

Abstract
Introduction
The current standard care of chronic hepatitis B fails to induce a durable off-drug control of HBV replication in the vast majority of treated patients. The primary reasons are its inability to eliminate the covalently closed circular (ccc) DNA, the nuclear form of HBV genome, and restoration of the dysfunctional host antiviral immune response against the virus.
Area covered
Accordingly, discovery and development of therapeutics to completely stop HBV replication, eliminate or functionally inactivate cccDNA as well as activate a functional antiviral immune response against HBV are currently the primary efforts toward finding a cure for chronic hepatitis B.
Expert opinion
We believe that through a consecutive or overlapping three-stage antiviral and immunotherapy program to (i) completely stop HBV replication and cccDNA amplification; _(ii) reduce viral antigen load and induce HBV surface antigen (HBsAg) seroclearance through eradication or inactivation of cccDNA and RNA interference-mediated degradation of viral mRNA and (iii) activate a functional antiviral immune response against HBV through therapeutic immunization or immunotherapy, a functional cure of chronic HBV infection can be achieved in the majority of chronic HBV carriers.
1. Introduction
Chronic hepatitis B virus (HBV) infection affects 240 million people worldwide 1. Approximately one-third of these individuals will develop severe liver diseases, including cirrhosis, hepatocellular carcinoma (HCC) and liver failure. The currently approved antiviral therapeutics are two formulations of alpha-interferon (IFN-α) and five nucleos(t)ide analogues (NUCs). While the NUCs inhibit HBV DNA polymerase activity with varying potencies and barriers to resistance, the antiviral activity of IFN-α against HBV may result from the combinatory effects of activation of host antiviral immune response as well as suppression of HBV covalently closed circular (ccc) DNA transcription and promotion of HBV nucleocapsid decay in hepatocytes 2–5. Clinically, sustained suppression of viral replication with a long-term NUC therapy or through a 48-week of pegylated IFN-α therapy has been associated with improvement of liver diseases, prevention of liver decompensation and reduction of hepatocellular carcinoma morbidity and mortality in a portion of treated patients 6, 7. However, HBV surface antigen (HBsAg) seroconversion, the hallmark of a successful immune response to HBV with complete and durable control of infection, or a “functional cure”, is rarely achieved with the current therapies 8, 9.
The reasons for the failure of the current antiviral regimens to achieve the functional cure of chronic HBV infection in the vast majority of treated patients are two folds. First, despite of significant suppression of viral DNA replication and progeny virion production, viral DNA polymerase inhibitors do not efficiently reduce the amounts of cccDNA, the nuclear form of HBV genome that is synthesized from the cytoplasmic relaxed circular (rc) DNA and exists as a minichromosome to transcribe HBV mRNAs 10–12. This notion is supported by the observation that cccDNA becomes the predominant form of HBV DNA replication intermediates in the livers of patients or animals after a long-term NUC treatment 13, 14. Second, despite potent suppression of HBV replication in the livers of treated patients, the dysfunction of HBV-specific antiviral immunity persists. Inability of the immune responses in chronic HBV carriers to recognize cells harboring HBV infection and cure or eliminate the cells actively producing virus is the fundamental reason for the chronicity of HBV infection and the big challenge for development of therapeutics to cure chronic hepatitis B 15. Therefore, the goals of current drug discovery and development efforts toward a functional cure of HBV infection are (i) complete suppression of HBV replication to prevent the spread of HBV infection to susceptible host cells and the amplification of cccDNA pool in infected cells; (ii) eradication or transcriptional inactivation of cccDNA from infected cells and (iii) restoration of host innate and adaptive immune responses against HBV to achieve a durable control of HBV replication. Because immunotherapeutic approaches to activate host innate and adaptive immune response against HBV have been extensively reviewed recently 16, 17, this article will focus on the development of antiviral agents targeting viral components and their interactions with host cellular proteins to achieve the therapeutic goals.
