

Abstract
Rice grain chalkiness is a highly complex trait involved in multiple metabolic pathways and controlled by polygenes and growth conditions. To uncover novel aspects of chalkiness formation, we performed an integrated profiling of gene activity in the developing grains of a notched-belly rice mutant. Using exhaustive tandem mass spectrometry-based shotgun proteomics and whole-genome RNA sequencing to generate a nearly complete catalog of expressed mRNAs and proteins, we reliably identified 38,476 transcripts and 3,840 proteins. Comparison between the translucent part and chalky part of the notched-belly grains resulted in only a few differently express genes (240) and differently express proteins (363), thus making it possible to focus on ‘core’ genes or common pathways. Several novel key pathways were identified as of relevance to chalkiness formation, in particular the shift of C and N metabolism, the down-regulation of ribosomal proteins and the resulting low abundance of storage proteins especially the 13 kDa prolamin subunit, and the suppressed photosynthetic capacity in the pericarp of the chalky part. Further, genes and proteins as transporters for carbohydrates, amino acid/peptides, proteins, lipids and inorganic ions showed an increasing expression pattern in the chalky part of the notched-belly grains. Similarly, transcripts and proteins of receptors for auxin, ABA, ethylene and brassinosteroid were also up-regulated. In summary, this joint analysis of transcript and protein profiles provides a comprehensive reference map of gene activity regarding the physiological state in the chalky endosperm.
Keywords: Chalkiness, Proteome, Rice, Transcriptome, White-belly mutant
Introduction
Improvement in rice grain quality has attracted attention worldwide. In developing countries such as China, where grain yield has been increased substantially by breeding and planting technologies, producing high quality rice is becoming a desired goal along with the rising standard of living (Zhang 2007). In addition, high temperature during grain filling proves to have a negative effect on rice quality, in particular grain chalkiness, suggesting the need for research on precautionary measures under the likely scenario of global warming (Zhao and Fitzgerald 2013). However, relatively slow progress has been made in unraveling the genetic background of grain quality traits, reflecting the complexity of their underlying mechanisms.
Chalkiness is the opaque part in rice endosperm. According to its location, chalkiness is grouped into white-belly, white-core, white-base, etc. Microscopic study reveals that the opaque tissue is loosely packed with storage-like starch granules and protein bodies (PBs), making the whole grain vulnerable to cracking during the polishing process (Xi et al. 2014, Sreenivasulu et al. 2015). Therefore, chalkiness is not only a cosmetic characteristic, but also a key factor influencing milling quality. Research showed that chalkiness formation was related to abnormal carbohydrate metabolism such as cell wall and starch biosynthesis. Dysfunction of GIF1 and UGPase1 genes in cell wall biosynthesis caused chalky endosperm (Wang et al. 2008, Woo et al. 2008). Mutations in genes participating in starch synthesis such as OsAPL2, SSIIIa and BEIIb and the regulators RSR1and OsbZIP58 resulted in a chalky phenotype (Tanaka et al. 2004, Fujita et al. 2007, Fu and Xue. 2010, Zhang et al. 2012, Wang et al. 2013). Perturbation in expression of genes involved in protein metabolism also leads to chalkiness, such as down-regulation of BiP1 (Wakasa et al. 2011) and PDI (Han et al. 2012) for protein folding, and mutation in OsRAB5A (Fukuda et al. 2013) and OsVPS9A (Liu et al. 2013) for protein sorting. Recently, the role of proteins in the occurrence of chalkiness has been evidenced by cloning and characterization of Chalk5, encoding a vacuolar H+-translocating pyrophosphatase that affects the endomembrane protein trafficking system and consequently the formation of PBs (Li et al. 2014).
Carbon (C) and nitrogen (N) metabolism are the main metabolic processes during rice grain filling, and their products, the storage of starch and proteins, dominate grain chemical compositions and build the foundation for quality traits such as chalkiness and eating quality. N and C metabolism interact at numerous points in plant metabolism. A large proportion of N in leaves is invested in the photosynthesis proteins, particularly Rubisico, the world’s most abundant protein. Decreases in N supply lead to reduction in proteins for photosynthesis, which in turn negatively affects C assimilation (Schmollinger et al. 2014). In grains, amino acid biosynthesis mainly depends on the C skeletons derived from glycolysis, the tricarboxylic acid cycle (TCA) and the pentose phosphate pathway (PPP). This interdependence of C and N metabolism creates problems when attempting to describe an independent contribution of either C or N to quality.
High-throughput profiling of transcripts or proteins is an efficient method for exploring the system atlas of complex biological processes, with the advantages of high resolution, deep coverage and dynamic landscapes (Lan et al. 2012). Technological advances have improved dramatically, making it possible to measure gene activity at a genome-wide scale, such as the iTRAQ (isobaric tags for relative and absolute quantitation) method to probe protein abundance, and RNA sequencing (RNA-Seq) to detect mRNA expression. Integrated measurement and interpretation of changes in protein and transcript abundance are mandatory for generating a complete inventory of gene networks, because of post-translational turnover and alternative translation efficiency (Lan et al. 2012). Joint analysis of multiple omics data increases the understanding of gene networks by offering a more holistic view of the target biological processes (Moreno-Risueno et al. 2010). In rice, the integrative approach has deepened our knowledge concerning the response of seedling growth to jasmonic acid and ozone (Cho et al. 2007, Cho et al. 2008), induced callus differentiation of mature seed (Yin et al. 2007) and mitochondrial arginine metabolism during germination under anaerobic conditions (Taylor et al. 2010). For grain chalkiness, transcriptomic (Yamakawa et al. 2007, Liu et al. 2010) and proteomic (Lin et al. 2005, Lin et al. 2014) studies showed that chalkiness occurrence is involved in multiple metabolic and regulatory pathways. However, there is little information concerning the parallel analysis of chalkiness formation at both the mRNA and protein levels.
Using ethyl methanesulfonate (EMS) as the chemical mutagen, we identified a notched-belly mutant with high occurrence of chalkiness, which predominantly occurs in the bottom part under the notched line (Lin et al. 2014; Fig. 1). In an attempt to identify pathways crucial for chalkiness formation, an integrated profiling of gene activity by RNA-Seq and iTRAQ was performed. We compared the differences in the abundance of mRNAs and proteins between the translucent part and chalky part of the developing notched-belly grains, and found several novel key pathways related to chalkiness formation, such as the shift of C and N metabolism, the down-regulation of ribosomal proteins and the resulting low abundance of storage proteins especially the 13 kDa prolamin subunit, the suppressed photosynthetic capacity in the pericarp as well as the up-regulation of transporters and phytohormone receptors. We argue that the joint analysis of gene and protein expression data provides a complete picture regarding the physiological state in chalky endosperm, and expands and deepens our insight into the regulation of the metabolic network in developing rice grain.
Fig. 1.

Developing grains at 5, 10, 15 and 20 days after flowering (DAF) of the notched-belly mutant.
Results
Characterization of developing rice grains
We sampled and observed morphological features of the developing notched-belly grains (also called caryopses) on the middle primary rachis at 5, 10, 15 and 20 days after flowering (DAF). The developing grains greatly increased in size from 5 to 10 DAF, and then had a slight increase and appeared to reach the size of mature seeds at 15 DAF (Fig. 1). The upper part above the notched line became translucent, while the bottom part under it appeared chalky at 20 DAF (Fig. 1). In general, the developmental changes in grain morphology are consistent with previous observations (Xu et al. 2008). According to the classification of Xu et al. (2008), we tentatively divided the development process from 5 to 20 DAF into early (5–10 DAF), mid (10–15 DAF) and late (15–20 DAF) stages.
Genome-wide changes in mRNA and protein abundance in notched-belly grains
The transcriptome in each sample was determined by RNA-Seq on the Illumina platform, and expression estimates were calculated as reads per kilobase per million reads (RPKM). Transcripts of 38,476 genes were detected. Replicate experiments showed a generally high overlap of the detected transcripts. Validation of transcriptome data was performed by quantitative real-time PCR (qRT-PCR). Twenty-four genes functioning in C and N metabolism were selected (Supplementary Table S1). Expression patterns determined by qRT-PCR were consistent with those by RNA-Seq, confirming the accuracy of the results of the latter (Supplementary Fig. S1).
