

Abstract
SimpleSearch provides access to a database containing information about T-DNA insertion lines of the GABI-Kat collection of Arabidopsis thaliana mutants. These mutants are an important tool for reverse genetics, and GABI-Kat is the second largest collection of such T-DNA insertion mutants. Insertion sites were deduced from flanking sequence tags (FSTs), and the database contains information about mutant plant lines as well as insertion alleles. Here, we describe improvements within the interface (available at http://www.gabi-kat.de/db/genehits.php) and with regard to the database content that have been realized in the last five years. These improvements include the integration of the Araport11 genome sequence annotation data containing the recently updated A. thaliana structural gene descriptions, an updated visualization component that displays groups of insertions with very similar insertion positions, mapped confirmation sequences, and primers. The visualization component provides a quick way to identify insertions of interest, and access to improved data about the exact structure of confirmed insertion alleles. In addition, the database content has been extended by incorporating additional insertion alleles that were detected during the confirmation process, as well as by adding new FSTs that have been produced during continued efforts to complement gaps in FST availability. Finally, the current database content regarding predicted and confirmed insertion alleles as well as primer sequences has been made available as downloadable flat files.
Keywords: Arabidopsis thaliana, knockout mutants, T-DNA integration, insertional mutagenesis, reverse genetics, genomics, systems biology and evolution
The nucleotide sequences in this paper have been submitted to ENA/GenBank/DDBJ with the accession numbers HE664175 to HE667735, HG967694 to HG969186, LM644484-LM644508, LN713374-LN713448 and LT615049-LT615076.
Introduction
The determination of functions of genes found in the genome of Arabidopsis thaliana is one of the key tasks of basic plant research since the genome sequence has been released in 2001 (The Arabidopsis Genome Initiative 2000). One method to gain insights into possible gene functions is reverse genetics, where the gene of interest is made non-functional in a so-called knockout mutant. Subsequently, the mutant homozygous for the respective knockout allele is characterized in comparison to wildtype to identify the phenotype caused by the defect gene.
Before the emergence of the CRISPR/Cas9 system (Cong et al. 2013, Gaj et al. 2013) for plant genome editing, the method of choice to achieve knockouts in A. thaliana was the use of the DNA integration mechanism provided by Agrobacterium tumefaciens. The transfer DNA (T-DNA) which is present on a plasmid in agrobacteria is transferred into plant cells and integrated into the plant genome (Gelvin 2000). The inserted T-DNA often disrupts the gene at the insertion site (Wang et al. 1984, Peralta and Ream 1985, Gelvin 1998, Krysan et al. 1999). While the exact mechanism of integration is still debated, a process mediated by plant repair pathways seems likely (Tzfira et al. 2004, Gelvin 2010, Kleinboelting et al. 2015). Due to the (largely) randomness of the insertion process with respect to the insertion position in the nuclear genome (Szabados et al. 2002, Li et al. 2006, Kim et al. 2007), a large number of mutants needs to be generated in order to achieve knockout lines for most of the genes. Nevertheless, this task has been accomplished to a good extent by combined action of several projects in which A. thaliana knockout mutants have been generated systematically (Alonso et al. 2003, Rosso et al. 2003, Kuromori et al. 2009, O’Malley and Ecker 2010, Li et al. 2014). Given that it is easy to check for available insertion mutants, but quite laborious to create a CRIPSR/Cas9 knockout, the insertion mutant collections are still heavily used.
There are several databases, websites and portals available to start searching for insertion mutants, each with specific advantages and disadvantages (Kuromori et al. 2009, Stracke et al. 2010, Bolle et al. 2011). Generally, the basis for these search options are flanking sequence tags (FSTs). FSTs are short sequences flanking the inserted T-DNA that represent the genome sequence either upstream or downstream of the insertion site (Bouchez and Höfte 1998, Parinov and Sundaresan 2000). One traditional start point for insertion mutant identification is T-DNA Express (http://signal.salk.edu/cgi-bin/tdnaexpress), which is part of the SIGnAL (SALK Institute Genomic Analysis Laboratory) website provided by the group of Joe Ecker at the SALK Institute which also hosts the largest T-DNA collection in A. thaliana worldwide (Alonso et al. 2003). The SIGnAL website contains a wealth of genomics data including comprehensive data addressing the A. thaliana transcriptome, methylome and epigenome as well as genomics data spanning from rice to human. T-DNA Express (Alonso et al. 2003, O’Malley and Ecker 2010) allows to identify FSTs and deduced insertion positions that indicate the existence of a potential insertion allele in essentially all sequence-indexed A. thaliana T-DNA insertion populations. However, details about lines and insertion alleles are not included in T-DNA Express, and since the database is FST-based some of the indicated insertions are not represented by real mutants.
The Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org) also provides options to search for and identify T-DNA insertion mutants (see Portals > Mutant and Mapping Resources > Find Mutants). Specifically, the catalog of the Arabidopsis Biological Resource Center (ABRC) is integrated into TAIR (Berardini et al. 2015). ABRC, located at the Ohio State University in Columbus (USA), and the European (Nottingham) Arabidopsis Stock Centre (NASC) located at Nottingham University (UK), represent the two largest stock centers which essentially mirror their seed collections (Scholl et al. 2000). The NASC stock catalog (http://arabidopsis.info) also covers T-DNA insertion mutants comprehensively. Together, ABRC and NASC serve the scientific community with seed stocks of publicly available T-DNA insertion mutants. In addition, the RIKEN BioResource Center (BRC) Experimental Plant Division in Japan (http://epd.brc.riken.jp/en/arabidopsis) offers some unique resources including T-DNA activation tagged lines (Nakazawa et al. 2003); however, this line collection has not been sequence-indexed with FSTs.
