is essential for MG function and homeostasis in adult mice. The results further indicate that we have created a mouse model with inducible MG atrophy that causes ocular symptoms similar to those of MGD in humans.
Figure 3.
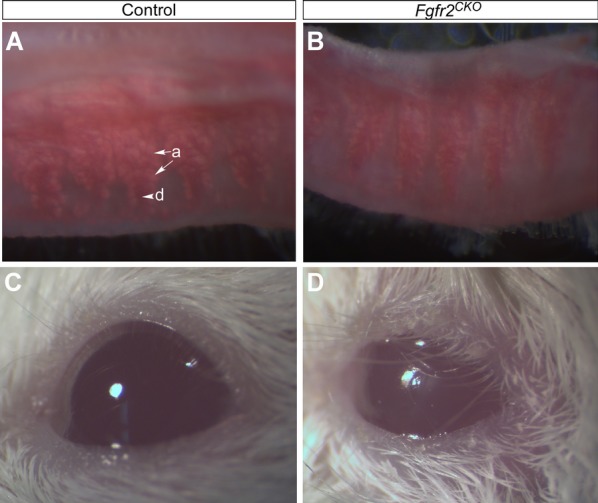
MG atrophy and signs of DED in Fgfr2CKO mice. (A, B) ORO lipid staining in control and Fgfr2CKO mice. Upper eyelids were dissected from control mice (fed regular chow) and Fgfr2CKO mice (fed Dox chow for 10 days) and subjected for whole-mount ORO staining to view lipid (meibum) production by MG acini (arrows labeled a). Meibum secreted into main duct (arrowhead labeled d) was stained in a dark-red color in control mice (A). Compared with control mice, Fgfr2CKO mice exhibited a reduction of MG acinar area and meibum production (B). (C, D) Compared with control mice (C), Fgfr2CKO mice showed signs of ocular irritation, including loss of corneal luster and macerated eyelids, after extended (2 to 3 weeks) Dox chow feeding (D). Such signs of ocular irritation were likely due to increased tear evaporation and tear film instability.
Histologic examination of MG atrophy after 1 week of Dox feeding revealed a moderate reduction in the number of MG acini in Fgfr2CKO mice compared with the number of MG acini in control mice (Fig. 4). The conjunctival and corneal epithelia looked similar in control and Fgfr2CKO mice. Immunostainings of mucin 5AC (Muc5AC) as a goblet cell marker and of Krt13 as a conjunctival epithelial marker (Supplementary Fig. S1) further substantiated the aforementioned findings. After 2 weeks of Dox feeding, Fgfr2CKO mice had developed severe MG atrophy. However, PAS staining revealed no discernible difference in goblet cell density between control and Fgfr2CKO mice. In contrast to the conjunctival epithelium, the corneal epithelial layer after 2 weeks of Dox feeding appeared to be thinner in Fgfr2CKO mice than in control mice (Figs. 4C, 4D).
Figure 4.

Histology of eyelid and ocular surface tissues in Fgfr2CKO and control mice. (A, B) H&E staining of frontal sections from control (no Dox) and Fgfr2CKO mice after 1 week of Dox feeding, showing a moderate reduction in the number of MG acini in mutant mice compared with the number of MG acini in control mice (broken line circled areas). The conjunctival (Conj) and corneal (Co) epithelium (epi) looked similar in the control and Fgfr2CKO mice. (C, D) PAS staining showed severe MG atrophy, with minimal acinar tissue, after 2 weeks of Dox feeding. There was no discernible difference in PAS-positive goblet cells (gb, arrowheads) between control and Fgfr2CKO mice. The thickness of the corneal epithelial layer (epi, arrows in D) was significantly reduced in Fgfr2 deletion mice. (E, F) ORO staining of cross sections of the upper eyelids in Fgfr2CKOand control mice showing a reduction MG acinar size (labeled a) and a decrease in meibum volume in the central duct (labeled d) in Fgfr2CKO mice fed Dox chow for 10 days. Sections were counterstained with hematoxylin. M, muscle; HF, hair follicle.
Cross sections of the upper eyelids of Fgfr2CKO and control mice were stained with ORO to correlate reduced meibum production with the morphologic and histologic changes of MG atrophy (Figs. 4E, 4F). Lipid staining confirmed that MG atrophy in Fgfr2CKO mice was associated with a severe decrease in acinar size and a reduction of meibum volume in the central duct. The results suggest that the degree of MG atrophy in this mouse model can be modulated by the duration of Dox feeding.