Changes in the proteome of developing notched-belly grains were quantitatively cataloged using the iTRAQ technology. The search algorithm Mascot was used to identify proteins. We identified 13,629 unique peptides from 363,174 spectra, corresponding to 4,646 proteins in the eight samples of developing grains. Quantitative information was obtained for 3,840 proteins. Robustness of the analysis was supported by multiple reaction monitoring (MRM) of eight proteins from the eight samples. Expression level changes for seven proteins showed similar trends to those by iTRAQ analysis, except that of UDP-GlcDH, thus validating the iTRAQ data (Supplementary Table S2).
Overall, transcripts were detected for 98.34% of the proteins (Fig. 2A). To explore the relationship between proteins and their cognate genes, we matched all expressed proteins with their cognate mRNAs at four time points and observed a weak correlation, with r value ranging from 0.09 at 5 DAF to 0.169 at 20 DAF (Fig. 3), indicating a strong post-translational regulation and the necessity of joint analysis of the transcriptome and proteome. The weak correlation between mRNA and proteins may be due to the fact that the majority of the detected proteins and genes are down-regulated during grain developing. As reported by Lan et al. (2012), in comparison with the up-regulated genes, a decreased abundance of proteins was not closely correlated with changes in the corresponding mRNAs.
Fig. 2.

Comparison of protein and transcript abundance in developing rice grains. (A) Congruency between the detected transcripts and proteins of rice endosperm. (B) Number of differentially expressed genes in relation to developmental and chalkiness effects (absolute value of log2Ratio ≥1 with probability ≥0.8); Ch5, Ch10, Ch15, and Ch20, chalky part at 5, 10, 15 and 20 days after flowering (DAF); Tr5, Tr10, Tr15 and Tr20, translucent part at 5, 10, 15 and 20 DAF. (C) Number of differentially expressed proteins in relation to developmental and chalkiness (occurence) effects (1.2-fold change with P-value <0.05). (D) Functional classification and distribution of the differentially expressed genes and proteins between the chalky and translucent parts at four time points via the Gene Ontology (GO) database.
Fig. 3.
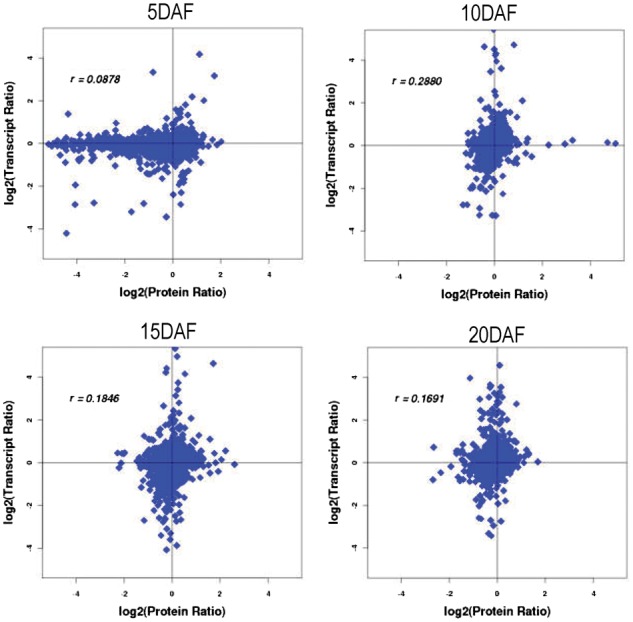
Concordance between changes in the abundance of mRNA and its encoded protein in the developing grains at four sampling times. Transcript ratio and Protein ratio, the fold changes of transcript and protein between the upper translucent part (Tr) and bottom chalky part (Ch) in rice endosperm, respectively. r, Pearson correlations coefficeint of the comparisons between fold changes of proteins and transcripts at each sampling time.
Developmental effect on transcriptome and proteome profiling
The differentially expressed genes (DEGs) were identified by using NOIseq and a strict algorithm of the Beijing Genomics Institute (BGI, Shenzhen), and differentially expressed proteins (DEPs) by a 1.2-fold change in combination with a P-value <0.05. Developmental stage showed a large effect on mRNA and protein abundance. Generally, DEGs and DEPs showed a diminishing trend along with developing progress (Fig. 2B, D). In comparison with the initial sampling time 5 DAF, the numbers of up-regulated DEGs in the upper translucent part of the notched-belly grain were 243, 521 and 678 at 10, 15 and 20 DAF, respectively; in contrast, there were 1,507, 5,049 and 5762 down-regulated DEGs, respectively. Similarly, in the upper part, there were 65, 112 and 115 up-regulated DEPs, while 130, 156 and 228, respectively, were down-regulated. The reduction in transcripts and proteins in grain coincides with the switch of the physiological state of developing grains, from cell division of the embryo and endosperm to storage accumulation (grain filling).
Chalkiness effect on transcriptome and proteome profiling
We evaluated the chalkiness effect by comparison of DEGs and DEPs between the upper translucent part (Tr) and bottom chalky part (Ch). Results revealed a relatively smaller effect of chalkiness (occurrence) on mRNA and protein abundance as compared with that of the developmental stage. Surprisingly, numbers of DEGs and DEPs between Ch and Tr were drastically lower than those among developmental stages. Only 240 genes (Supplementary Table S3) and 363 proteins (Supplementary Table S4) were identified as expressed differently between Tr and Ch at at least one time point, with 15 pairs of genes/protein detected in both protocols. The notable reduction in gene and protein number makes it easier to highlight common or ‘core’ pathways related to grain chalkiness formation, indicating the practical value of the notched-belly mutant.
Gene Ontology (GO) analysis classified the DEGs and DEPs into 18 functional groups (Fig. 2D), i.e. carbohydrate metabolism; photosynthesis; energy metabolism; cell wall biosynthesis; amino acid metabolism; lipid metabolism; nucleotide metabolism; secondary metabolism; transcription; protein synthesis, assembly and degradation; plant hormone metabolism; signaling; transportation; development; seed storage protein; redox homeostasis; stress response; and function unknown gene/protein. Among them, function unknown was the largest group and stress response was the second largest group in DEGs, while protein synthesis, assembly and degradation was the largest group and stress response was the second largest group in DEPs. In addition, the majority of DEGs were consistently up-regulated across the four time points, except for those participating in photosynthesis and storage protein accumulation. However, no clear trend was observed for DEPs, and only proteins for cell synthesis were consistently decreased during grain development.
Chalkiness-related genes and proteins in C metabolism
Reduced energy production in the TCA cycle and oxidative phosphorylation
Glycolysis, PPP, the TCA cycle and oxidative phosphorylation are core metabolic pathways in endosperm, providing energy and the C skeleton for other metabolic pathways. Three DEGs and 21 DEPs were indentified in the above pathways (Fig. 4; Supplementary Tables S3, S4). In glycolysis and the PPP, up-regulation of the AEP gene and AEP protein at 10, 15 and 20 DAF was detected, while G6P-E was down-regulated. ALDO and FBA proteins were more abundant at 10 and 15 DAF, whereas 6-PGL was lower at the two time points. In the TCA cycle, ACLA-2, IDHC and SDH6 responsible for generating NADH and FADH2 exhibited a downward trend at four time points, except the up-regulation of ACLA-2 and SDH6 (at 5 DAF), SDHB (at 10 and 15 DAF) and ACOC (at 15 and 20 DAF). In oxidative phosphorylation, NDUB9 protein of complex I was mainly down-regulated. SDHB in complex II was decreased at 5 and 20 DAF but increased at 15 DAF. The COX6A gene and COX6A protein in complex IV were also less abundant at most of the time points. Similarly, five ATPases in complex V were down-regulated, but not VATE (at 5 and 20 DAF), ATPO (at 5 DAF) and V-PPase (at 15 DAF). Collectively, these findings indicate that pathways for energy production were inhibited in the chalky part of the notched-belly grain.
Fig. 4.