GABI-Kat is the second largest collection worldwide for T-DNA insertion mutants in A. thaliana and originally comprised 92,657 (almost 974 times 96) lines. The lines were initially represented by T1 plants, of which 89,705 produced T2 seed and yielded T1 DNA for the generation of FSTs. GABI-Kat FSTs were produced using PCR-based methods. Shortly, amplicons flanking the T-DNA were generated on genomic DNA from each individual line (Strizhov et al. 2003), followed by sequencing of the amplicons and subsequent mapping of the reads to the genome sequence. Insertion sites were predicted on the basis of the mapping position of FSTs (Li et al. 2003, Kleinboelting et al. 2012). The predicted insertion alleles were classified as a ‘gene hit’ if the predicted insertion position was located between 300 bp upstream of the ATG and 300 bp downstream of the stop codon of an annotated gene (if there are no UTRs annotated), or if the predicted insertion position is located between transcription start and transcript end of an annotated gene (for genes with annotated UTRs), and also if the insertion is positioned between transcription start and transcript end (TS2TE) of an RNA-encoding gene. It should be noted that this operational definition ignores the correct textbook definition of a eukaryotic gene, which for sure includes regulatory regions outside of the transcribed part of a gene. The best prospects of causing a NULL allele of a protein coding gene, which usually is the best starting point for gene function searches, have T-DNA insertions between ATG and STOP in genomic DNA (CDS plus introns). We refer to these cases (for protein coding genes only) as ‘CDSi hit’ (Kleinboelting et al. 2012).
The databases mentioned above, e.g. T-DNA Express, NASC or TAIR/ABRC, link to the GABI-Kat database and the web interface SimpleSearch (http://www.gabi-kat.de/db/genehits.php) for details of GABI-Kat lines. These details include, for example, segregation data and the deduced number of T-DNA loci that a given line may contain, so-called confirmation sequences of insertion alleles confirmed in the T2 generation (Li et al. 2007), as well as valuable data on resolved so-called ‘contamination groups.’ We have introduced contamination groups to deal with the fact that in a significant number of cases, insertions were predicted in multiple lines at very narrow positions (Kleinboelting et al. 2012). This is often due to contaminations that occurred during the PCR-based high-throughput FST generation process. SimpleSearch directs users to the correct and confirmed line within a group of different lines with the same predicted insertion allele. Since 2005, when the first lines that have been confirmed by PCR and amplicon sequencing were donated to NASC, SimpleSearch serves as an entry point for selecting GABI-Kat insertion alleles for an order from the stock center.
Users can also search for GABI-Kat insertions directly in SimpleSearch and submit seed orders (Li et al. 2007) for lines that are not (yet) available from the stock centers. Using primers specific for the insertion allele to be studied, the junction region between genome and the T-DNA insertion site is amplified and sequenced. The sequences resulting from this process are called ‘confirmation sequences’ since they are used to verify the predicted insertion by BLAST analyses. If the sequences confirm the prediction, T2 seeds of lines containing those insertions are sent to the user. Subsequently, each confirmed line is donated to NASC as a T3 set. A T3 set consists of seeds from usually 12 sulfadiazine resistant (i.e. T-DNA containing, see Rosso et al. 2003) T2-plants of which statistically four (1/3 since homozygous wildtype T2 plants will die) should be homozygous for the insertion allele if the line contained the T-DNA at a single locus. As mentioned above, confirmed GABI-Kat lines can be ordered from NASC (Scholl et al. 2000), and after transfer from NASC to ABRC in the US also from there.
The GABI-Kat website and the search interface SimpleSearch has grown over time since it was launched in June 2002. In addition to the search for knockout alleles in AtYFG’s (your favorite gene; Li et al. 2003) and the seed order functions, the database of the DUPLO project (Bolle et al. 2013) that addressed the systematic construction of double mutants in pairs of closely related genes, was integrated. In 2014, an automatic online primer design tool for the primers required for confirmation and genotyping of T-DNA insertion lines was incorporated (Huep et al. 2014).
With respect to annotation, the initial GABI-Kat FST dataset was interpreted based on TIGR annotation version 5 (Haas et al. 2003, Wortman et al. 2003), in which the genome sequence was represented by individual BAC sequences. In 2011, we replaced the TIGR5 sequence basis by the TAIR9 sequence which represents the genome with contiguous pseudochromosomes. In addition, the TAIR10 annotation dataset was integrated which allowed predicting an insertion position on the pseudochromosomes for each insertion (Kleinboelting et al. 2012). In June 2016, a new A. thaliana genome sequence annotation was published by the Arabidopsis information portal Araport (Krishnakumar et al. 2015) which was named Araport11 (Cheng et al. 2016). The new Araport11 annotation, which is still based on the TAIR9 genome sequence, contains an increased number of nuclear protein coding genes (27,445 in Araport11 vs 27,206 in TAIR10). In addition to almost 240 new genes, about 500 genes gained in Araport11 were compensated by about 500 genes lost from TAIR10. About 5,000 structures of protein coding genes have been updated. Also, a large number of RNA-encoding genes (about 3,500) and genomic features like small RNA loci (about 36,000) have been added. Obviously, easy access to GABI-Kat insertion alleles of genes that have been changed or added in Araport11 requires the incorporation of Araport11 data into SimpleSearch.