To evaluate the efficiency of Dox induced gene deletion, we examined the levels of Cre and FGFR2 in the eyelids of control and Fgfr2CKO mice (Supplementary Fig. S2). After 4 days of Dox feeding, Cre was expressed in most MG acinar cell nuclei but with variable amounts (Supplementary Fig. S2B). The expression of Cre in ductal cells was delayed, but high levels were detected after 6 days of Dox feeding (Supplementary Fig. S2C). In response to Cre expression, FGFR2 level was reduced significantly in the MG acini of FGFR2CKO mice fed with Dox chow compared with that in control mice (Supplementary Figs. S2D–SE′). Immunofluorescence of FGFR2 was diminished in both MG acinar and ductal cells but was still detectable in the conjunctival epithelium of Fgfr2CKO mice after 10 days of Dox induction (Supplementary Figs. S2F, S2F′).
To further evaluate the phenotype of MG atrophy in Fgfr2CKO mice, we examined the immunofluorescence of Krt14 expression in different MG structures, such as the acinus, ductule, and the central duct (Fig. 5). As the diagram in Figure 5 illustrates, cross sections of the upper eyelid in the distal MG area and of the lower eyelid close to MG orifice were selected to investigate any changes in acini around the ductules and acini around central duct. MG acinar atrophy was readily visible in Fgfr2CKO mice fed with Dox for 1 week compared with the control mice. The ductal structures in mutant mice were less affected than acinar tissue. The acellular area (occupied by meibum) within the ductule or central duct was reduced or nearly absent in Fgfr2CKO mice, a phenotype consistent with reduced meibum synthesis and secretion and ductal obstruction with exfoliated epithelial plugs known in human MGD.
Figure 5.

Immunofluorescence of Krt14. Cross sections of the distal acinar area of the upper eyelid (A, B) and the central ductal area of lower eyelid (C, D), as illustrated in the diagram on left, were examined for Krt14 immunofluorescence. (A, B) MG acinar atrophy was seen in Fgfr2CKO mice fed with Dox for 7 days, as shown by the reduction of Krt14 staining compared with control mice. In contrast to acinar tissue, the ductal structures in mutant mice were not affected significantly (compare areas marked by dotted lines in B and D versus A and C). (C, D) The acellular area (occupied by meibum) within the ductule or central duct (white arrows, labeled d) can be readily seen in control mice, but is either visibly reduced or nearly absent in Fgfr2CKO mice because of decreased meibum production.
PPARγ, Krt10, and Krt16 Expression by Immunohistochemistry
In normal MGs, meibocytes within an MG acinus undergo a pattern of differentiation, starting from undifferentiated proliferating basal cells in the periphery and progressing to suprabasal nucleated meibocytes that synthesize lipids. During this differentiation process, peroxisome proliferator–activated receptor gamma (PPARγ), a lipid-activated hormone receptor that regulates lipid synthesis, is up-regulated in newly differentiated meibocytes and then down-regulated in mature or terminally differentiated meibocytes.22,23 Compared with control mice, Fgfr2CKO mice fed Dox chow for 1 week exhibited a significant decrease in the number of PPARγ-positive meibocytes, which correlates with the finding of acinar atrophy (Figs. 6A, 6B).
Figure 6.

PPARγ, Krt10, and Krt16 expression in lower eyelids detected by immunohistochemistry. (A) In control mice, within an acinus, PPARγ is not expressed in undifferentiated basal cells (small arrow in A). PPARγ expression (brown nuclear staining) is up-regulated in differentiated meibocytes (large arrow) and then down-regulated in mature meibocytes (white arrow). Meibocytes inside the duct (labeled D; red broken-line circle) were negative for PPARγ. (B) In Fgfr2CKO mice fed with Dox chow for 1 week, MG acinar atrophy was noted, with a reduction of PPARγ-positive meibocytes in MG acinus. Additionally, although ductal cells in mutant mice were PPARγ negative, they did not appear to be fully mature (as shown in the nucleated cells inside red circle in Fig. 5B). (C, D) In both control and Fgfr2CKO mice, Krt16 expression levels were higher in the ductules (d) and main duct (labeled D) than in acini. Severe MG acinar atrophy was seen in Fgfr2CKO mice, but ductal tissues looked intact. (E, F) Krt10 expression was found in cells within central duct (red broken-line circles) in both control and Fgfr2CKO mice. In Fgfr2CKO mice, a reduction of meibum volume and the presence of nucleated cell debris were seen (F). HF, hair follicles.