Expression patterns of genes and proteins for carbohydrate metabolism in the upper translucent part (Tr) and the bottom chalky part (Ch) of the notched-belly grains. Annotations of genes and proteins are available in Supplementary Tables S4 and S5. Black characters with yellow background are genes, whereas white characters with blue background are proteins. The four squares under the names of genes or proteins indicate abundance change of Ch5/Tr5, Ch10/Tr10, Ch15/Tr15 and Ch20/Tr20, respectively. A red square indicates up-regulation, whereas a green square indicates down-regulation.
Increased degradation of storage and structure substances
Starch is one of the main storage compounds in rice endosperm. We detected four DEGs and four DEPs involved in sucrose and starch metabolism (Fig. 4). The abundance of UGPase for sucrose synthesis was higher in the chalky part at four time points, while SuS3 for sucrose catabolism was suppressed except at 20 DAF. ADPase and SSII in starch synthesis and AMY3E and AMY3E for starch degradation were both increased. In addition, ISA3, an isoamylase-type starch debranching enzyme, also demonstrated a decreasing trend except at 5 DAF. Further, GLU36 and GLU31 for polysaccharide catabolism were also suppressed.
Meanwhile, increased degradation in the structural substances of the cell wall was observed, with eight DEGs and 11 DEPs identified (Fig. 4; Supplementary Tables S3, S4). mRNAs encoding UXS and GRP1 for cell wall synthesis were down-regulated at four time points, while those for cell wall degradation (PME and PE) were up-regulated, except Talin, PGIP1 and Lectin. The 11 DEPs participating in cell wall synthesis were also less abundant in the chalky part, excluding AEP and PRPL1 at 15DAF.
Preference for short-chain fatty acid synthesis
Three DEGs and eight DEPs were identified in lipid metabolism (Fig. 4). The gene (SDR) and proteins including ENR and ALDR for glycerate and short-chain fatty acid synthesis showed an increase at 15 and 20 DAF. At the same time, the abundances of mRNA of KCS1 and CUT1 and protein of LACS2 for hexadecanoate or long-chain fatty acid synthesis were decreased within the transcriptome and proteome of the chalky part. Further, ACOX for β-oxidation of hexadecanoate was up-regulated.
Chalkiness-related genes and proteins in amino acid metabolism
Increased threonine biosynthesis
There existed no clear trend for the amino acids asparagine, glutamine, arginine and proline. Transcripts of GLN12 and proteins of GLN11, GLN12 for glutamine synthesis increased their abundance at 10 and 15 DAF (Fig. 5). Proteins of ASNS2 for asparagine synthesis were also induced at 20 DAF. In the pathway of arginine and proline synthesis, P5CS, OAT and PIP were up-regulated at different time points, except P5CS at 10 DAF. Notably, threonine synthesis in the chalky part tended to be promoted, as THRB and THRC were up-regulated at the four time points.
Fig. 5.

Expression patterns of genes and proteins for amino acid metabolism in the upper translucent part and the bottom chalky part of the notched-belly grains. Annotations of genes and proteins are available in Supplementary Tables S4 and S5. Black characters with yellow background are genes, whereas white characters with blue background are proteins. The four squares under the names of genes or proteins indicate abundance change of Ch5/Tr5, Ch10/Tr10, Ch15/Tr15 and Ch20/Tr20, respectively. A red square indicates up-regulation, whereas a green square indicates down-regulation.
Active SMM cycle
Metabolites of the S-methylmethionine (SMM) cycle enter the ethylene, nicotianamine and polyamine biosynthetic pathways and provide the methyl group for the majority of methylation reactions required for plant growth and development. Two DEGs and three DEPs were detected, with NAAT for methionine synthesis and ACCO for ethylene synthesis increasing at four time points. Similarly, proteins of MTN and ARD were also up-regulated. In addition, MTNA was only abundant at 10 DAF (Fig. 5).
Enhanced aromatic amino acid synthesis, accompanied by reduced lignin synthesis
Aromatic amino acids serve as precursors of numerous natural products, such as pigments, alkaloids, hormones and cell wall components. MIFH catalyzing tyrosine and phenylalanine catabolism were also up-regulated at 5 and 15 DAF (Fig. 5). Expression of genes (NAAT and PheA) and proteins (PAT, AspAT and TRPC) was increased at four time points. Three proteins (ACSF3, ROMT and SOHCT) functioning in lignin synthesis were down-regulated, indicating that derivatives of aromatic amino acids may be shifted to synthesize other natural products, such as pigments or alkaloids, rather than lignins.
Chalkiness-related genes and proteins in protein synthesis and degradation
Higher abundance of transcripts and proteins for transcription
The abundance of genes and proteins for transcription was higher in the chalky bottom part of the notched-belly grain. A total of five and 15 DEGs were mapped to the pathway of genome organization and transcription, respectively (Fig. 6). Interestingly, they were all up-regulated in the chalky part, excluding one gene (MADS1). A total of four, seven and 10 DEPs were mapped to the pathway of genome organization, RNA splicing and transcription, respectively. Similarly, they showed a growing trend, but there were some exceptions such as RH52A, SYMPK and NRAP (Fig. 6). the function of the majority of these transcription factors in rice is still largely unknown. However, some of them were identified as playing a key role in plant development and its response to abiotic stress, such as Ramy1 as a trans-acting protein which is probably involved in the gibberellin-induced expression of the rice α-amylase gene (Peng et al. 2014), OsMADS1 as a regulator of rice panicle development (Prasad et al. 2001), VIP1 as a regulator of osmosensory signaling in Arabidopsis (Tsugama et al. 2012) and OsCOIN as an enhancer of tolerance of rice to chilling, salt and drought (Liu et al. 2007).
Fig. 6.

Expression patterns of genes and proteins for protein assembly and degradation in the upper translucent part and the bottom chalky part of the notched-belly grains. Annotations of genes and proteins are availabble in Supplementary Tables S4 and S5. Black characters with yellow background are genes, whereas white characters with blue background are proteins. The four squares under the names of genes or proteins indicate an abundance change of Ch5/Tr5, Ch10/Tr10, Ch15/Tr15 and Ch20/Tr20, respectively. A red square indicates up-regulation, whereas a green square indicates down-regulation.
Perturbed mRNA surveillance and degradation
Despite enhancing transcription activities, Rae1L involved in mRNA transport though the nuclear pore complex was less abundant in the chalky part (Fig. 6). Supporting this, PABP1, encoding a polyadenylate-binding protein involved in the RNA surveillance pathway, was up-regulated. Meanwhile, three proteins for mRNA surveillance, namely SF3b, RNP1 and GRP RU17, increased their expression at 5 DAF and then decreased. We also identified four proteins, CSL4, EDC4, EXD1 and LSM4, in the exosome that plays a key role in RNA degradation, but they showed no recognizable trend.
Decrease in ribosomal proteins
A total of 25 ribosomal proteins were detected in ribosomes, including 13 large subunit proteins (60S), 11 small subunit proteins (40S) and RNP1 (Fig. 6). Notably, all these proteins were less abundant in the chalky part, except RNP, S21 or S27 at some time points. Two aminoacyl-tRNA synthetase, Leu-RS and Lys-RS, were decreased at 10 DAF, but tended to increase at other time points. In addition, we identified and mapped five eukaryotic translation initiation factors (IF1A, eRF3A, IF4E1, IF4B and IF4E1) and three elongation factors (EF2, EF1G1 and EFTs), but they were affected differently by the chalkiness effect.
Enhanced protein degradation
Protein ubiquitination plays a vital role in cellular processes which mainly functions as a signal for 26S proteasome degradation. In the ubiquitin ligase complex, seven DEPs were identified, with UFM1, URM1, SUMO1 and SUMO2 having an upward but Ubc12, SUMoAE and SKP1L-1A showing a downward trend in the chalky part (Fig. 6). In the 26S proteasome, the abundance of five proteins was higher for most of the time points. Interestingly, 12 DEGs mapped to phagolysosomes were more abundant in the chalky part, with the exception of HADH. Ten DEPs mapped to phagolysosomes responded differently to chalkiness, with half of them increased and the other half decreased in abundance.