Results and Discussion
Since 2011, when the status of the GABI-Kat database and the SimpleSearch front end was last summarized (Kleinboelting et al. 2012), several improvements have been implemented: (i) FSTs, insertion predictions, confirmation sequences, and insertion site annotations have been updated to the Araport11 genome annotation, (ii) exact data on the position of confirmed insertions have been integrated, (iii) additional insertion alleles detected during the confirmation process as well as from new FSTs have been made available, (iv) literature information with a focus on the allele designations assigned to a given GABI-Kat T-DNA allele has been incorporated, and (v) the visualization component has been improved to provide additional information, especially on confirmed insertions. Also, the left menu of the website has been updated (Supplemental Fig. 1). Taken together, these enhancements led to a significantly better access to more GABI-Kat insertion alleles via SimpleSearch.
Update to Araport11
The existing predicted insertion positions obtained for each GABI-Kat FST were used for the re-annotation because the genome sequence version (TAIR9) underlying the Araport11 annotation did not change. Consequently, the number of ‘genome hits’ that is based on the TAIR9 pseudochromosomes stayed the same. These directly sequence-based features are unaffected by changes in structural gene annotation. Table 1 shows what the GABI-Kat database gained from the update to Araport11 annotation in comparison to TAIR10, on the basis of the same FST content. The number of lines which have at least one GABI-Kat ‘gene hit’ has increased from 52,206 to 56,114. Although the number of protein-coding nuclear genes went up in Araport11 by only about 240, the quite large increase in ‘gene hits’ for the GABI-Kat population was explained by the big increment of RNA-encoding genes. Araport11 covers 5,611 nuclear RNA-encoding genes, including long intergenic non-coding RNA (lincRNA) genes, natural antisense transcript (NAT) genes as well as novel transcribed regions. Before the update to Araport11, SimpleSearch contained 1,290 nuclear RNA-encoding genes from TAIR10 and neither lincRNA nor NAT genes. The Araport11 update caused an increase from 1,147 to 4,742 GABI-Kat lines with gene hits in RNA-encoding genes (Supplemental Fig. 2). The number of protein-coding genes for which at least one insertion allele is predicted for the GABI-Kat population increased from 20,235 to 20,697, reflecting the increase in protein-coding genes on the one hand and the extended UTR coverage in Araport11 on the other.
Table 1.
Comparison of insertion allele annotation based on TAIR10 to that based on Araport11
| TAIR10 (2016‐08‐14)a | Araport11 (2016‐08‐15)a | |
|---|---|---|
| Number of T1 plants selected and planted | 92,657 | 92,657b |
| Number of lines with intact T1 DNA and T2 seeds | 89,705 | 89,705b |
| Total number of lines with genome hits | 77,034 | 77,034b |
| Number of lines with gene hits | 52,206c | 56,114c |
| Lines with gene hits, protein coding genes only | 49,062d | 51,620d |
| Lines with hits in RNA-encoding genes | 1,147d | 4,742d |
| Lines with gene hits, pseudogenes only | 1,006d | 1,062d |
| Number of lines with hits in transposable elements | 6,681 | 6,646 |
| Total number of lines with CDSi hits | 47,360 | 47,515 |
| Number of genes with at least one hit | 22,337 | 23,582 |
| Number of protein coding genes with at least one hit | 20,235 | 20,697 |
| Number of CDSi with at least one hit | 14,137 | 14,235 |
a Numbers from immediately before and after update to Araport11.
b Since the data content has not been changed, these numbers are the same.
c Pseudogenes, protein coding and RNA-encoding genes counted.
d These numbers do not add up to the ‘Number of lines with gene hits’ value because a single line can contain hits of different types.
Overall, the number of hits in all genome features has increased due to the larger number of items in the new annotation. These additional hits can now be accessed directly when searching for the corresponding AGI code in the SimpleSearch web interface. Links from external databases like T-DNA Express that have also been updated to Araport11 find their correctly annotated counterparts in SimpleSearch also for hits that were affected by the Araport11 update. Besides the increased overall number of gene hits, the more important fact is that the Araport11 annotation contains more reliable gene structures due to the inspection of large amounts of RNA-Seq data (Cheng et al. 2016). Therefore, also conclusions about the effect of an insertion into a gene become more reliable. According to our experience this is of special relevance for insertions in the area around transcription start of a given gene, because hits in the 5′UTR often have stronger effects than hits in the promoter region.
At some time in the future, probably after a significantly improved version of the A. thaliana Col-0 reference genome sequence has been generated using long read sequencing technology like PacBio (see e.g. VanBuren et al. 2015), a new mapping of all FSTs will be required. A heavy drawback of such a new reference genome sequence is that each and every data point related to a genome position will be affected. On the other hand, it would allow identification of T-DNA insertions in repetitive regions of the genome.