2. Development of novel HBV replication inhibitors
2.1 Inhibition of HBV entry
HBV infection begins by attachment to the surface of hepatocyte via the interaction of its large envelope protein (HBL) and cellular heparan sulfate proteoglycan (HSPG) 18, which is followed by the specific binding of the pre-S1 region of HBL to sodium taurocholate cotransporting polypeptide (NTCP), a liver-specific bile salt transporter and the recently identified cellular receptor of HBV 19, 20 (Figure 1). HBV binding to its receptor triggers a caveolin-1-mediated endocytosis to internalize the virus into hepatocytes 21. Recently, using a high-throughput infectious cell culture model enabling RNA interference-mediated loss-of-function screening of hepatitis delta virus (HDV) entry and infection, Verrier and colleagues identified glypican 5 as an additional host cell entry factor for HBV and HDV 22. Apparently, specific disruption of the interactions between HBL and host cellular receptor and entry factors will selectively inhibit HBV infection. For instance, myrcludex B, a synthetic HBV preS1 domain-derived lipopeptide, binds to NTCP and efficiently inhibits HBV and HDV infection of hepatocytes in culture and in vivo in humanized chimeric uPA mouse model 23, 24. In a phase I clinical trial, myrcludex B showed excellent tolerability and dose-dependent pharmacokinetics 25. In a phase Ib/IIa clinical trial in chronically HDV infected patients, myrcludex B monotherapy for 24 weeks significantly reduced HDV RNA serum levels and induced ALT normalization. Furthermore, combination therapy with pegylated IFN-α demonstrated synergistic effects in reducing HDV RNA and HBV DNA serum levels 26. Hence, as a first-in-class HBV and HDV entry inhibitor, myrcludex B is very promising for further clinical development.
Figure 1. HBV replication cycle in hepatocyte and targets of currently available antiviral therapeutics.
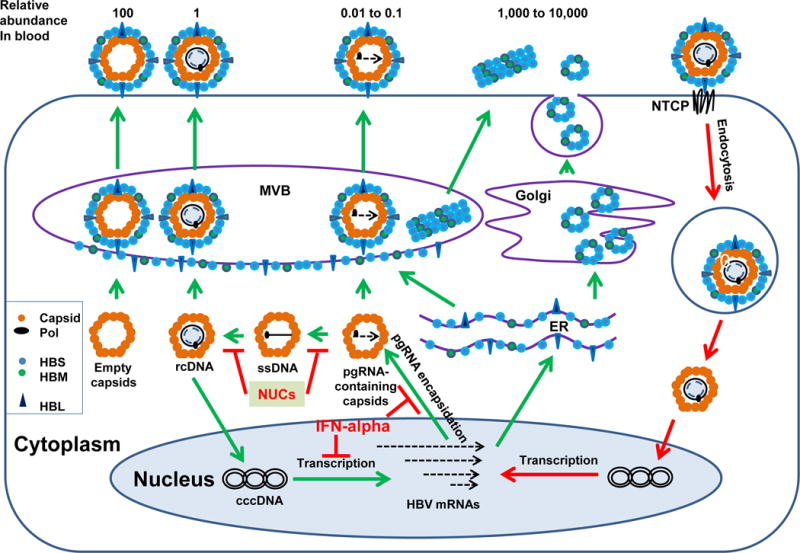
Briefly, HBV infects hepatocyte by binding to its cellular receptor NTCP and entering into the cells via endocytosis. Upon arriving at the cytoplasm, nucleocapsid delivers viral relaxed circular (rc) DNA into the nucleus to be converted into cccDNA by cellular DNA repair machinery. The cccDNA serves as a transcriptional template for production of all viral RNAs, which are subsequently exported to the cytoplasm to translate viral proteins and serve as a pgRNA to be packaged into nucleocapsid. Within the nucleocapsids, pgRNA is reverse transcribed into single-stranded (ss) DNA and then rcDNA. In addition, capsid protien can also assemble into viral RNA/DNA-free empty capsids. Interestingly, while rcDNA-containing mature nucleocapsids can be enveloped and secreted as virions via multi-vesicle bodies (MVB), pgRNA containing nucleocapsids as well as empty capsids can also been enveloped and secreted as virion-like particles. In the blood of HBV infected individuals, the number of empty capsid-containing virion-like particles is approximately 100-fold more than virions. However, the number of pgRNA-containing virions is approximately 10 to 100-fold less than virions. In addition to virion-like particles, HBV-infected hepatocytes also secrete empty envelope particles or filaments in the amounts of 1000 to 10,000-fold more than virions.
In addition to myrcludex B, several classes of structurally distinct small molecules, including bile salt analogues and cyclosporin A derivatives, have been shown to bind NTCP and inhibit HBV and HDV infection of hepatocytes 27, 28. However, their antiviral efficacy has not been demonstrated in animal models in vivo. A major concern regarding the use of NTCP substrate analogues to inhibit HBV/HDV infection is the potential interference with the physiological function of NTCP in bile acid enterohepatic circulation. Interestingly, since the essential amino acid residues in NTCP for HBV/HDV infection and bile acid transportation are overlapped, but not identical, it is thus possible to develop NTCP-targeting drugs that specifically inhibit HBV and HDV infection without compromising the function of NTCP to transport bile acid 29, 30.