Reduced prolamin accumulation
Prolamin is the major component of PBI in rice endosperm. In this study, 11 genes and two proteins were detected, and the majority of them were down-regulated in the chalky part (Fig. 6). Notably, two genes encoding 13 kDa prolamin were decreased across all developmental stages, as has been seen in the chalky grains caused by high temperature (Yamakawa et al. 2007, Yamakawa and Hakata 2010). These results in combination suggest that weak accumulation of prolamin, in particular the 13 kDa subunit, plays a key role in the formation of chalkiness.
Photosynthetic capacity of the chalky part of the notched-belly grains
In pigment synthesis, Chl a and Chl b synthesis was enhanced by up-regulation of UROD at 5 DAF and of SRD at 10, 15 and 20 DAF (Fig. 7). Eight genes and nine proteins for photosynthetic electron transport were expressed differently between translucent and chalky parts. Notably, most of them were down-regulated in the chalky part, with two exceptions of PetC in Cyt b6f and Lhcb4 at 5 and 10 DAF. Further, ATPD of ATP synthase was mainly down-regulated at 10, 15 and 20 DAF, but it decreased at 5 DAF. In photosynthetic C assimilation, Rubisco increased its gene activity at all time points (Fig. 4). However, two Rubisco proteins varied differently when compared between the translucent and chalky parts. Collectively, our results show relatively lower photosynthetic capacity in the chalky part.
Fig. 7.

Expression patterns of genes and proteins for photosynthesis in the upper translucent part (Tr) and the bottom chalky part (Ch) of the notched-belly grains. Annotations of genes and proteins are available in Supplementary Tables S4 and S5. Black characters with yellow background are genes, whereas white characters with blue background are proteins. The four squares under the names of genes or proteins indicate abundance change of Ch5/Tr5, Ch10/Tr10, Ch15/Tr15 and Ch20/Tr20, respectively. A red square indicates up-regulation, whereas a green square indicates own-regulation.
Transporters and receptors in the chalky part of the notched-belly grains
Transporters are important in the movement of materials in and out of cells. We identified 21 genes and 10 proteins as transporters for carbohydrates, amino acids/peptides, proteins, lipids, inorganic ions and ADP/ATP (Supplementary Table S5). In general, all these transporters were enhanced in the chalky part. There were no doubt some exceptions of down-regulation, such as three genes (nsLTP3, nsLTP1 and LTPL124) and three proteins (nsLTP1, nsLTP2 and LTPL7) for lipid transportation, and three genes (MT-L1, MtN21 and NODULIN-21) for inorganic ion transportation.
Plant hormones (phytohormones) regulate plant growth and development. In this experiment, 14 genes and three proteins were identified as receptors of auxin, ABA, ethylene and brassinosteroid (Supplementary Table S6). In general, all these receptors were increased in the chalky part, except PIN1 the auxin receptor, and AWPM-19L the receptor of ABA.
Discussion
The notched-belly mutant and its application in dissecting metabolic pathways involved in grain chalkiness
Grain chalkiness is a complex trait controlled by polygenes and affected greatly by various environmental conditions. Slow advances has been made in the genetic foundation of chalkiness, reflected by fewer genes cloned despite a large number of quantitative trait loci (QTLs) mapped (Li et al. 2014, Sreenivasulu et al. 2015, Zhao et al. 2016). This is partially due to its sophisticated physiological mechanism, which is still unclear up to now, in particular as to the underlying metabolic pathways. Omics technology such as transcriptomic, proteomic and metabolomic approaches has been employed to uncover the metabolic atlas for chalkiness formation between near-isogenic lines (Liu et al. 2010) or under conditions of dry wind (Wada et al. 2014) and high temperature (Yamakawa et al. 2007, Yamakawa and Hakata 2010, Li et al. 2011, Mitsui et al. 2013), by which our understanding concerning chalkiness has been broadened. However, comparative analysis of omics data in these studies was made between cultivars or near-isogenic lines, which showed a contrasting response of chalkiness to growing conditions. These comparisons cannot perfectly exclude the influence of genetic background and environmental conditions. As reported, even for grains within a panicle, a difference in anthesis time causes large variations in occurrence of chalkiness (Zhang et al. 2014).
We identified a notched-belly mutant with high occurrence of white-belly by using a chemical mutagen (Lin et al. 2014). During the grain filling stage, the notched line is visible at 5 DAF, partly due to the restriction by the palea and lemma of caryopsis elongation (Fig. 1), thereby dividing the endosperm into two compartments, the upper part being translucent but the bottom part mainly opaque, which can be observed at about 20 DAF. Thus the formation of the notched-line makes it possible to investigate the process of chalkiness formation. Using the upper part as control, we compared the variations in abundance of the transcriptome and proteome between the translucent and chalky parts. Notably, this comparison was made within the same genetic background, and is different from those comparisons conducted either between varieties or between chalky grains and perfect grains for a given variety. Importantly, a large number of genes (38,476) and proteins (3,840) were identified in the developing grains, but only a small part of them were expressed differently between the translucent and chalky part, with the number of chalkiness-related DEGs and DEPs being 240 and 363, respectively. Among these, several genes and proteins have been identified as being related to chalkiness formation, such as UGPase1 (Woo et al. 2008), Amy3E (Hakata et al. 2012), ADPGase (OsAPL2; Zhang et al. 2012, Wu et al. 2015) and 13 kDa prolamin (Yamakawa and Hakata 2010). Interestingly, the protein V-PPase encoded by the gene chalk5, the only one named after chalkiness so far (Li et al. 2014), is on the list of the 363 DEPs. Therefore, we can focus on ‘core’ genes or common pathways responsible for chalkiness formation by using this notched-belly mutant to minimize the influence of genetic background and environmental conditions. In addition, the findings of this study, in particular the switch of C metabolism, low photosynthetic capacity and reduced prolamin accumulation, are of value for future studies on grain chalkiness, serving as a platform for candidate gene identification.
Notably, position effect within grains should be considered when explaining the data of this study, as the bottom chalky part is attached to the embryo and is vulnerable to the influence of the embryo. Thus the difference between the upper and lower parts was mainly associated with the combined effect of the chalkiness and the embryo, as reported in our previous works (Lin et al. 2014, Lin et al. 2016). Technically, it is impossible to recognize perfect grains or chalky grains at an early stage; this compound effect cannot be dissected at the present time. Thus, this study has limitations in explaining the underlying mechanisms of grain chalkiness, with the role of the embryo still needing to be fully examined.
Switch of C metabolism
C metabolism is central to the cellular process during grain development, supplying metabolites for starch synthesis, C skeletons for amino acid synthesis and intermediates for secondary metabolism. Starch dominates the chemical profiling of rice grains, comprising about 90% of grain dry matter and therefore being a major factor influencing grain quality traits such as chalkiness (Duan et al. 2005). So far, numerous reports have accumulated regarding the role of starch synthesis or degradation in chalkiness formation. Genomic surveys have identified a lot of genes or QTLs related to chalkiness, such as SSIIIa (Fujita et al. 2007), UGPase1 (Woo et al. 2008), FLO6 (Peng et al. 2014), FLO2 (Wu et al. 2015) and ISA1 (Sun et al. 2015), indicating the importance of starch for chalkiness.
Meanwhile, functional genomics studies showed a rather complicated mechanism underlying the influence of C metabolism on chalkiness formation. Yamakawa and Hakata (2010) presented an atlas showing the effect of high temperature on chalkiness formation by joint analysis of metabolites and gene expression in the central C metabolic pathways. They found that the inhibition of starch deposition might be associated with down-regulation of sucrose import/degradation and starch biosynthesis, and/or up-regulation of starch degradation, as well as inefficient ATP production by an inhibited cytochrome respiration chain. Recently, we profiled and compared the metabolomes between the upper translucent part and the bottom chalky part of developing grains from the notched-belly mutant, using the composite platforms of UPLC/MS/MS and GC/MS. Generally, in comparison with the translucent upper part, the chalky endosperm had lower levels of metabolites regarding C and N metabolism for synthesis of storage starch and protein (data not shown). Similarly, in the current study, we observed perturbed C metabolism in the chalky part of the notched-belly mutants, especially the simultaneously increased activity of the AMY3E gene and AMY3E protein, and the reduced abundance of mRNA and proteins functioning in oxidative phosphorylation, supporting that starch hydrolysis by α-amylase is implicated in the formation of chalky tissue. In addition, we detected high abundance of ALDR at 15 and 20 DAF, indicative of the switch of glycolysis to bypass the pathway of alcoholic fermentation, which is essential for starch accumulation at the mid to late stage of grain development (Xu et al. 2008).