Improved data on the exact position of insertion after confirmation
Due to the high-throughput process of FST generation that results in relatively low sequence read quality of the FSTs, the predicted insertion position from FST data is often inaccurate. The confirmation sequences obtained during the validation steps at GABI-Kat while confirming insertion predictions in the T2 generation are generally of much better quality. These data allow a more precise deduction of the exact insertion position (Kleinboelting et al. 2015). The established method in SimpleSearch and other FST databases was to keep the insertion position predicted on the basis of the original FST. This practice was used in SimpleSearch initially even after confirmation of the insertion allele by amplicon sequencing. Now, we use the more accurate position deduced separately from the confirmation sequences to correct the initial prediction and provide that position in SimpleSearch along with the details on confirmation sequences (Supplemental Fig. 3). Since GABI-Kat is the only A. thaliana T-DNA insertion population that performs in-house confirmation of insertion alleles for quality control, the confirmation sequence data used are a resource unique to GABI-Kat. For this reason, the insertion position information from the specialized GABI-Kat database is more reliable for the insertion alleles available from the stock centers than predictions available from other sources.
Additional insertion alleles detected during the confirmation process
The FSTs generated for the GABI-Kat lines do not detect all T-DNA insertions within the lines examined, although several FSTs have been produced for each line. This is true also for other insertion populations because FST generation is generally not exhaustive. Often, a single FST of good quality is selected for each line. There are GABI-Kat lines which cover up to three confirmed insertion alleles at fully independent loci (see e.g. 011F01 which contains three confirmed insertion alleles at the loci At3g56580/Chr3:20962189, At5g05180/Chr5:1535520 and F26P21/Chr4:15944202). Also, the GABI-Kat lines at NASC (13,967) contain significantly more confirmed insertion alleles (14,280) than the pure number of lines suggests. Nevertheless, there are some hidden insertions in the population. When mapping sequences of amplicons from the confirmation process to the genome sequence, we observed in some cases a good fit to a locus different to and far away from the predicted insertion position(s). Examination of these cases indicated that the ‘wrong mapping’ was due to annealing of the gene-specific primer by chance close to a T-DNA insertion that was not initially predicted by FSTs. If we detected such a case, the hint from the confirmation sequence was used to predict an additional insertion site that is addressed subsequently by another confirmation process. If confirmed, the confirmation sequence (or the reverse complement, if the sequence was obtained with the gene-specific primer) is copied to serve as a new FST and the additional insertion becomes available to users. Currently, 150 additional FSTs have been generated in that way of which 119 are gene hits (see Supplemental Table 1 containing a list of all 5,177 new FSTs submitted to ENA/GenBank/DDBJ, FST type ‘FST derived from confirmation sequence indicating an additional insertion’).
An example case for an insertion allele confirmed in that way occurred with line 380H11. The line was predicted to contain an insertion in At1g16705 (coding for ‘p300/CBP acetyltransferase-related protein-like protein’). A gene-specific primer was designed (5′-ATTACTGCTTTTTGGCTCTGTGAT-3′) to confirm this insertion, and the confirmation PCR resulted in an amplicon. However, the confirmation sequence was not mapping to the locus At1g16705, but mapped close to Chr2:14514000 at the locus At2g34380. We addressed the insertion position deduced from the ‘wrong’ confirmation sequence, and finally confirmed an insertion at Chr2:14514399 in the 3′-UTR of At2g34380 (coding for ‘putative adipose-regulatory protein (Seipin)’). A (pseudo-) FST with the AccNo HG967727 for an insertion allele of At2g34380 was derived from the confirmation sequence which is available in SimpleSearch. We observed such cases essentially only if the initially designed amplimer could not be realized by PCR because the predicted T-DNA was not present close to the ideal annealing position of the gene-specific primer addressing the initially assumed insertion allele.
Overall, 5,177 additional FSTs generated at GABI-Kat have been submitted to ENA/GenBank/DDBJ since 2012 which became gradually accessible in SimpleSearch. Table 2 presents the increase in terms of data which have been added to SimpleSearch in the last 5 years. Of the 5,177 new FSTs, 4,281 were regular FSTs, 746 were FSTs derived from a confirmation sequence made for the second border of the insertion to determine both junctions between T-DNA and the genome (see Kleinboelting et al. 2015), and the remaining ones were the FSTs generated from confirmation sequences as mentioned above. A list of all submitted FSTs along with the predicted insertion site is given in Supplemental Table 1.
Table 2.
Summary of data added to the GABI-Kat SimpleSearch database since 2011
| Data type | Number of entriesa (2011‐09‐15) | Number of entries (2016‐08‐15) |
|---|---|---|
| GK FSTs | ∼133,000 | 143,601 |
| Lines | 71,235b | 77,034c |
| with segregation data | 15,289 | 20,037 |
| available at NASC | 9,644 | 13,967 |
| Insertion alleles (predicted genome hits) | 88,580d | 95,233d |
| analyzed with final result | 16,081e | 26,319e |
| delivered to individual users | 6,816 | 7,819 |
| confirmed and available at NASC | 9,653f | 14,280f |
| Distinct genes covered | 21,005 | 24,789 |
| protein coding genes | 19,120 | 20,697 |
| RNA-encoding genes | 182 | 988 |
| pseudogenes | 420 | 481 |
| transposable element genes | 1,283 | 1,416 |
| Distinct CDSi covered | 13,037 | 14,235 |
a Numbers as of September 15, 2011, taken from (Kleinboelting et al. 2012).