2.2. Targeting the multi-functional HBV polymerase protein to inhibit HBV DNA replication
Unlike classical retroviral reverse transcriptases that consist of a reverse transcriptase (RT) domain and a ribonuclease H (RNase H) domain, HBV polymerase has two additional domains, a terminal protein domain (TP) and a spacer domain, at its N-terminal region. The unique structure features additional function of HBV polymerase in viral DNA replication 31. Specifically, HBV polymerase binds a stem-loop structure epsilon (ε) at the 5′ region of the viral pregenomic (pg) RNA to facilitate the pgRNA and polymerase itself being packaged into nucleocapsid. Within nucleocapsid, polymerase primes the synthesis of the minus strand of viral DNA by catalyzing the formation of a phosphodiester linkage between a hydroxyl residue of a tyrosine located in its TP domain and dGMP with the sequence of 5′-UUC-3′ located at the bulge of ε as a template. The priming reaction terminates following the synthesis of only 3 nucleotides, which are subsequently transferred to the 3′ end of pgRNA at DR1 sequence to continue the elongation of the minus strand DNA toward the 5′ terminus of pgRNA. The pgRNA is simultaneously degraded by the RNase H domain of the polymerase, except for its capped 5´ terminal region including 5´ DR1, which is translocated to the DR2 region of the minus strand DNA to prime the synthesis of plus strand DNA. Apparently, HBV DNA polymerase plays essential roles in packaging pgRNA, priming and elongating viral DNA synthesis as well as degrading viral pgRNA. Upon activation by cellular nucleoside kinases, the NUCs can be incorporated into DNA primer and elongating DNA chains by competition with the natural nucleotide substrates and terminate viral DNA synthesis. However, the NUCs do not inhibit polymerase binding of pgRNA and RNase H activity.
Due to the great success of the approved NUCs in treatment of chronic hepatitis B, development of novel NUCs against HBV is not a high demand. Currently, two tenofovir prodrugs with reduced renal toxicity than that of tenofovir, GS-7340 (tenofoviralafenamide fumarate) and CMX157, sponsored by Gilead Science and Contravir, are at phase III or Phase II clinical trials, respectively 32. Despite the great antiviral potency, accumulating evidence suggests that NUC treatment does not completely inhibit HBV DNA synthesis in hepatocytes 13, 33. Hence, development of non-NUC allosteric HBV DNA polymerase inhibitors as well as drugs that disrupt polymerase binding of pgRNA, priming of minus-strand DNA synthesis or inhibit RNase H activity to completely suppress HBV genome replication, either as monotherapeutic agents or in combination with NUCs, is highly desired 34. Particularly, Tavis and colleagues recently developed biochemical and cell-based assays for discovery of HBV RNase H inhibitors and found that some of the human immunodeficiency virus (HIV) RNase H and integrase inhibitors, including napthyridinone derivative, β-thujaplicinol 35, 36, 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) derivatives 37 and α-hydroxytropolones (αHT) 38, were active against HBV RNase H and inhibited HBV DNA replication by chelating the divalent cations in the RNaseH active site 39.
2.3. Inhibition of nucleocapsid assembly
As mentioned above, HBV DNA replication occurs inside of nucleocapsids. Therefore, inhibition of nucleocapsid assembly will preclude viral DNA replication. Interestingly, assembly of a replication competent nucleocapsid relies on cellular HSP90 chaperone complex-assisted HBV DNA polymerase folding and binding to pgRNA, which triggers the recruitment of 120 copies of core protein dimmers to package the pgRNA-polymerase complex into an icosahedral nucleocapsid. Accordingly, inhibition of either HSP90 function or disrupting the interactions between viral polymerase, pgRNA and core proteins should inhibit nucleocapsid assembly. Indeed, it has been demonstrated that inhibition of HSP90 ATPase activity can efficiently inhibit pgRNA encapsidation into capsids 40. More importantly, several classes of structurally distinct small molecules have been shown to accelerate capsid assembly kinetics via modulation of capsid protein interaction, which results in altered nucleocapsid assembly pathway and formation of either empty capsids or non-capsid polymers.
As illustrated in Figure 2, five chemotypes of nucleocapsid assembly inhibitors have been reported thus far. While heteroaryldihydropyrimidines (HAPs), such as Bay 41-4109 and GSL-4, misdirect capsid assembly to form non-capsid polymers of core proteins 41–44, all other nucleocapsid inhibitors induce the formation of morphologically “normal” capsids devoid of viral pgRNA and DNA polymerase 45–48. Interestingly, crystal structure analyses of HAPs or phenylpropenamides (PPAs) in complex with core protein and capsids revealed that both compounds bind a hydrophobic pocket, designated as HAP pocket, at the dimer-dimer interface near the C-terminal of core protein subunits, with contribution from two neighboring core protein dimmers. Binding of these molecules in the HAP pocket induces large scale allosteric conformational changes in core protein subunits and results in quaternary and/or tertiary structure changes of capsids 49, 50. Moreover, a V124W mutation fills the HAP pocket and renders resistance to the inhibition of nucleocapsid assembly by HAPs and PPAs 51. Intriguingly, it was demonstrated recently that NZ-4, a derivative of bis-heterocycle tandem pairs, induces the formation of morphologically normal, but genome-free capsids in a core protein C-terminal arginine-rich domain-dependent manner 47. Pharmacologically, as anticipated, by specifically targeting core protein interaction, all the reported nucleocapsid inhibitors showed excellent antiviral activity against the NUC-resistant strains of HBV 45, 48. Thus far, several nucleocapsid assembly inhibitors, such as, GLS4, NVR 3-778 and AB-423, have been shown to inhibit HBV replication in mice models and are currently under preclinical or clinical development 44, 52.