Causative factors for low abundance of prolamins
Proteins are stored in PBs in rice grains, with PBI mainly containing prolamins whereas PBII contains glutelins, globulins and albumins. Next to starch, protein is the second most abundant component in rice grain, accounting for approximately 8% of grain dry matter (Duan et al. 2005). Increasing evidence proves that accumulation of proteins is of relevance to chalkiness occurrence, supported by findings from microscopic observations (Xi et al. 2014), genomic studies (Li et al. 2014) and functional genomic research (Lin et al. 2005, Yamakawa et al. 2007, Yamakawa and Hakata 2010, Lin et al. 2014). In addition, the decrease in prolamin accumulation, especially of the 13 kDa prolamin subunit, has been seen in the chalky grains when ripening under high temperatures (Yamakawa et al. 2007, Yamakawa and Hakata 2010).
Prolamins in rice are composed of cysteine-rich 10 kDa (CysR10), 14 kDa (CysR14) and 16 kDa (CysR16) molecular species, and a cysteine-poor 13 kDa (CysP13) polypeptide (Ushijima et al. 2011). Individual prolamins have distinct functions in the formation of PBs. Kawakatsu et al. (2010) examined the effect of prolamin fractions on rice quality using knockdown lines and have examined their effects on seed quality. They found that reduction in 13 kDa prolamins not only reduced the size of PBs but also altered the rugged peripheral structure; however, severe suppression of 10 or 16 kDa prolamins did not lead to this phenotype. On the other hand, Nagamine et al. (2011) suggest that CysR10 is required for tight packaging of the proteins into a compact spherical structure. Nevertheless, changes in the structure of PBs would result in loose packing of PBs and starch granules, and consequently the occurrence of chalkiness, as has been verified by the characterization of Chalk5 (Li et al. 2014).
Unsurprisingly, a low abundance of transcripts and proteins involved in prolamin accumulation was identified in the current study. This agreed with the findings of our metabolomic analysis showing that chalky parts contained lower levels of the majority of protein amino acids (except glycine, aspartate and arginine) than translucent parts, indicating the relevance of low protein accumulation to grain chalkiness (data not shown). Further, our joint analysis of transcriptome and proteome profiling of the developing notched-belly grains uncovered the possible mechanism underlying the reduction in protein accumulation. In the chalky part, despite the enhanced gene activity of transcription, ribosomal proteins were significantly down-regulated, and protein degradation, in particular non-ubiquitin-mediated, in the phagolysosome was enhanced. This may partly explain the reduction in the low abundance of proteins (in particular the prolamins) in the chalky tissues as reported by Yamakawa et al. (2007) and Yamakawa and Hakata (2010). In addition, we observed that one gene encoding globulin (11S globulin) was up-regulated in the chalky part. This reverse relationship between prolamin and globulin may be associated with the compensating effect between storage proteins which needs further investigation (Kawakatsu et al. 2010).
Role of pericarp photosynthesis
Pericarp photosynthesis is a source of photosynthate supplying the developing grains, in particular at its early stage when cell division in the endosperm and embryo are active. The contribution of ear photosynthesis to grain filling in wheat and barley was estimated to be between 10% and 76%, and, of this, 33– 43% has been attributed to pericarp photosynthesis (Caley et al. 1990). In wheat, photosynthetic activity peaked at about 15–20 DAF, coinciding with maximum Chl content in the pericarp green layer (Caley et al. 1990, Xiong et al. 2013). In rice, pericarp photosynthesis was reported in the grains of transgenic rice plants with the C4-Pdk gene of maize (Fukayama et al. 2001). However, there is little information regarding the significance of pericarp photosynthesis in rice grain development, and its physiological properties are unclear.
In this study, grain samples were de-embryoed and cut into two parts along the notched-line, with the pericarp remaining together with the endosperm in the upper and bottom parts. Thus it is natural that we detected transcripts and proteins responsible for photosynthesis. Notably, insufficient photosynthesis capacity was observed for the chalky part, in particular genes and proteins functioning in photosynthetic electron transport, suggesting the potential role of pericarp in the formation of chalky endosperm. Interestingly, a recent study showed that down-regulated expression of a lipid transporter, OsLTPL36, which was exclusively expressed in developing seed pericarp and endosperm, led to a decreased seed setting rate and caused chalky endosperm (Wang et al. 2015), supporting the importance of pericarp in studies on chalkiness. Future work dissecting the endosperm and pericarp tissues is required to elucidate the contribution of photosynthesis to grain filling, the results of which should also deepen our understanding of rice yield.
Communication between embryo and endosperm
Rice grain mainly consists of three tissues, embryo, endosperm and pericarp. Endosperm basically serves to nourish the developing and germinating embryo. Co-ordination between embryo and endosperm is required for normal grain development. Evidence has shown that intercellular signal transduction pathways play major roles in the communication between embryo and endosperm (Fiume and Fletcher 2012). In Arabidopsis, nearly 400 kinases are expressed in embryos (Nodine et al. 2011), and among these, several receptor-like kinases (RLKs) have been shown to be required for proper endosperm and/or embryo development. Phytohormones constitute other types of these signals; gradients of hormones are generated in the different seed compartments, and their ratios comprise the signals that induce/inhibit particular processes in seed development (Locascio et al. 2014). In this study, we observed two kinds of possible communications between embryo and endosperm, the transporters for metabolites and the signal messengers of hormones. A total of 21 genes and 10 proteins as transporters for carbohydrates, amino acids/peptides, proteins, lipids, inorganic ions and ADP/ATP were detected. Notably, they showed an increasing trend in the chalky part. We also noted 14 genes and three proteins which were identified as receptors of auxin, ABA, ethylene and brassinosteroid, with a similar up-regulation in the chalky part. So far, the mechanism of action of transporters and hormones as signals for the communication between the embryo and the endosperm is not perfectly understood. Thus the role of the embryo should be exhaustively examined in a future study on rice chalkiness.
Conclusions
Integration of omics approaches is a promising route to obtain a multilayered picture of cellular regulatory processes. This study aims to identify key pathways involved in chalkiness formation by joint analysis of the transcriptome and proteome. Interestingly, a notched-belly mutant was employed as the material with which we performed a novel comparison of the upper translucent part and bottom chalky part within the same grain, resulting in very few but important genes and proteins participating in key pathways related to chalkiness formation. These include the shift of C and N metabolism, the down-regulation of ribosomal proteins and the resulting low abundance of storage proteins especially the 13 kDa prolamin subunit, and the suppressed photosynthetic capacity in the pericarp. In addition, genes and proteins of transporters or phytohormone receptors were also up-regulated in the chalky endosperm. Collectively, the joint analysis of gene and protein expression profiles provides a complete picture regarding the physiological state in chalky tissue. Despite the complex mechanisms underlying grain chalkiness, these findings afford novel clues for full understanding of the metabolic network involved in the occurrence chalkiness. In addition, roles of pericarp photosynthesis and embryo development should be accurately interpreted in future studies.
Materials and Methods
Plant materials and growth
The notched-belly mutant (DY1102) was obtained by EMS treatment of the M2 population of a japonica rice cultivar Wuyujing3 (Lin et al. 2014). It had a high ratio of notched-belly grains, ranging from 66.58% (greenhouse) to 75.67% (ambient temperature) in 2014. White-belly occurs mainly on the bottom part, i.e. 85.12% under greenhouse and 93.13% under high night temperature treatments (Supplementary Table S7). Notably, grains on the middle primary rachis have a higher ratio of white-belly, and were sampled for transcriptome and proteome analysis.