Normally, as meibocytes proceed through their usual holocrine differentiation, the acinar cells increase in size as a consequence of accumulating lipid droplets, and then rupture and release their lipid cargo into the ductule space. Before the lipid (meibum) in the central duct is delivered to the ocular surface to form the outer layer of tear film, most of the cellular remnants are disintegrated. In the control mice, this process was evident by the lack of PPARγ expression in mature meibocytes with high lipid content inside the duct (Fig. 6A). In Fgfr2CKO mice, the meibocytes in the central duct contained more densely packed and nucleated cell debris, suggesting that FGFR2 deletion inhibits differentiation and maturation of meibocytes and disrupts the normal holocrine mechanism (Fig. 6B).
To confirm the observation that ductal structures are less or minimally affected compared with acini in Fgfr2CKO mice, we examined by immunohistochemistry the expression of Krt16, a marker for ductal epithelia (Figs. 6C, 6D). In control mice, Krt16 was expressed at a much higher level in MG ductules and central duct than in acini. In Fgfr2CKO mice, despite severe MG acinar atrophy, the intensity of Krt16 immunostaining in the ductal tissues was at a similar level as in control mice (compare Fig. 6D with 6C). This result suggests that, in contrast to MG acinar cells, ductal epithelial cells are not significantly affected by FGFR2 deletion.
Jester et al.24 previously demonstrated that expression of Krt1 (which pairs with Krt10) extends from the fully keratinized epidermis (skin) into the ductal epithelium at the MG orifice in young adult mice and that Krt1 expression extends posteriorly toward the conjunctiva. Krt10 expression in the MG central duct was also shown by Call et al.25 recently. We found that Krt10 was expressed in ductal epithelial cells in both control and Fgfr2CKO mice (Figs. 6E, 6F). However, the central ducts of Fgfr2CKO mice showed a reduction of meibum volume and an increase of cellular debris (Fig. 6F). These results suggest that MG acinar cell differentiation and maturation are FGFR2 dependent. In contrast, ductal epithelial cells are not significantly affected by the loss of FGFR2.
Decrease of MG Basal Cells in Fgfr2CKO Mice
When apoptosis was examined by TUNEL assay, a few positive nuclei (indicating apoptosis) were only found in the duct, but not in the acini, in both control and Fgfr2CKO mice, suggesting that MG atrophy in mutant mice is not caused by cell death (data not shown). To further explore the pathogenic mechanisms of MG atrophy in Fgfr2CKO mice, we examined the number of basal cells in MG acini using p63 as a marker. Transcription factor p63 is a member of the p53 family and a key regulator of epithelial cell fate.21,26–29 We found that basal cell density in MG acini of Fgfr2CKO mice was largely reduced after 4 days of Dox induction compared with the control mice (compare Fig. 7A with 7B). We then investigated the expression pattern of another cell proliferation marker, proliferating cell nuclear antigen (PCNA) (Figs. 7C, 7D). Immunofluorescence of PCNA (red) was colocalized with Krt14 immunofluorescence (green) to demarcate the MGs in eyelids. In control mice, PCNA-positive nuclei were found in basal epithelial cells of MG acini and ducts, suggesting that these cells are in the proliferative state. In Fgfr2CKO mice fed Dox chow for 1 week, PCNA-positive nuclei were found in ductal basal cells but rarely seen in acinar basal cells. We further confirmed that cell proliferation in MG ductal epithelium of Fgfr2CKO mice by colocalizing PCNA immunofluorescence with Krt17. Krt17, similar to Krt 16 (as shown in Figs. 6C, 6D), is also preferentially expressed in MG ductal cell (Supplementary Fig. S3). Taken together, these results suggest that FGFR2 deletion primarily inhibits proliferation of the MG acinar basal cells.
Figure 7.