Figure 2. Five chemotypes of representative HBV capsid assembly inhibitors.
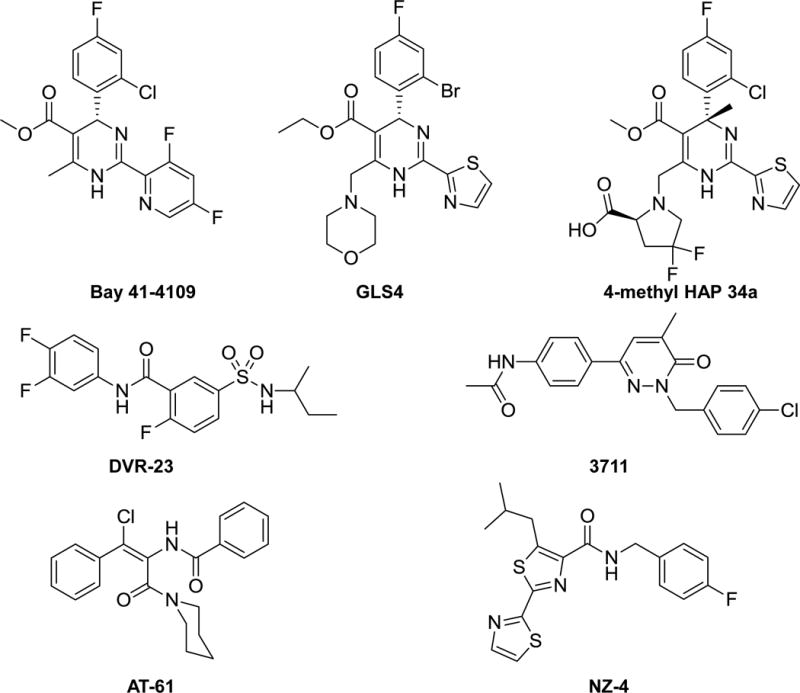
Bay 41-4109, GSL4 and 4-metnyl HAP 34a are heteroaryldihydropyrimidine derivatives. DVR-23 is a sulfamoylbenzamide. AT-61 is a phenylpropenamide. NZ-4 is an isothiafludine. Compound 3711 is a pyridazinone.
3. Eradication or functional inactivation of cccDNA is essential for a functional cure
cccDNA exists as a minichromosome in the nucleus of hepatocyte to serve as a transcription template for viral mRNAs, but cannot replicate itself via semi-conservative replication. Instead, cccDNA is synthesized from viral relaxed circular (rc) DNA from infecting virions or progeny cytoplasmic nucleocapsids (Figure 1). The cytoplasmic nucleocapsid rcDNA is delivered into the nucleus via importin-β-mediated nuclear transportation pathway and converted into cccDNA, presumably by cellular DNA repair enzymes and other host factors 53. The exact copy number of cccDNA may vary among infected cells and in response to different pathophysiological conditions. The average copy number of cccDNA in a HBV-infected hepatocyte is approximately 10. Due to the lack of metabolic labeling studies, the stability or rate of turn-over of cccDNA in infected hepatocytes is unknown. However, using NUCs to inhibit new cccDNA formation in cultured cells and infected animals in vivo, the amounts of cccDNA in non-dividing primary hepatocytes is steady for a six-week culture period, but declined with apparent half lives of 9–14, 33–50 and 35–57 days in HBV-infected chimpanzees. WHV-infected woodchuck and DHBV-infected ducks, respectively 15. However, clinical studies demonstrated that although NUC monotherapy for 48 to 52 weeks reduced circulating viremia by 4.7 to 5.5 log10 and cytoplasmic HBV DNA levels in the hepatocytes by approximately 2 log10, reduction of cccDNA was much less pronounced, only by 0.11 to 1.0 log10 54–56. Although the mechanisms underlying the apparent stability of cccDNA in vivo remains elusive, it is clearly evident from the decade-long clinical experience that prolonged NUC monotherapy is unlikely to eradicate cccDNA in the vast majority of chronic HBV carriers and therapeutic regimens that directly attack cccDNA are in pressing need to achieve a functional cure.