In 2014, five plants of DY1102 were hand transplanted into a plastic pot filled with 15 kg of clay soil, 30 cm in height and 34 cm in diameter. After growth under natural environmental conditions until 2–3 d before flowering, pots were transferred to a 28°C/23°C plant chamber (12 h day–12 h night cycles; Model FYS-10; Hengyu), with light intensity >75% of ambient light conditions and relative humidity 70 ± 5% (Shi et al. 2015). Flowering dates of caryopsis on the middle primary rachis were marked. Sampling time points were 5, 10, 15 and 20 DAF, conducted at 09:00 h. Grains were frozen by liquid nitrogen and then stored at − 80 °C until analysis. For RNA-Seq and iTRAQ analyses, the developing grains were dehusked, and the embryos were removed. Subsequently, the remaining endosperms were cut into upper parts (defined as translucent, Tr) and bottom parts (defined as chalky, Ch) along the notched line. Each sample was measured with three biological replicates.
Gene expression profiling using RNA-Seq
Total RNA extraction
Total RNA for RNA-Seq and qRT-PCR analyses was isolated from about 0.3 g of sample with TRIzol reagent (Invitrogen) using the standard protocol, with DNA removed by digestion with DNase I (Ambion). Total RNA was dissolved in 100 μl of RNase-free water and was quantified using a NanoDrop spectrophotometer (Thermo Scientific). RNA quality was evaluated using the 6000 Pico LabChip of the Agilent 2100 Bioanalyzer (Agilent).
cDNA synthesis and Illumina sequencing
The mRNA was enriched by using oligo(dT) magnetic beads. Mixed with the fragmentation buffer, mRNA was fragmented into short fragments (about 200 bp). Then the first-strand cDNA was synthesized using a random hexamer primer employing the mRNA fragments as templates, and the second strand was synthesized using buffer, dNTPs, RNase H and DNA polymerase I. The double-stranded cDNA was purified with magnetic beads. End repair and 3'-end single nucleotide (adenine) addition was then performed. Finally, the fragments were ligated to sequencing adaptors and enriched by PCR amplification for library construction. The qualification and quantification of the sample library were performed using an Agilent 2100 Bioanaylzer and ABI StepOnePlus Real-Time PCR System. The library products were sequenced with Illumina HiSeq™ 2000.
Bioinformatics analysis
Raw reads were transferred from original image data by base calling. Clean reads were the high quality sequences after data cleaning from raw reads, on which all the following analyses are based. The statistics of clean reads for each sample are shown in Supplementary Fig. S2. The clean reads were then analyzed as follows. (i) Clean reads were mapped to reference sequences and/or a reference gene set using SOAPaligner/SOAP2 with Burrows–Wheeler transformation algorithms (Li et al. 2009), allowing no more than two mismatches in the alignment. (ii) The gene expression level was calculated by the RPKM method (Mortazavi et al. 2008). (iii) The DEGs between two groups and samples were identified by NOIseq and a strict algorithm of BGI-Shenzhen developed from the method of Audic and Claverie (1997). In addition, we used a false discovery rate (FDR) ≤1% and an absolute value of log2Ratio ≥1 as the threshold to judge the significance of gene expression difference in addition to probability ≥0.8 among three biological replicates. Functional annotation of the DEGs was performed using the GO database, and the metabolic pathway analysis was conducted according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (http://www.genome.jp/kegg/pathway.html) with the KAAS server (Moriya et al. 2007).
Protein identification and quantification via iTRAQ analysis
Protein extraction
Total proteins for iTRAQ and MRM analyses were extracted from endosperms of the developing grains in DY1102. Samples were ground into powder in liquid nitrogen, and 0.2 g was suspended with chilled trichloroacetic acid/acetone solution [10% (w/v) trichloroacetic acid in acetone, 10 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF)] at –20 °C overnight. The supernatant was discarded after centrifugation at 12,000×g for 30 min at 4°C. The pellet was air-dried and dissolved in lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS, 40 mM Tris–HCl, pH 8.5). The suspension was sonicated at 200 W for 5 min and then centrifuged at 4°C, 30,000×g for 15 min. The supernatant was mixed well with 5 vols. of chilled acetone for 2 h at –20 °C to precipitate proteins, and then the precipitate was obtained by centrifugation. Finally, the pellet was dissolved in 500 μl of 0.5 M tetraethylammonium bromide (TEAB; Applied Biosystems), and the suspension was centrifuged at 4°C, 30,000×g for 15 min. The supernatant was transferred to a new tube and quantified using a Bradford kit (Bio-Rad).
iTRAQ labeling and SCX fractionation
The protein profiling by iTRAQ was performed by the BGI. Total protein (100 μg) of each sample solution was digested with Trypsin Gold (Promega) at 37 °C for 16 h. After digestion, peptides were freeze-dried by vacuum centrifugation. Peptides of eight samples in a biological replicate were reconstituted in 0.5 M TEAB and then labeled with iTRAQ 8-plex kits according to the manufacturer’s manual (AB Sciex Inc.) as follows: Ch5, 113; Tr5, 114; Ch10, 115; Tr10, 116; Ch15, 117; Tr15, 118; Ch20, 119; and Tr20, 121. The labeled peptides in a biological replicate were pooled as a set and resolved into 12 fractions using an Ultremex SCX column (Phenomenex) in an LC-20AB HPLC system (Shimadzu). The eluted fractions were then desalted using a Strata X C18 column (Phenomenex) and dried under vacuum.
LC-MS/MS analysis
Each fraction was resuspended in buffer (2% acetonitrile, 0.1% formic acid), and a portion of a 2.5 μg sample was loaded on nanoUPLC (LC-20AD, Shimadzu) for separation, and then subjected to tandem mass spectrometry (Q-EXACTIVE, Thermo Fisher Scientific) coupled online to the nanoUPLC. The data acquisition was operated according to Shen et al. (2015). Briefly, MS scan spectra were acquired in the Orbitrap at a mass resolution of 70,000 with the range m/z 350–2,000. The 15 most intense peaks above 20,000 in the MS survey scan were fragmented in the high-energy collision dissociation (HCD) mode with a normalized collision energy setting of 27(±12%), and MS2 scan spectra were obtained in the Orbitrap at a mass resolution of 17,500 with the range m/z 100–1,800.
Bioinformatic analysis
All raw files from a biological replicate were searched against the RAP-DB Os-Nipponbare database (40,011 sequences; http://rapdb.dna.affrc.go.jp/download/irgsp1.html) and UniProt database with MASCOT2.3.02 software (Matrix Science).
For protein identification, the database searching parameters including: (i) allowing one max missed cleavage in the trypsin digests; (ii) a mass tolerance of 20 p.p.m. was permitted for intact peptide masses and 0.05 Da for fragment mass tolerance; (iii) Gln→pyro-Glu (N-terminal Q), oxidation (M) and iTRAQ 8-plex (Y) are chosen in variable modifications, while carbamidomethyl (C), iTRAQ 8-plex (N-term) and iTRAQ 8-plex (K) are chosen in fixed modifications; (iv) the peptide charge was set to + 2 and +3, and monoisotopic mass was chosen; and (v) an automatic decoy database strategy was set as the reverse of the target database. All protein and peptide identifications were based on 99% confidence, and determined by an FDR ≤1%.
For protein quantification, the filters were set as follows: (i) ‘median’ was chosen for the protein ratio type; (ii) the minimum precursor charge was set to 2+ and minimum peptides were set to 2, and only unique peptides were used for quantification; and (iii) normalization by median intensities, and outliers were removed automatically. The peptide threshold was set as above for identity. A 1.2-fold change in all of three biological replicates was set to identify significant DEPs in addition with a P-value <0.05. Functional annotation was performed using the GO database, and the metabolic pathway analysis was conducted according to the KEGG pathway database.
Validation of transcriptome and proteome data using qRT-PCR and MRM analysis
Quantitative RT-PCR analysis
First-strand cDNA was synthesized with purified total RNA using the PrimScript™ RT reagent Kit (TAKARA). Real-time PCR was performed by SYBR Premix Ex Taq™ (TAKARA) in a LightCycler 480 real-time PCR system (Roche) with specific primers (Supplementary Table S1). The relative quantification analysis was performed by relative standard curve according to threshold values (CT) generated. ACTIN1 was used as internal control to normalize all data.