Expression of basal and proliferating cell markers. (A, B) Transcription factor p63 in MGs detected by immunohistochemistry on crysections. The basal cells in MG acini (dark brown nuclei indicated by arrows in A) and ducts (data not shown) express p63 (A). In FGFR2CKO mice fed with Dox for 4 days, the number of basal cells around the MG acini was markedly reduced (B). (C, D) Immunofluorescence of Krt14 (green) and proliferative cell nuclear antigen (PCNA) (red). Krt14 expression marks the areas of MGs in eyelids. In control mice (C), intense PCNA immunofluorescence was found in acinar (a) and ductal (d) basal epithelial cell nuclei (red as indicated by white arrow in C), suggesting that these cells are in the proliferative state. Within a MG acinus, PCNA immune intensity decreases when basal cells are differentiated into meibocytes (arrowhead in C). In Fgfr2CKO mice induced by Dox for 1 week, PCNA-positive nuclei were rarely seen in acinar basal cells, but detected in ductal basal layer (white broken-line circle in D). (E, F) BrdU-labeled cells were found in MG acinar and ductal basal cell layer in control mice (arrows in E). In Fgfg2CKO mice fed with Dox for 6 days, BrdU incorporation was seen in MG ductal basal cells (arrows in F), but rarely detected in the acini. HF, hair follicle.
BrdU incorporation assay was performed to examine the effect of FGFR2 on cell cycle progression in MG acinar basal cells (Figs. 7E, 7F). In control mice, BrdU-positive nuclei, indicative of cells at S-phase of cell cycle, were found in the basal cell layers of MG acini and in central ducts. Compared with the control mice, a drastic reduction in BrdU-labeled cells was noted in MG acini of Fgfr2CKO mice, suggesting that FGFR2 deletion blocks cell cycle progression in MG acinar basal cells. In contrast, BrdU incorporation in the ductal basal cells of Fgfr2CKO mice did not seem to be affected. The BrdU labeling index in ductal epithelium was 7.31 ± 0.65% in control and 5.90 ± 0.55% in Fgfr2CKO (P = 0.08), indicating the difference was not statistically significant. The findings of BrdU labeling are consistent with those of PCNA immunofluorescence. Taken together, our cell proliferation studies imply that FGFR2 signaling is crucial for acinar basal cell proliferation to maintain MG homeostasis in adult mice.
To explore the downstream signaling pathways of FGFR2, we further examined the levels of ERK1/2 (or ERK) proteins and their active form, the phosphorylated ERK (or pERK), in the MGs of control and Fgfr2CKO mice fed with Dox for 4 days (Supplementary Fig. S4). We found that ERK proteins were present in both MG acinar and ductal cells in control mice (Supplementary Figs. S4A, S4C), but pERK (the active form) was mostly localized in the nuclei of MG acini. Despite MG atrophy in Fgfr2CKO mice, pERK could still be found in the nuclei of MG acinar cells of both control and Fgfr2CKO mice (Supplementary Figs. S4B′, S4D′), suggesting that FGFR2 may regulate MG basal cell proliferation via ERK-independent pathways.
Discussion
By definition, MGD is a chronic, diffuse MG abnormality, commonly characterized by terminal duct obstruction and/or qualitative/quantitative changes in glandular secretion.5,7,30 This condition may result in alterations of the tear film on the ocular surface, symptoms of eye irritation, clinically apparent inflammation, and ocular surface morbidities.2,31,32 MGD is the leading cause of DED in the United States and elsewhere in the developed world, affecting 5% to 30% of the population aged 50 or older.3,33 Age-related changes in human MGs include primary acinar atrophy and/or inspissation of MG orifices leading to decreased meibum secretion. MG atrophy and dropout can apparently occur as early as 25 years of age, and the extent of these pathogenic changes increases significantly and approximately linearly with age.33 At present, the underlying mechanism of age-related MG acinar atrophy is poorly understood. In this study, we developed a novel mouse model of inducible MG atrophy by conditional deletion of FGFR2. We demonstrate for the first time in mice that FGFR2 plays an essential role in MG acinar maintenance and homeostasis. Importantly, the observed MG atrophy in our mouse model as a result of loss of FGFR2 is in striking resemblance to age-related MG atrophy in humans.34 Because FGFR2 proteins are present at high levels in both mouse and human MGs (Fig. 1), our current findings thus imply that reduced or altered FGFR2-signaling activity could be one of the underlying mechanisms that lead to age-related MG acinar atrophy and glandular dropouts in humans.