3.1. Targeting cccDNA by sequence-specific DNA-editing technologies
In order to specifically eradicate or functionally inactivate cccDNA minichromosomes, the therapeutic agents must be able to recognize and target the structural features that are unique to cccDNA, but not host cellular chromatins. Obviously, the most unique structure feature of cccDNA minichromosome is its DNA sequence. Therefore, several sequence-specific DNA editing technologies, such as zinc finger nucleases, meganucleases, transcription activator-like effector nuclease (TALENs) and the clustered regularly interspaced short palindromic repeats (CRISPR)/CAS9 57–62, have been explored as potential therapeutic approaches to functionally inactivate or eliminate cccDNA. Recently, using next generation sequencing (NGS) technology, Seeger and Sohn determined the complete spectrum of mutations in cccDNA following Cas9 cleavage and repair by nonhomologous end joining (NHEJ) DNA repair pathway 63. The authors elegantly demonstrated that over 90% of HBV cccDNA was edited by Cas9. Among the mutated cccDNA molecules, approximately 67% had a single nucleotide deletion at position −4 with respect to the protospacer adjacent motif (PAM) sequence. About 10% had a single nucleotide insertion at the cleavage site and 16% exhibited deletions ranging from 2 to 6 nucleotides in length. Moreover, approximately 6% of the cleavage cccDNA had deletions of 3 and 6 nucleotides which maintain the open reading frame of HBx protein. If the one or two amino acid deletion does not compromise HBx function, the results suggest that in order to completely inactivate cccDNA function, more than one sgRNAs targeting cccDNA sequences encoding critical residues of HBV proteins are desired 63. Moreover, double cleavage of cccDNA by Cas9 may not only more efficiently inactivate cccDNA function, but also make cccDNA repair more difficult and thus favor its elimination 64. However, despite all these exciting progresses, several key technical issues, particularly the potential off-target cleavage of host cellular DNA and efficient delivery to all the HBV infected cells, need to be resolved. Considering the fast development in gene editing technologies, clinical applicable gene editing therapeutics for the cure of chronic hepatitis B may be eventually achievable.
3.2. Targeting HBV core or X protein to inactivate cccDNA function
As an episomal minichromosome, cccDNA transcription is subjected to epigenetic regulation. It was showed that cccDNA bound H3/H4 hyperacetylation is correlated with high viral replication in HBV infected patients 65. Furthermore, IFN-α suppresses cccDNA transcription by reducing its H3K9 and H3K27 acetylation, but not increasing H3K9 and H3K27 trimethylation 2, 66. Although the HBV genome-wide epigenetic analysis did not reveal any histone modification that is unique to cccDNA minichromosome 66, many studies did suggest that HBV core and HBx proteins are recruited to cccDNA minicromosome and regulate its transcriptional activity. Therefore, selectively disrupting the interactions between the viral proteins and their host cellular partners for cccDNA regulation may specifically inactivate cccDNA transcription or destabilize cccDNA minichromosomes.
The first direct evidence suggesting that HBV core protein binds to cccDNA is from an electron microscopic and biochemical investigation of the architecture of HBV minichromosome. The studies revealed that core protein is a structural component of viral cccDNA minichromosome and its binding to cccDNA reduces the nucleosomal spacing of the minichromosome and may thus regulate its transcriptional activity 67. Recent studies further suggested that core protein promotes an epigenetic permissive state of HBV cccDNA by binding on CpG islands of cccDNA 68. Intriguingly, Lucifora and colleagues found that cytokine-induced DNA cytosine deaminase, APOBEC3A, was recruited to cccDNA minichromosome by interaction with core protein 69. This remarkable finding implies that core protein can also be hijacked by the host immune system to destabilize and eliminate cccDNA. Hence, pharmacological disruption of core-core and core-host protein interaction may alter cccDNA stability and transcription. It will be interesting to investigate whether the current available nucleocapsid assembly effectors that specifically modulate core protein subunit interaction in capsid assembly would also have effects on the recruitment and function of core protein on cccDNA minichromosomes. It is entirely possible that the cccDNA binding and transcriptional regulation by core protein rely on distinct core-core and/or core-host protein interaction and thus novel antiviral agents are required to inhibit those functions.
HBx is a 17 kDa viral protein essential for viral DNA replication, which is supported by the facts that HBx-deficient HBV fails to replicate in cultured primary hepatocytes or human hepatocytes in the livers of humanized uPA-chimeric mice in vivo 70, 71. Although HBx may promote HBV DNA replication in the cytoplasm by modulating cytosolic calcium signaling, cell cycle progression and capsid protein phosphorylation 72–76, the recent studies highlight a key role of HBx in activation of cccDNA minichromosome transcription70, 77.