MRM analysis
Eight DEPs were selected for MRM analysis (Supplementary Table S2). Samples were digested as described in iTRAQ analysis and spiked with 1 pmol of bovine serum albumin (BSA) for data normalization. MRM analyses were performed on a QTRAP 5500 mass spectrometer (AB SCIEX) equipped with Waters nano Acquity Ultra Performance LC system. Multiple MRM transitions were monitored using unit resolution in both Q1 and Q3 quadrupoles to maximize specificity. Skyline software was used to integrate the raw file generated by QTRAP 5500 (AB SCIEX), and an iRT strategy was operated to define chromatography of a given peptide against a spectral library. All transitions of each peptide were used for quantification unless interference from the matrix was observed. A spike of BSA was used for label-free data normalization. MSstats with the linear mixed-effects model was used to compare protein abundance between different samples, and the P-values were adjusted to control the FDR at a cut-off of 0.05.
Supplementary data
Supplementary data are available at PCP online.
Funding
This work was supported the National Natural Science Foundation of China [grants 31171485 and 31470086]; the National High Technology Research and Development Program of China [grant 2014AA10A605]; and the National Science and Technology Supporting Program of China (grants 2012BAD04B08 and 2013BAD07B09).
Supplementary Material
Acknowledgments
We thank Ms Zhenzhen Wu of BGI for her help in iTRAQ and RNA-Seq analysis.
Disclosures
The authors have no conflicts of interest to declare.
Glossary
Abbreviations
- C
carbon
- DAF
days after flowering
- DEG
differently expressed gene
- DEP
differentially expressed protein
- EMS
ethyl methanesulfonate
- FDR
false discovery rate
- GO
Gene Ontology
- iTRAQ
isobaric tags for relative and absolute quantitation
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MRM
multiple reaction monitoring
- N
nitrogen
- PB
protein body
- PPP
pentose phosphate pathway
- qRT-PCR
quantitative real-time PCR
- QTL
quantitative trait locus
- RNA-Seq
RNA sequencing
- RPKM
reads per kilobase per million reads
- TCA
tricarboxylic acid
References
- Audic S., Claverie J.M. (1997) The significance of digital gene expression profiles. Genome Res. 7: 986–995. [DOI] [PubMed] [Google Scholar]
- Caley C.Y., Duffus C.M., Jeffcoat B. (1990) Photosynthesis in the pericarp of developing wheat grains. J. Exp. Bot. 41: 303–307. [Google Scholar]
- Cho K., Agrawal G.K., Shibato J., Jung Y.H., Kim Y.K., Nahm B.H., et al. (2007) Survey of differentially expressed proteins and genes in jasmonic acid treated rice seedling shoot and root at the proteomics and transcriptomics levels. J. Proteome Res. 6: 3581–3603. [DOI] [PubMed] [Google Scholar]
- Cho K., Shibato J., Agrawal G.K., Jung Y.H., Kubo A., Jwa N.S., et al. (2008) Integrated transcriptomics, proteomics, and metabolomics analyses to survey ozone responses in the leaves of rice seedling. J. Proteome Res. 7: 2980–2998. [DOI] [PubMed] [Google Scholar]
- Duan M., Sun S.S. (2005) Profiling the expression of genes controlling rice grain quality. Plant Mol. Biol. 59: 165–178. [DOI] [PubMed] [Google Scholar]
- Fiume E., Fletcher J.C. (2012) Regulation of Arabidopsis embryo and endosperm development by the polypeptide signaling molecule CLE8. Plant Cell 24: 1000–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu F.F., Xue H.W. (2010) Coexpression analysis identifies Rice Starch Regulator1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiol. 154: 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N., Yoshida M., Kondo T., Saito K., Utsumi Y., Tokunaga T., et al. (2007) Characterization of SSIIIa-deficient mutants of rice: the function of SSIIIa and pleiotropic effects by SSIIIa deficiency in the rice endosperm. Plant Physiol. 144: 2009–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukayama H., Tsuchida H., Agarie S., Nomura M., Onodera H., Ono K., et al. (2001) Significant accumulation of C4-specific pyruvate, orthophosphate dikinase in a C3 plant, rice. Plant Physiol. 127: 1136–1146. [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Wen L., Satoh-Cruz M., Kawagoe Y., Nagamura Y., Okita T.W., et al. (2013) A guanine nucleotide exchange factor for Rab5 proteins is essential for intracellular transport of the proglutelin from the Golgi apparatus to the protein storage vacuole in rice endosperm. Plant Physiol. 162: 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Wang Y., Liu X., Jiang L., Ren Y., Liu F., et al. (2012) The failure to express a protein disulphide isomerase-like protein results in a floury endosperm and an endoplasmic reticulum stress response in rice. J. Exp. Bot. 63: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakata M., Kuroda M., Miyashita T., Yamaguchi T., Kojima M., Sakakibara H., et al. (2012) Suppression of alpha-amylase genes improves quality of rice grain ripened under high temperature. Plant Biotechnol. J. 10: 1110–1117. [DOI] [PubMed] [Google Scholar]
- Kawakatsu T., Hirose S., Yasuda H., Takaiwa F. (2010) Reducing rice seed storage protein accumulation leads to changes in nutrient quality and storage organelle formation. Plant Physiol. 154: 1842–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan P., Li W., Schmidt W. (2012) Complementary proteome and transcriptome profiling in phosphate-deficient Arabidopsis roots reveals multiple levels of gene regulation. Mol. Cell. Proteomics 11: 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Chen Z., Hu M., Wang Z., Hua H., Yin C., et al. (2011) Different effects of night versus day high temperature on rice quality and accumulation profiling of rice grain proteins during grain filling. Plant Cell Rep. 30: 1641–1659. [DOI] [PubMed] [Google Scholar]
- Li R., Yu C., Li Y., Lam T.W., Yiu S.M., Kristiansen K., et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- Li Y., Fan C., Xing Y., Yun P., Luo L., Yan B., et al. (2014) Chalk5 encodes a vacuolar H(+)-translocating pyrophosphatase influencing grain chalkiness in rice. Nat. Genet. 46: 398–404. [DOI] [PubMed] [Google Scholar]
- Lin S.K., Chang M.C., Tsai Y.G., Lur H.S. (2005) Proteomic analysis of the expression of proteins related to rice quality during caryopsis development and the effect of high temperature on expression. Proteomics 5: 2140–2156. [DOI] [PubMed] [Google Scholar]
- Lin Z.M., Zhang X.C., Yang X.Y., Li G.H., Tang S., Wang S.H., et al. (2014) Proteomic analysis of proteins related to rice grain chalkiness using iTRAQ and a novel comparison system based on a notched-belly mutant with white-belly. BMC Plant Biol. 14: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.M., Zheng D.Y., Zhang X.C., Wang Z.X., Lei J.C., Liu Z.H., et al. (2016) Chalky part differs in chemical composition from translucent part of japonica rice grains as revealed by a notched-belly mutant with white-belly. J. Sci. Food Agric. 96: 3937–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Ren Y., Wang Y., Peng C., Zhou K., Lv J., et al. (2013) OsVPS9A functions cooperatively with OsRAB5A to regulate post-Golgi dense vesicle-mediated storage protein trafficking to the protein storage vacuole in rice endosperm cells. Mol. Plant 6: 1918–1932. [DOI] [PubMed] [Google Scholar]
- Liu K., Wang L., Xu Y., Chen N., Ma Q., Li F., et al. (2007) Overexpression of OsCOIN, a putative cold inducible zinc finger protein, increased tolerance to chilling, salt and drought, and enhanced proline level in rice. Planta 226: 1007–1016. [DOI] [PubMed] [Google Scholar]