It is well known that the MG acinar cells (meibocytes) secrete meibum via a holocrine mechanism.7,35 During this secretion process, the meibocytes undergo a well-defined program of cell differentiation and maturation, which can be defined morphologically as undifferentiated (basal), differentiating, mature, and hypermature meibocytes. A single MG acinus consists of a contiguous layer of proliferative basal cells in the acinar periphery that surrounds a cluster of lipid-filled meibocytes undergoing differentiation and maturation.36 Lysis of hypermature meibocytes releases meibum via a small canal (ductule) to a central excretory duct that opens at the lid margin near the mucocutaneous junction. To maintain meibum production and secretion continuously, the meibocytes are constantly undergoing renewal that is sustained by division of the basal cells in the peripheral acinus. Our current data from the control mice (Fig. 7) also support such a notion on MG homeostasis. However, how does FGFR2 signaling control MG homeostasis at cellular level? Our findings in the Fgfr2CKO mice with Dox-induced MG atrophy suggest that cell proliferation markers, including PCNA expression and BrdU incorporation, are drastically reduced in the MG acinar basal cells (Fig. 7). Deletion of FGFR2 induced by Dox results in the loss of MG acinar basal cells, as shown by the number of p63-expressing cells (Fig. 7B) and disruption of the continuous replenishment of meibocytes and regeneration of MG clusters, thus leading to MG atrophy. This finding further suggests that FGFR2 signaling plays an essential role in maintaining acinar basal cell proliferation in MGs. Alternatively, it is also possible that FGFR2 activity is required for the maintenance of MG acinar progenitor/stem cell population, which was recently demonstrated by Parfitt et al.37 Dox-induced FGFR2 deletion may result in a depletion of MG acinar progenitor cells in Fgfr2CKO mice. Nonetheless, whether FGFR2-signaling activity plays a similar role in human MGs and whether such activity is altered in aged human MGs await further investigation.
In obstructive MGD, hyperkeratinization of the MG orifice is thought to lead to cystic ductal dilation and downstream disuse atrophy of the MG acini.7,31,38,39 However, ductal hyperkeratinization was not observed in a mouse model of age-related MGD.24 In our current mouse model, we did not see ductal dilation in Fgfr2CKO mice, and the K16 and K10 expression patterns were not affected by MG atrophy (Fig. 6). By immunofluorescence, a low level of FGFR2 was still detectable in MG ductal epithelial cells in Fgfr2CKO mice fed Dox for 6 days (data not shown). The differential deletion efficiency between MG acinar and ductal cells potentially could result from a higher FGFR2 level (Fig. 1B) and a slower cell turnover rate in ductal epithelial cells than in acinar meibocytes.37 However, the acellular areas (indicative of meibum volume) in MG ducts were visibly reduced, and the pyknotic nuclei were increased in the ductules and central ducts of Fgfr2CKO mice (Figs. 5–7). These observations suggest that meibocyte differentiation and maturation was compromised in Fgfr2CKO mice. We speculate that such an accumulation of cellular/nuclear debris in the ductal system could be a contributing factor, eventually leading to pathogenic ductal obstruction.
Clinically, MGD is often associated with an inflammatory response. The causal relationship between MGD and inflammation remains somewhat debatable.9,40 In our Fgfr2CKO mouse model, inflammation was not readily evident when the mutant mice developed severe MG atrophy after 2 weeks of Dox treatment (Fig. 3D). Because Dox has been shown to have anti-inflammatory effects and is frequently used clinically for treating MGD,41,42 it is possible that Dox is suppressing inflammation in the Fgfr2CKO mice. Alternative conditional deletion systems have been investigated,43 and we are currently breeding another transgenic model using tamoxifen (instead of Dox) to turn on Cre expression. Nonetheless, we can take advantage of our current Dox inducible model to explore the pathogenic mechanisms of glandular atrophy in MGD independent of inflammation-mediated pathways.
Conventionally, DED is often considered to be a disorder of tear film, accompanied by changes in several ocular surface tissues, including the conjunctiva, goblet cells and corneal epithelium.44–46 Similar to MGs, these ocular surface epithelial tissues also express Krt14 and are subject to FGFR2 deletion in our mouse model. However, the deletion efficiency varies among these tissues, depending on several factors such as the K14-promoter activity level, accessibility to Dox, and cell turnover rate. When FGFR2 protein level was examined by immunofluorescence in ocular surface tissues of Fgfr2CKO mice fed with Dox for 10 days, the signal intensity was reduced but still detectable in conjunctival and corneal epithelial cells (data not shown). In this study, we could not examine whether prolonged Dox exposure results in complete depletion of FGFR2 in these tissues, because the general health of Fgfr2CKO mice started to deteriorate after 2 weeks of feeding with Dox.