HBx is known to be a trans-activating protein with the ability to stimulate the transcription of episomal DNA, including its own cccDNA minichromosome 78. CHIP analyses indicated that HBx was recruited to the cccDNA minichromosomes and recruiting epigenetic modifiers to set up a transcription permissive cccDNA epigenetic status altered cccDNA-associated histone modification 77, 79, 80. Specifically, HBx had been shown to promote cccDNA-associated histone acetylation as well as recruit histone lysine demethylase-1 (LSD1) to relieved SETDB1-mediated H3K9me3 and HP1 induced condensation of cccDNA minichromosomes77. In addition, HBx was also demonstrated to enhance cccDNA transcription by interacting with transcriptional repressor Spindlin1 to prevent its recruitment to cccDNA minichromosome 81 or inhibiting PP1 phosphatase activity to extend the half-life of transcription factor p-CREB 82. HBx was also reported to interact with a cellular hydroxylse, jumonji C-domain-containing 5 (JMJD5), to promote the transcription of key hepatocyte enriched transcription factors, such as HNF4A, CEBPA, and FOXA3, to enhance cccDNA transcription 73. Remarkably, Decorsière and colleagues reported recently that cellular “structural maintenance of chromosomes” (Smc) complex Smc5/6 suppressed the transcription of cccDNA minichromosome and HBx activates the transcription by hijacking DDB1-containing E3 ubiquitin ligase to target the Smc5/6 for degradation 83.
In summary, accumulating evidence suggests that both core and HBx proteins can be recruited to cccDNA minichromosome and modulate its transcription activity via multiple mechanisms. Interestingly, a screening of FDA approved drug library found that erbinafine, a antifungal medicine, suppressed HBx-mediated HBV transcription via unknown mechanism 84. Further investigation on the mechanism of core and HBx recruitment to and regulation on cccDNA transcription should reveal and validate molecular targets for the discovery of antivirals to selectively inactivate cccDNA function.
3.3. Phenotypic screening of compounds targeting cccDNA
In addition to rationally discovering compounds that selectively target viral proteins and their interacting cellular partners essential for cccDNA function, recent technical development in HBV infection cell culture systems 19, 20, establishment of a HepG2-derived stable cell line expressing epitope-tagged HBeAg in a cccDNA-dependent manner 85 and production of recombinant HBV reporter viruses 86 will allow for high throughput screening of compound libraries to phenotypically identify compounds that destabilize or functionally inactivate cccDNA.
4. Induction of HBsAg seroclearance and seroconversion
Clinical studies indicate that while resolution of an acute HBV infection is associated with a polyclonal and vigorous HBV-specific T-cell response, chronic HBV infection is always associated with a weaker or barely detectable HBV-specific T-cell response 87. Failure to induce a functional antiviral immune response in chronic HBV infection is due to the prolonged exposure of T cells to the high quantities of viral antigens in the liver and high levels of HBsAg in circulation, which results in depletion or functional exhaustion of HBV-specific T cells. The exhausted T cells are characterized by the increased expression of negative co-stimulatory molecules and dysregulation of co-stimulatory pathways. Accordingly, lowering intrahepatic viral antigen load and/or reducing HBsAg antigenemia may be essential for the recovery of functional antiviral immune response to durably control HBV infection. However, how much the reduction of viral antigen load or HBsAg antigenemia is required for a spontaneous or treatment-induced recovery of anti-HBV immune response in chronic carriers remains to be determined. Clinical studies of spontaneous and treatment-induced HBsAg seroclerance/anti-HBs seroconversion and a recent experimental study in mice may be helpful to answer this important question, at least in part.