- Liu X., Guo T., Wan X., Wang H., Zhu M., Li A., et al. (2010) Transcriptome analysis of grain-filling caryopses reveals involvement of multiple regulatory pathways in chalky grain formation in rice. BMC Genomics 11: 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locascio A., Roig-Villanova I., Bernardi J., Varotto S. (2014) Current perspectives on the hormonal control of seed development in Arabidopsis and maize: a focus on auxin. Front. Plant Sci. 5: 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui T., Shiraya T., Kaneko K., Wada K. (2013) Proteomics of rice grain under high temperature stress. Front. Plant Sci. 4: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Risueno M.A., Busch W., Benfey P.N. (2010) Omics meet networks—using systems approaches to infer regulatory networks in plants. Curr. Opin. Plant Biol. 13: 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M. (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35: 182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Nagamine A., Matsusaka H., Ushijima T., Kawagoe Y., Ogawa M., Okita T.W., et al. (2011) A role for the cysteine-rich 10 kDa prolamin in protein body I formation in rice. Plant Cell Physiol. 52: 1003–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodine M.D., Bryan A.C., Racolta A., Jerosky K.V., Tax F.E. (2011) A few standing for many: embryo receptor-like kinases. Trends Plant Sci. 16: 211–217. [DOI] [PubMed] [Google Scholar]
- Peng C., Wang Y., Liu F., Ren Y., Zhou K., Lv J., et al. (2014) FLOURY ENDOSPERM6 encodes a CBM48 domain-containing protein involved in compound granule formation and starch synthesis in rice endosperm. Plant J. 77: 917–930. [DOI] [PubMed] [Google Scholar]
- Peng R., Yao Q., Xiong A., Fan H., Li X., Peng Y., et al. (2004) A new rice zinc-finger protein binds to the O2S box of the alpha-amylase gene promoter. Eur. J. Biochem. 271: 2949–2955. [DOI] [PubMed] [Google Scholar]
- Prasad K., Sriram P., Kumar S., Kushalappa K., Vijayraghavan U. (2001) Ectopic expression of rice OsMADS1 reveals a role in specifying the lemma and palea, grass floral organs analogous to sepals. Dev. Genes Evol. 211: 281–290. [DOI] [PubMed] [Google Scholar]
- Schmollinger S., Muhlhaus T., Boyle N.R., Blaby I.K., Casero D., Mettler T., et al. (2014) Nitrogen-sparing mechanisms in Chlamydomonas affect the transcriptome, the proteome, and photosynthetic metabolism. Plant Cell 26: 1410–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Zhang Y., Zou J., Meng J., Wang J. (2015) Comparative proteomic study on Brassica hexaploid and its parents provides new insights into the effects of polyploidization. J. Proteomics 112: 274–284. [DOI] [PubMed] [Google Scholar]
- Shi P., Tang L., Lin C., Liu L., Wang H., Cao W., et al. (2015) Modeling the effects of post-anthesis heat stress on rice phenology. Field Crops Res. 177: 26–36. [Google Scholar]
- Sreenivasulu N., Butardo V.M. Jr., Misra G., Cuevas R.P., Anacleto R., Kavi Kishor P.B. (2015) Designing climate-resilient rice with ideal grain quality suited for high-temperature stress. J. Exp. Bot. 66: 1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., Zhou Q., Yao Y., Qiu X., Xie K., Yu S. (2015) Identification of genomic regions and the isoamylase gene for reduced grain chalkiness in rice. PLoS One 10: e0122013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N., Fujita N., Nishi A., Satoh H., Hosaka Y., Ugaki M., et al. (2004) The structure of starch can be manipulated by changing the expression levels of starch branching enzyme IIb in rice endosperm. Plant Biotechnol. J. 2: 507–516. [DOI] [PubMed] [Google Scholar]
- Taylor N.L., Howell K.A., Heazlewood J.L., Tan T.Y., Narsai R., Huang S., et al. (2010) Analysis of the rice mitochondrial carrier family reveals anaerobic accumulation of a basic amino acid carrier involved in arginine metabolism during seed germination. Plant Physiol. 154: 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugama D., Liu S., Takano T. (2012) A bZIP protein, VIP1, is a regulator of osmosensory signaling in Arabidopsis. Plant Physiol. 159: 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushijima T., Matsusaka H., Jikuya H., Ogawa M., Satoh H., Kumamaru T. (2011) Genetic analysis of cysteine-poor prolamin polypeptides reduced in the endosperm of the rice esp1 mutant. Plant Sci. 181: 125–131. [DOI] [PubMed] [Google Scholar]
- Wada H., Masumoto-Kubo C., Gholipour Y., Nonami H., Tanaka F., Erra-Balsells R., et al. (2014) Rice chalky ring formation caused by temporal reduction in starch biosynthesis during osmotic adjustment under foehn-induced dry wind. PLoS One 9: e110374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakasa Y., Yasuda H., Oono Y., Kawakatsu T., Hirose S., Takahashi H., et al. (2011) Expression of ER quality control-related genes in response to changes in BiP1 levels in developing rice endosperm. Plant J. 65: 675–689. [DOI] [PubMed] [Google Scholar]
- Wang E., Wang J., Zhu X., Hao W., Wang L., Li Q., et al. (2008) Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 40: 1370–1374. [DOI] [PubMed] [Google Scholar]
- Wang J.C., Xu H., Zhu Y., Liu Q.Q., Cai X.L. (2013) OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J. Exp. Bot. 64: 3453–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhou W., Lu Z., Ouyang Y., Yao J. (2015) A lipid transfer protein, OsLTPL36, is essential for seed development and seed quality in rice. Plant Sci. 239: 200–208. [DOI] [PubMed] [Google Scholar]
- Woo M.O., Ham T.H., Ji H.S., Choi M.S., Jiang W., Chu S.H., et al. (2008) Inactivation of the UGPase1 gene causes genic male sterility and endosperm chalkiness in rice (Oryza sativa L.). Plant J. 54: 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.P., Pu C.H., Lin H.Y., Huang H.Y., Huang Y.C., Hong C.Y., et al. (2015) Three novel alleles of FLOURY ENDOSPERM2 (FLO2) confer dull grains with low amylose content in rice. Plant Sci. 233: 44–52. [DOI] [PubMed] [Google Scholar]
- Xi M., Lin Z.M., Zhang X.C., Liu Z.H., Li G.H., Wang Q.S., et al. (2014) Endosperm structure of white-belly and white-core rice grains shown by scanning electron microscopy. Plant Prod. Sci. 17: 285–290. [Google Scholar]
- Xiong F., Yu X.R., Zhou L., Wang F., Xiong A.S. (2013) Structural and physiological characterization during wheat pericarp development. Plant Cell Rep. 32: 1309–1320. [DOI] [PubMed] [Google Scholar]
- Xu S.B., Li T., Deng Z.Y., Chong K., Xue Y., Wang T. (2008) Dynamic proteomic analysis reveals a switch between central carbon metabolism and alcoholic fermentation in rice filling grains. Plant Physiol. 148: 908–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa H., Hakata M. (2010) Atlas of rice grain filling-related metabolism under high temperature: joint analysis of metabolome and transcriptome demonstrated inhibition of starch accumulation and induction of amino acid accumulation. Plant Cell Physiol. 51: 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa H., Hirose T., Kuroda M., Yamaguchi T. (2007) Comprehensive expression profiling of rice grain filling-related genes under high temperature using DNA microarray. Plant Physiol. 144: 258–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L., Tao Y., Zhao K., Shao J., Li X., Liu G., et al. (2007) Proteomic and transcriptomic analysis of rice mature seed-derived callus differentiation. Proteomics 7: 755–768. [DOI] [PubMed] [Google Scholar]
- Zhang D., Wu J., Zhang Y., Shi C. (2012) Phenotypic and candidate gene analysis of a new floury endosperm mutant (osagpl2-3) in rice. Plant Mol. Biol. Rep. 30: 1303–1312. [Google Scholar]
- Zhang Q. (2007) Strategies for developing green super rice. Proc. Natl. Acad. Sci. USA 104: 16402–16409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.C., Lin Z.M., Liu Z.H., Li G.H., Wang Q.S., et al. (2014) Analysis of variations in white-belly and white-core rice kernels within a panicle and the effect of panicle type. J. Integr. Agric. 13: 1672–1679. [Google Scholar]
- Zhao X., Fitzgerald M. (2013) Climate change: implications for the yield of edible rice. PLoS One 8: e66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Daygon V.D., McNally K.L., Hamilton R.S., Xie F., Reinke R.F., et al. (2016) Identification of stable QTLs causing chalk in rice grains in nine environments. Theor. Appl. Genet. 129: 141–153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.