When conjunctival epithelia of control mice and Fgfr2CKO mice fed with Dox chow for 2 weeks were carefully examined by histology, we did not observe any major changes in cell morphology or goblet cell density (Fig. 4; Supplementary Fig. S1). However, the corneal epithelial layer of Fgfr2CKO mice was notably thinner than that of control mice (Fig. 4D, pointed out by arrows). Because corneal surface damage can occur in MGD patients,45 the corneal changes in Fgfr2CKO mice could be secondary to the severe MG atrophy induced after 2 weeks of Dox feeding. Alternatively, corneal epithelial cells can be directly affected by FGFR2 deletion, as we have previously shown that FGFR2 plays a critical role in control of cell proliferation during early corneal development.47 To further explore whether FGFR2 is directly involved in maintaining normal corneal epithelium homeostasis, an inducible Cre system driven by the cornea-specific Krt12 promoter can be used in future studies to eliminate confounding factors such as the interference from eyelids and other ocular surface tissues.48
In conclusion, our novel mouse model with conditional deletion of FGFR2 clearly demonstrates that the FGFR2-signaling pathway is critical for MG maintenance and homeostasis in adult mice. Because human MGs also express high levels of FGFR2 and because MGD is the most common cause of evaporative DED, it would be exciting to further substantiate that age-related MG atrophy and glandular dropouts in aging humans are a result of deficient FGFR2 signaling. Understanding the underlying mechanism of MGD can potentially lead to the development of effective management of this condition. The animal model of MG atrophy we developed not only should allow us to elucidate a previously unexplored disease process, but would also facilitate the development of novel therapeutic strategies specific for MGD, such as modulation of FGFR2 signaling in MGs and ocular surface tissues.
Supplementary Material
Acknowledgments
The authors thank Bethany Irlmeier, Alyssa Labonte, and Abby Lueckenotte for technical assistance and Sharon Morey for editorial assistance.
Supported by National Institutes of Health R01 Grant EY24221 (LWR).
Disclosure: L.W. Reneker, None; L. Wang, None; R.T. Irlmeier, None; A.J.W. Huang, None
References
- 1. Management and therapy of dry eye disease: report of the Management and Therapy Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007; 5: 163–178. [DOI] [PubMed] [Google Scholar]
- 2. Baudouin C,, Messmer EM,, Aragona P,, et al. Revisiting the vicious circle of dry eye disease: a focus on the pathophysiology of meibomian gland dysfunction. Br J Ophthalmol. 2016; 3: 300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nichols KK,, Bacharach J,, Holland E,, et al. Impact of dry eye disease on work productivity, and patients' satisfaction with over-the-counter dry eye treatments. Invest Ophthalmol Vis Sci. 2016; 57: 2975–2982. [DOI] [PubMed] [Google Scholar]
- 4. The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007; 5: 75–92. [DOI] [PubMed] [Google Scholar]
- 5. Nelson JD,, Shimazaki J,, Benitez-del-Castillo JM,, et al. The international workshop on meibomian gland dysfunction: report of the definition and classification subcommittee. Invest Ophthalmol Vis Sci. 2011; 52: 1930–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nichols KK,, Foulks GN,, Bron AJ,, et al. The international workshop on meibomian gland dysfunction: executive summary. Invest Ophthalmol Vis Sci. 2011; 52: 1922–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knop E,, Knop N,, Millar T,, Obata H,, Sullivan DA. The international workshop on meibomian gland dysfunction: report of the subcommittee on anatomy, physiology, and pathophysiology of the meibomian gland. Invest Ophthalmol Vis Sci. 2011; 52: 1938–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green-Church KB,, Butovich I,, Willcox M,, et al. The international workshop on meibomian gland dysfunction: report of the subcommittee on tear film lipids and lipid-protein interactions in health and disease. Invest Ophthalmol Vis Sci. 2011; 52: 1979–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lim A,, Wenk MR,, Tong L. Lipid-based therapy for ocular surface inflammation and disease. Trends Mol Med. 2015; 21: 736–748. [DOI] [PubMed] [Google Scholar]