b Database release version 24.
c Database release version 28 from August 15, 2016.
d Insertion alleles are different from lines, because a line can contain several insertions. An insertion is expected to be different from another one in the same line if the distance between the two predicted insertion positions is at least 20 kbp (Kleinboelting et al. 2015). The gain of 6,653 predicted insertion alleles (from 88,580 (September 15, 2011) to 95,233 (August 15, 2016)) is in part due to data from the Ecker group (O’Malley et al. in preparation). Selected GK-lines were analyzed by TDNA-Seq using Illumina technology (NCBI accession numbers KG779961 to KG787552), and the resulting predictions have been included in SimpleSearch. In addition, 119 cases are derived from ‘composite FSTs’ as described (Huep et al. 2014).
e A final result can be ‘confirmed’, but also ‘failed to confirm’ or ‘part of a contamination group’; see (Kleinboelting et al. 2012).
f For each confirmed insertion there are confirmation sequences available which are generated from the amplicon that spans the T-DNA/genome sequence junction. For about 1,400 insertions there are data from both (the ‘north’ and the ‘south’) junction of the inserted T-DNA sequences (Kleinboelting et al. 2015).
Lines from the SALK population have also been exploited by the group of Joe Ecker for additional insertion alleles at SALK, and the results have constantly been incorporated into T-DNA Express (see http://signal.salk.edu/Source/AtTOME_Data_Source.html). These ongoing efforts contribute to an increasing saturation of the A. thaliana gene space with knockout alleles.
Incorporation of literature information on allele designations
A large number of insertion alleles from GABI-Kat have been used in research for gene functions, and quite some of these results have been published. We collect the literature data, also as a proof that the public money that funded GABI-Kat for many years was well-spent, and record the PubMed-ID (see http://www.gabi-kat.de/publications.html). Paper entries can be linked to the line containing the insertion allele, and we also capture the allele designation assigned by the first publication to the respective GABI-Kat T-DNA allele. Extraction of the allele-related information requires manual curation, and capturing this data is work in progress. The data can be accessed in SimpleSearch in two ways; publications listed on the ‘Publications’ page of the website mention the PubMed-ID and the GABI-Kat line-ID or the allele designation (if this information has already been extracted). When a link between paper and insertion allele has been established, the respective paper is mentioned on the ‘Line and FST details’ page of SimpleSearch. An example is the GABI-Kat allele 290E08/At1g65480 that has been described by Yoo et al. (2005) as an allele of FLOWERING LOCUS T (FT) and was designated ft-10. This small bibliographic exercise should by no means be an attempt to collect paper data about A. thaliana research comprehensively, as it is done for example at TAIR (Berardini et al. 2015), but it might be a useful contribution to detecting synonyms of allele designations.
Updated visualization component
An important feature of SimpleSearch is the visualization of insertion sites where users can visually inspect regions of interest for predicted insertions, and now also of confirmed insertion sites. Furthermore, the visualization component has been updated to support Araport11 annotation data, providing the most recent annotation context to users checking for suitable insertion alleles. Although already partially included in the TAIR10 annotation data set which was the basis for the GK release v24 of 2011-03-07, the visualization component now also displays features like RNA-encoding genes. Different types of genes that are either protein-coding, RNA-encoding, transposable elements or pseudogenes, are now displayed in different colors (Fig. 1).
Fig. 1.

Summary of features of the updated visualization component. There are several ways to access the visualization component, usually via a click on one of the triangles somewhere in the SimpleSearch content. There are two views to the visualization: the ‘Line view’ (top) and the ‘Insertion view’ (bottom). The ‘Line view’ can be accessed when coming from inspecting the details of a specific line. In this case, only primers, confirmation sequences and insertion predictions (triangles) are displayed that correspond to the line selected (1). The user can navigate in the visualization by entering a position or an AGI locus code (2), or by scrolling through the genome using the arrowheads (3). Further information is provided by tooltips (4), and by links to pages within SimpleSearch (a click on the symbols for primers, FSTs, confirmation sequences or the triangles representing lines causes calling the respective information) or to Araport for genes. Since annotation units/BACs are not supported by Araport11, these are linked to TAIR. To access the ‘Insertion view,’ the respective button can be used (5). This view shows all insertions within the selected range without FSTs, confirmation sequences and primers that are related to a specific line/insertion allele. When there are multiple insertions close to each other, they are displayed in a larger triangle. These larger triangles might show multiple colors if there are confirmed and failed insertion alleles (originating from a contamination) at nearby positions (6); other color combinations in the larger triangles are also possible (7). When the visualization was initially started in the line view, it is possible to switch back (8). It is also possible to switch to the primer design tool (Huep et al. 2014) to generate primers for the selected position (9).
As mentioned in the introduction, we have combined insertions that were predicted in multiple lines at very narrow positions into contamination groups—essentially a group of different lines with the same predicted insertion (Kleinboelting et al. 2012). The updated visualization component now displays a larger triangle for several lines at very narrow positions, and upon mouse-over links to all relevant lines are presented. The normal triangles are color-coded for confirmed (blue), failed (red) and unchecked insertions (green, see Supplemental Fig. 4). This color code has been transferred to the larger triangles. For example, a red and blue multi-colored triangle is displayed if there are failed insertion predictions as well as a confirmed insertion allele available for essentially the same position. In most cases, such red/blue multi-color triangles represent resolved contamination groups where only one of the lines truly contains the insertion allele. Multi-triangles may resolve into individual triangles by zooming in, or may combine from individual triangles when zooming out.