Spontaneous HBsAg seroclearance, defined by the loss of serum HBsAg on two occasions at least six months apart and remaining absent up to the last visit, is rare, but consistently occurs at an annual of spontaneous HBsAg seroclearance varied from 0.12 to 2.38% in Asian countries and 0.54 to 1.98% in Western countries 88. Prolonged NUC therapy reduces viral load in the vast majority of treated patients by more than 4.5 log, but does not increase the rate of HBsAg seroclearance. However, 48 to 52 weeks of pegylated IFN-α therapy increases the incidence of HBsAg seroclearance by 3 to 6 folds 89. Clinically, spontaneous and treatment-induced HBsAg loss/seroconversion is associated with the stable control of HBV infection and excellent outcome, if without pre-existing cirrhosis and viral superinfection. However, only 17%, 56% and 76% of the carriers have detectable anti-HBs at 1, 5 and 10 years after HBsAg seroclearance 90. The observed slow anti-HBs seroconversion after spontaneous and treatment-induced HBsAg seroclearance in chronic HBV carries suggests that (i) seroclearance, but not lowering the level of HBsAg, may be required to restore the antiviral immune response; (ii) spontaneous restoration of effective antiviral immune response to sustained control HBV infection after HBsAg seroclearance may not immediately occur; and (iii) therapeutic vaccination/immunotherapeutics may be required to accelerate the activation of effective antiviral immune response after HBsAg seroclearance. Moreover, a recent study demonstrated that the immune tolerance status to HBV in AAV-HBV infected mice is attributed to the level and duration of circulating HBsAg 91. Intriguingly, depletion of circulating HBsAg by a monoclonal anti-HBsAg in tolerant mice could gradually reduce tolerance and reestablish B cell and CD4+ T cell responses to subsequent Engerix-B vaccination 91. Furthermore, HBsAg-specific CD8+ T cells induced by the addition of a TLR agonist resulted in clearance of HBV in both peripheral blood and liver 91. These results re-enforce the notion that circulating HBsAg plays an important role in induction of immune tolerance to HBV and HBsAg seroclearance with following immunization or immune agonist therapy is required to promote the restoration of humoral and T cell immune response to achieve a durable control of HBV infection, or a functional cure.
Concerning the therapeutic approaches to lower HBV antigen load and particularly, induce HBsAg seroclearance, we consider that the most efficient way is to reduce viral protein expression in infected cells by eradication or inactivation of cccDNA with the technologies discussed in the previous section or RNA interference (RNAi)-mediated degradation of viral mRNA. Due to the improvement in RNA chemistry and hepatocyte-targeted delivery technologies in recent years, RNAi technologies, such as ARB-1467, ARC-520, AVV/HB-BB-331 and ALN-HBV, have now been demonstrated in preclinical animal models and human clinical trials to efficiently suppress HBV protein expression and reduces viral load 92. Their effects on immune restoration by decreasing viral antigen load are under investigation. In addition to reducing viral antigen expression, compounds that selectively inhibit HBsAg secretion from hepatoma cells have also been reported 93, 94. However, their mode of action and in vivo efficacy in reducing HBsAg levels remain to be determined. Interesting, nucleic acid polymers (NAPs), sequence-independent phosphoro-thioated oligonucleotides, was reported to inhibit HBV entry and HBsAg secretion. REP9-AC (REP 2055), a 40- nucleotide poly-cytidine amphipathic DNA polymer, have been demonstrated to efficiently reduce viral load and surface antigen of duck hepatitis B virus and induce antibodies against surface antigen in duck 95. REP 2139 in combination therapy with pegylated IFN alpha-2a is in clinical trial for chronic HBV and HDV co-infection 96.
5. Conclusions
To achieve a functional cure of chronic hepatitis B, i.e., HBsAg seroclerance and anti-HBs ceroconversion, in the vast majority of treated patients, therapeutic agents targeting multiple unexplored viral and host cellular functions, particularly nucleocapsid assembly, cccDNA metabolism as well as innate and adaptive immunity against HBV, are currently under preclinical or clinical development. A complete and periodically updated list of HBV antiviral and immunotherapeutic agents under preclinical and clinical development can be found in the “Drug Watch” program of Hepatitis B Foundation 97. Currently, the major hurdles for the development of curative therapeutics are the lack of cell culture and animal models supporting robust HBV infection and high throughput assays to efficiently identify and evaluate therapeutic agents in biologically relevant systems. Immune competent animal models supporting persistent HBV infection is particularly important for development of immunotherapeutics to induce a functional antiviral immune response that durably controls HBV infection. Obviously, development of the in vitro and in vivo HBV infection models will also facilitate the investigation of HBV replication, cccDNA biology and immunopathology, which should provide basis for rational development of therapeutics to reach the clinical endpoints of chronic hepatitis B treatment.
6. Expert opinions
Ideally, cure of HBV infection should be the sterilizing elimination or eradication of the virus from infected individuals and return the patients to their pre-infection risk of death from liver diseases. However, it is well established that even persons with naturally resolved acute HBV infection have a risk of death from hepatocellular carcinoma (HCC) three times greater than those who have never been infected. Moreover, the facts that persistence of HBV cccDNA can be detected even in the liver of patient resolved HBV infection and HBV replication can be reactivated by immune suppression therapies in individuals with naturally resolved acute hepatitis B decades ago suggest that “cure” of HBV infection by host immune responses is at least not always associated with the clearance of the virus, but most likely through a tight immune surveillance of the virus to minimize its pathological consequences. It is, therefore, unrealistic or even unnecessary to develop therapeutics to completely eradicate HBV (cccDNA). Instead, we believe that the overall goal of antiviral therapy for chronic HBV infection is to induce a durable off-drug control of HBV infection, which has been associated with improvement of liver diseases, prevention of liver decompensation and reduction of hepatocellular carcinoma morbidity and mortality 6, 7. The key to achieve a durable off-drug suppression of HBV replication is the therapeutic activation of a functional antiviral immune response, which is practically indicated as HBsAg to anti-HBs seroconversion in treated patients. Hence, we consider that HBsAg seroclearnce and anti-HBs seroconversion are the realistic endpoints for novel anti-HBV therapies.