- 10. Geerling G,, Tauber J,, Baudouin C,, et al. The international workshop on meibomian gland dysfunction: report of the subcommittee on management and treatment of meibomian gland dysfunction. Invest Ophthalmol Vis Sci. 2011; 52: 2050–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parfitt GJ,, Geyfman M,, Xie Y,, Jester JV. Characterization of quiescent epithelial cells in mouse meibomian glands and hair follicle/sebaceous glands by immunofluorescence tomography. J Invest Dermatol. 2015; 135: 1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Niemann C,, Horsley V. Development and homeostasis of the sebaceous gland. Semin Cell Dev Biol. 2012; 23: 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Melnik BC,, Schmitz G,, Zouboulis CC. Anti-acne agents attenuate FGFR2 signal transduction in acne. J Invest Dermatol. 2009; 129: 1868–1877. [DOI] [PubMed] [Google Scholar]
- 14. Grose R,, Fantl V,, Werner S,, et al. The role of fibroblast growth factor receptor 2b in skin homeostasis and cancer development. EMBO J. 2007; 26: 1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li X,, Wang C,, Xiao J,, McKeehan WL,, Wang F. Fibroblast growth factors, old kids on the new block. Semin Cell Dev Biol. 2016; 53: 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carter EP,, Fearon AE,, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015; 25: 221–233. [DOI] [PubMed] [Google Scholar]
- 17. Melnik BC. Role of FGFR2-signaling in the pathogenesis of acne. Dermatoendocrinology. 2009; 1: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garcia CM,, Huang J,, Madakashira BP,, et al. The function of FGF signaling in the lens placode. Dev Biol. 2011; 351: 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang J,, Dattilo LK,, Rajagopal R,, et al. FGF-regulated BMP signaling is required for eyelid closure and to specify conjunctival epithelial cell fate. Development. 2009; 136: 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fromm L,, Shawlot W,, Gunning K,, Butel JS,, Overbeek PA. The retinoblastoma protein-binding region of simian virus 40 large T antigen alters cell cycle regulation in lenses of transgenic mice. Mol Cell Biol. 1994; 14: 6743–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ng GY,, Yeh LK,, Zhang Y,, et al. Role of SH2-containing tyrosine phosphatase Shp2 in mouse corneal epithelial stratification. Invest Ophthalmol Vis Sci. 2013; 54: 7933–7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jester JV,, Brown DJ. Wakayama Symposium: peroxisome proliferator-activated receptor-gamma (PPARgamma) and meibomian gland dysfunction. Ocul Surf. 2012; 10: 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nien CJ,, Paugh JR,, Massei S,, Wahlert AJ,, Kao WW,, Jester JV. Age-related changes in the meibomian gland. Exp Eye Res. 2009; 89: 1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parfitt GJ,, Xie Y,, Geyfman M,, Brown DJ,, Jester JV. Absence of ductal hyper-keratinization in mouse age-related meibomian gland dysfunction (ARMGD). Aging (Albany NY). 2013; 5: 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Call M,, Fischesser K,, Lunn MO,, Kao WW. A unique lineage gives rise to the meibomian gland. Mol Vis. 2016; 22: 168–176. [PMC free article] [PubMed] [Google Scholar]
- 26. Yoh K,, Prywes R. Pathway regulation of p63, a director of epithelial cell fate. Front Endocrinol (Lausanne). 2015; 6: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng CC,, Wang DY,, Kao MH,, Chen JK. The growth-promoting effect of KGF on limbal epithelial cells is mediated by upregulation of DeltaNp63alpha through the p38 pathway. J Cell Sci. 2009; 122: 4473–4480. [DOI] [PubMed] [Google Scholar]
- 28. Parsa R,, Yang A,, McKeon F,, Green H. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Invest Dermatol. 1999; 113: 1099–1105. [DOI] [PubMed] [Google Scholar]