For confirmed insertions, the updated visualization component displays the blue triangle at the insertion position deduced from the confirmation sequences (see above), the original FST(s), the confirmation sequences as well as the primers for a visual validation of the confirmed insertion allele. If the confirmed position differs significantly from the predicted position (and also depending on the zoom), a light blue triangle is displayed for comparison indicating the position of the ‘outdated insertion prediction’ (Fig. 2). Depending on the distance between the positions of the outdated insertion prediction and the updated insertion position, as well as on the position of the insertion relative to an affected gene, the differences can be significant. There were cases in which the type of the insertion changed from ‘genome hit’ to ‘gene hit’ (Supplemental Fig. 5A), or even from one gene to another (Supplemental Fig. 5B). The deviation between initial prediction and confirmed insertion position is a chance or a risk that individual scientists have to face for newly confirmed insertion alleles. Compared to other insertion populations, this risk can be reduced by checking SimpleSearch for confirmed GABI-Kat T-DNA insertion alleles.
Fig. 2.
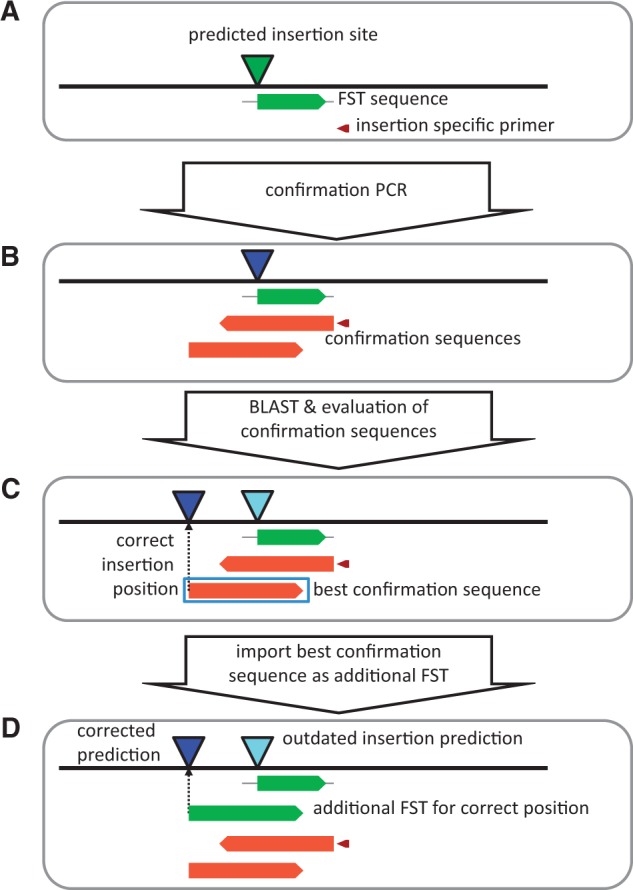
Correction of predicted insertion site using confirmation sequences. For the confirmation of a predicted insertion site, a position specific primer is generated (A) and the resulting amplicon is sequenced from both directions to generate confirmation sequences (B). These are evaluated using BLAST, and if the evaluation yields a positive result the best confirmation sequence is used to correct the predicted insertion position (C). If there is a significant distance between the original insertion position and the confirmed insertion position, we import the confirmation sequence as FST in addition, to make the insertion better accessible in external public databases (D).
Conclusion
During the last five years, the database content available via SimpleSearch has increased in terms of numbers and quality in several areas. At first, more FSTs have been either newly generated or derived from ‘not fitting’ confirmation sequences. This led to more predictions and access to knockout alleles of additional genes. Second, more accurate position information of confirmed insertion sites allows a more reliable prognosis of the effect that an insertion may have on gene function. In contrast to other databases like T-DNA Express (O’Malley and Ecker 2010) or TAIR (Berardini et al. 2015), SimpleSearch provides this confirmation data to users. Third, the incorporation of Araport11 data keeps the GABI-Kat database up to date with recent improvements of the genome annotation which is necessary for reliable identification of insertion mutants. It is also a necessary step to be compatible with other databases relying on the A. thaliana genome annotation. Fourth, the improved visualization component displays more reliable and more up-to-date data for GABI-Kat insertions than before. Finally, we hope that users of GABI-Kat, even if they order the insertion mutants from NASC or ABRC, always check SimpleSearch to avoid redundant data generation and to gain the most from existing information.
Materials and Methods
The Araport11 annotation (Cheng et al. 2016) was obtained from https://www.araport.org/ as gff3 file and imported into a MySQL database (GK-LIMS, internal GABI-Kat laboratory information management system) using a custom Perl script. To reduce complexity we have focused on the first annotated splice variant, which is also the largest and covers all possible splice variants of a protein coding gene. FSTs as well as confirmation sequences were mapped to the TAIR9 genome sequence using BLAST (Altschul et al. 1990, Camacho et al. 2009), the corresponding insertion positions and insertion classes were determined as described before (Kleinboelting et al. 2012).
After confirmation, the insertion position derived from the confirmation sequence was used to update the initial FST-based prediction. The method is essentially the same that has been used to study insertion site structures (Kleinboelting et al. 2015). In short, to determine a position for a junction between T-DNA and genome (insertion position), a MegaBLAST (Zhang et al. 2004) versus the TAIR9 genome sequence was performed. If there were several confirmation sequences with different insertion positions, the best among the confirmation sequences was chosen based on (i) the presence of T-DNA sequence, (ii) the e-value of the MegaBLAST hit and (iii) the primer used. A fasta file containing all FSTs, a list of confirmed insertion junctions as well as a file with all confirmation primers can be downloaded from http://www.gabi-kat.de/download/expert-download.html.
The visualization in SimpleSearch has been realized using the php GD module and uses data from the GK-LIMS MySQL database. The data is stored in several database tables for the annotation, FST and confirmation sequences, BLAST hits and corresponding gene hits, confirmation status, and primers. The visualization itself is embedded in a HTML form where the position or other features can be selected and updated. After submitting the HTML form, the visualization image in PNG format is repainted with the updated information. Mouse-overs and links to other parts of SimpleSearch from within the visualization are realized using an image map.
Supplementary Data
Supplementary data are available at PCP online.
Funding
This work was supported by the German Federal Ministry for Education and Research (BMBF, Foerderkennzeichen 0313855 and 031A533A). In addition, institutional funding provided by Bielefeld University/Faculty of Biology to the Chair of Genome Research has been used.
Supplementary Material
Acknowledgments
We thank Yong Li, Mario Rosso, the MPI for Plant Breeding Research and all former co-workers for their contribution to GABI-Kat, Prisca Viehoever and Ann-Christin Polikeit for high-quality Sanger sequencing, Helene Schellenberg, Ute Buerstenbinder and Andrea Voigt for technical assistance, and Benedikt Brink, Andreas Klötgen and Daniel Blume for database and visualization programming. In addition, the authors wish to thank all members of the Genome Research Team at Bielefeld University as well as the Bioinformatics Resource Facility of CeBiTec (Center for Biotechnology at Bielefeld University) for their excellent assistance and support. We also thank BMBF and PTJ for funding of GABI-Kat until the end of 2014.
Disclosures
The authors have no conflict of interest to declare.
Glossary
Abbreviations
- T-DNA
transfer DNA
- LB
left border
- RB
right border
References
- Alonso J.M., Stepanova A.N., Leisse T.J., Kim C.J., Chen H., Shinn P., et al. (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301: 653–657. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Berardini T.Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., et al. (2015) The Arabidopsis Information Resource: Making and mining the ‘Gold Standard’ annotated reference plant genome. Genesis 53: 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolle C., Huep G., Kleinbolting N., Haberer G., Mayer K., Leister D., et al. (2013) GABI-DUPLO: a collection of double mutants to overcome genetic redundancy in Arabidopsis thaliana. Plant J. 75: 157–171. [DOI] [PubMed] [Google Scholar]
- Bolle C., Schneider A., Leister D. (2011) Perspectives on systematic analyses of gene function in Arabidopsis thaliana: new tools, topics and trends. Current Genomics 12: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchez D., Höfte H. (1998) Functional Genomics in Plants. Plant Physiol. 118: 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009) BLAST+: architecture and applications. BMC Bioinform. 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C-Y., Krishnakumar V., Chan A., Thibaud-Nissen F., Schobel S., Town C.D. (2016) Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 10.1111/tpj.13415. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T., Gersbach C.A., Barbas C.F.r. (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in Biotech. 31: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelvin S.B. (1998) The introduction and expression of transgenes in plants. Curr. Opin. Biotech. 9: 227–232. [DOI] [PubMed] [Google Scholar]
- Gelvin S.B. (2000) Agrobacterium and plant genes involved in T-DNA transfer and integration. Ann. Rev. Plant Physiol. Plant Mol. Biol. 51: 223–256. [DOI] [PubMed] [Google Scholar]
- Gelvin S.B. (2010) Finding a way to the nucleus. Curr. Opin. Microbiology 13: 53–58. [DOI] [PubMed] [Google Scholar]
- Haas B.J., Delcher A.L., Mount S.M., Wortman J.R., Smith R.K., Jr, Hannick L.I., et al. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nuc. Acids Res. 31: 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huep G., Kleinboelting N., Weisshaar B. (2014) An easy-to-use primer design tool to address paralogous loci and T-DNA insertion sites in the genome of Arabidopsis thaliana. Plant Meth. 10: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Veena, Gelvin S. (2007) Genome-wide analysis of Agrobacterium T-DNA integration sites in the Arabidopsis genome generated under non-selective conditions. Plant J. 51: 779–791. [DOI] [PubMed] [Google Scholar]