- 29. Yang A,, Schweitzer R,, Sun D,, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999; 398: 714–718. [DOI] [PubMed] [Google Scholar]
- 30. Bron AJ,, Tomlinson A,, Foulks GN,, et al. Rethinking dry eye disease: a perspective on clinical implications. Ocul Surf. 2014; 12: S1–S31. [DOI] [PubMed] [Google Scholar]
- 31. Bron AJ,, Tiffany JM. The contribution of meibomian disease to dry eye. Ocul Surf. 2004; 2: 149–165. [DOI] [PubMed] [Google Scholar]
- 32. Cuevas M,, Gonzalez-Garcia MJ,, Castellanos E,, et al. Correlations among symptoms, signs, and clinical tests in evaporative-type dry eye disease caused by Meibomian gland dysfunction (MGD). Curr Eye Res. 2012; 37: 855–863. [DOI] [PubMed] [Google Scholar]
- 33. Yeotikar NS,, Zhu H,, Markoulli M,, Nichols KK,, Naduvilath T,, Papas EB. Functional and morphologic changes of meibomian glands in an asymptomatic adult population. Invest Ophthalmol Vis Sci. 2016; 57: 3996–4007. [DOI] [PubMed] [Google Scholar]
- 34. Arita R,, Morishige N,, Koh S,, et al. Increased tear fluid production as a compensatory response to meibomian gland loss: a multicenter cross-sectional study. Ophthalmology. 2015; 122: 925–933. [DOI] [PubMed] [Google Scholar]
- 35. Butovich IA. Tear film lipids. Exp Eye Res. 2013; 117: 4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nien CJ,, Massei S,, Lin G,, et al. The development of meibomian glands in mice. Mol Vis. 2010; 16: 1132–1140. [PMC free article] [PubMed] [Google Scholar]
- 37. Parfitt GJ,, Lewis PN,, Young RD,, et al. Renewal of the holocrine meibomian glands by label-retaining, unipotent epithelial progenitors. Stem Cell Rep. 2016; 7: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim HM,, Eom Y,, Song JS. The relationship between morphology and function of the meibomian glands. [published online ahead of print October 13, 2016] Contact Lens. doi:10.1097/ICL.0000000000000336. [DOI] [PubMed] [Google Scholar]
- 39. Nakayama N,, Kawashima M,, Kaido M,, Arita R,, Tsubota K. Analysis of meibum before and after intraductal meibomian gland probing in eyes with obstructive meibomian gland dysfunction. Cornea. 2015; 34: 1206–1208. [DOI] [PubMed] [Google Scholar]
- 40. Suzuki T,, Teramukai S,, Kinoshita S. Meibomian glands and ocular surface inflammation. Ocul Surf. 2015; 13: 133–149. [DOI] [PubMed] [Google Scholar]
- 41. Zhang Z,, Yang WZ,, Zhu ZZ,, et al. Therapeutic effects of topical doxycycline in a benzalkonium chloride-induced mouse dry eye model. Invest Ophthalmol Vis Sci. 2014; 55: 2963–2974. [DOI] [PubMed] [Google Scholar]
- 42. Doughty MJ. On the prescribing of oral doxycycline or minocycline by UK optometrists as part of management of chronic meibomian gland dysfunction (MGD). Cont Lens Anterior Eye. 2016; 39: 2–8. [DOI] [PubMed] [Google Scholar]
- 43. Stratis A,, Pasparakis M,, Markur D,, et al. Localized inflammatory skin disease following inducible ablation of I kappa B kinase 2 in murine epidermis. J Invest Dermatol. 2006; 126: 614–620. [DOI] [PubMed] [Google Scholar]
- 44. Gipson IK. Age-related changes and diseases of the ocular surface and cornea. Invest Ophthalmol Vis Sci. 2013; 54: ORSF48–ORSF53. [DOI] [PubMed] [Google Scholar]
- 45. Bron AJ,, Argueso P,, Irkec M,, Bright FV. Clinical staining of the ocular surface: mechanisms and interpretations. Prog Retin Eye Res. 2015; 44: 36–61. [DOI] [PubMed] [Google Scholar]
- 46. Gipson IK. Goblet cells of the conjunctiva: a review of recent findings. Prog Retin Eye Res. 2016; 54: 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J,, Upadhya D,, Lu L,, Reneker LW. Fibroblast growth factor receptor 2 (FGFR2) is required for corneal epithelial cell proliferation and differentiation during embryonic development. PLoS One. 2015; 10: e0117089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chikama T,, Liu CY,, Meij JT,, et al. Excess FGF-7 in corneal epithelium causes corneal intraepithelial neoplasia in young mice and epithelium hyperplasia in adult mice. Am J Pathol. 2008; 172: 638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.