- Kleinboelting N., Huep G., Appelhagen I., Viehoever P., Li Y., Weisshaar B. (2015) The structural features of thousands of T-DNA insertion sites are consistent with a double-strand break repair-based insertion mechanism. Molecular Plant 8: 1651–1664. [DOI] [PubMed] [Google Scholar]
- Kleinboelting N., Huep G., Kloetgen A., Viehoever P., Weisshaar B. (2012) GABI-Kat SimpleSearch: new features of the Arabidopsis thaliana T-DNA mutant database. Nucl. Acids Res. 40: D1211–D1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar V., Hanlon M.R., Contrino S., Ferlanti E.S., Karamycheva S., Kim M., et al. (2015) Araport: the Arabidopsis information portal. Nucl. Acids Res. 43: D1003–D1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysan P.J., Young J.C., Sussman M.R. (1999) T-DNA as an insertional mutagen in Arabidopsis. Plant Cell 11: 2283–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromori T., Takahashi S., Kondou Y., Shinozaki K., Matsui M. (2009) Phenome analysis in plant species using loss-of-function and gain-of-function mutants. Plant Cell Physiol. 50: 1215–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Dreher K., Knee E., Brkljacic J., Grotewold E., Berardini T.Z., et al. (2014) Arabidopsis database and stock resources. Methods Molec. Biol. 1062: 65–96. [DOI] [PubMed] [Google Scholar]
- Li Y., Rosso M.G., Strizhov N., Viehoever P., Weisshaar B. (2003) GABI-Kat SimpleSearch: a flanking sequence tag (FST) database for the identification of T-DNA insertion mutants in Arabidopsis thaliana. Bioinformatics 19: 1441–1442. [DOI] [PubMed] [Google Scholar]
- Li Y., Rosso M.G., Ulker B., Weisshaar B. (2006) Analysis of T-DNA insertion site distribution patterns in Arabidopsis thaliana reveals special features of genes without insertions. Genomics 87: 645–652. [DOI] [PubMed] [Google Scholar]
- Li Y., Rosso M.G., Viehoever P., Weisshaar B. (2007) GABI-Kat SimpleSearch: an Arabidopsis thaliana T-DNA mutant database with detailed information for confirmed insertions. Nucl. Acids Res. 35: D874–D878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa M., Ichikawa T., Ishikawa A., Kobayashi H., Tsuhara Y., Kawashima M., et al. (2003) Activation tagging, a novel tool to dissect the functions of a gene family. Plant J. 34: 741–750. [DOI] [PubMed] [Google Scholar]
- O’Malley R.C., Ecker J.R. (2010) Linking genotype to phenotype using the Arabidopsis unimutant collection. Plant J. 61: 928–940. [DOI] [PubMed] [Google Scholar]
- Parinov S., Sundaresan V. (2000) Functional genomics in Arabidopsis: large-scale insertional mutagenesis complements the genome sequencing project. Curr. Opin. Biotech. 11: 157–161. [DOI] [PubMed] [Google Scholar]
- Peralta E.G., Ream L.W. (1985) T-DNA border sequences required for crown gall tumorigenesis. In Proceedings of the National Academy of Sciences of the United States of America, vol. 82, pp. 5112–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso M.G., Li Y., Strizhov N., Reiss B., Dekker K., Weisshaar B. (2003) An Arabidopsis thaliana T-DNA mutagenised population (GABI-Kat) for flanking sequence tag based reverse genetics. Plant Molec. Biol. 53: 247–259. [DOI] [PubMed] [Google Scholar]
- Scholl R.L., May S.T., Ware D.H. (2000) Seed and molecular resources for Arabidopsis. Plant Physiol. 124: 1477–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracke R., Huep G., Weisshaar B. (2010) Use of mutants from T-DNA insertion populations generated by high-throughput screening. In The Handbook of Plant Mutation Screening. Edited by Meksem K., Kahl G. pp. 31–54. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- Strizhov N., Li Y., Rosso M.G., Viehoever P., Dekker K.A., Weisshaar B. (2003) High-throughput generation of sequence indexes from T-DNA mutagenized Arabidopsis thaliana lines. BioTech. 35: 1164–1168. [DOI] [PubMed] [Google Scholar]
- Szabados L., Kovacs I., Oberschall A., Abraham E., Kerekes I., Zsigmond L., et al. (2002) Distribution of 1000 sequenced T-DNA tags in the Arabidopsis genome. Plant J. 32: 233–242. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Tzfira T., Li J., Lacroix B., Citovsky V. (2004) Agrobacterium T-DNA integration: molecules and models. Trends Genet. 20: 375–383. [DOI] [PubMed] [Google Scholar]
- VanBuren R., Bryant D., Edger P.P., Tang H., Burgess D., Challabathula D., et al. (2015) Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527: 508–511. [DOI] [PubMed] [Google Scholar]
- Wang K., Herrera-Estrella L., Van Montagu M., Zambryski P. (1984) Right 25 bp terminus sequence of the nopaline T-DNA is essential for and determines direction of DNA transfer from agrobacterium to the plant genome. Cell 38: 455–462. [DOI] [PubMed] [Google Scholar]
- Wortman J.R., Haas B.J., Hannick L.I., Smith R.K., Jr., Maiti R., Ronning C.M., et al. (2003) Annotation of the Arabidopsis Genome. Plant Physiology 132: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S.K., Chung K.S., Kim J., Lee J.H., Hong S.M., Yoo S.J., et al. (2005) CONSTANS activates SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 through FLOWERING LOCUS T to promote flowering in Arabidopsis. Plant Physiol. 139: 770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Schwartz S., Wagner L., Miller W. (2004) A greedy aligning DNA sequences. J. Comp. Biol. 7: 203–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.