As illustrated in Figure 3, in our opinion, the therapeutic endpoints can be achieved through three consecutive or overlapping stages of therapies. The goal of the first stage is to completely inhibit HBV replication to stop the spread of infection and amplification of cccDNA pool in virally infected cells. This goal can be achieved with combination therapies of NUCs and novel HBV replication inhibitors currently under preclinical and clinical development. For patients with stronger residual immune response to HBV, complete suppression of HBV replication with the combination therapeutic regimens may result in gradual elimination of infected cells by immune response and hepatocyte turnover, which will reduce antigen load and favor the restoration of antiviral immune response and ultimately achieve a functional cure. However, for the majority of the chronic carriers, complete inhibition of HBV replication may not be able to reduce the number of virally infected cells. Therefore, the goal of the second stage therapy is to reduce viral antigen load and induce HBsAg seroclearance. This goal can be achieved by inhibition of viral protein expression, such as elimination or functional inactivation of cccDNA and/or siRNA-mediated degradation of viral mRNA. Due to the important role of secreted HBsAg in the induction of immune tolerance to HBV, therapeutics to induce intracellular degradation, inhibit secretion or accelerate the clearance from circulation of HBsAg will also be helpful. By reducing viral antigen load and particularly clearing HBsAg from circulation, the exhausted HBV-specific immune response may recover spontaneously. Because spontaneous anti-HBs seroconversion may take years after HBsAg seroclearance, the goal of the third stage therapy is to accelerate the recovery of exhausted immune response against HBV by therapeutic immunization and/or immunotherapy. Currently, several therapeutic immunization regimens are in preclinical and clinical development. TLR7 agonists and checkpoint blockade therapy with antibodies against PD1 is in clinical trial 98. Taking together, therapeutic agents to more potently suppress HBV replication, reduce viral antigen load and restore host HBV-specific antiviral immune response are currently under development. In our opinion, combination application of the three categories of therapeutic agents should finally provide a functional cure for the vast majority of chronic HBV carriers.
Figure 3. Illustration of the three-stage therapeutic program of chronic HBV infection.
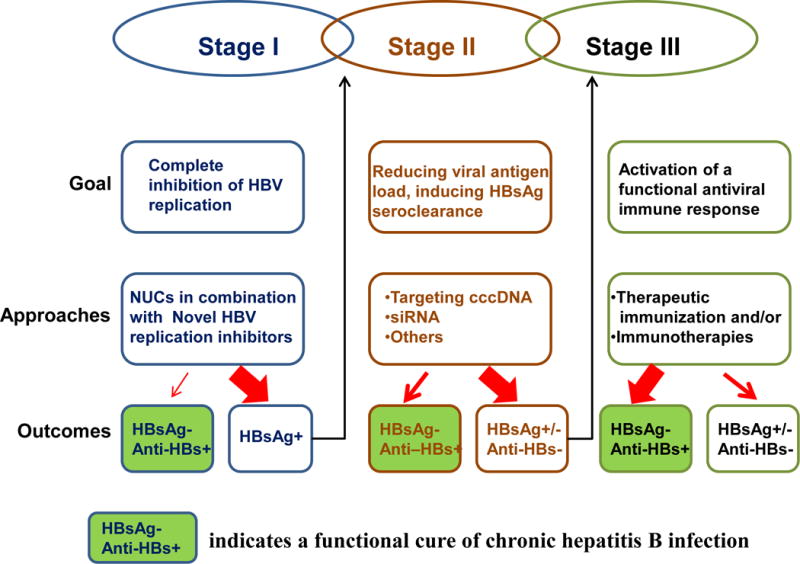
The contribution of current drug discovery efforts on the cure of chronic hepatitis B is highlighted in the approach boxes. The thickness of the arrows indicates the likelihood of the outcomes.
References
• Of outstanding interest
- 1.Ott JJ, Stevens GA, Groeger J, et al. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30:2212–9. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Liu F, Campagna M, Qi Y, et al. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog. 2013;9:e1003613. doi: 10.1371/journal.ppat.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu C, Guo H, Pan XB, et al. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol. 2010;84:9332–40. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belloni L, Allweiss L, Guerrieri F, et al. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122:529–37. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thimme R, Dandri M. Dissecting the divergent effects of interferon-alpha on immune cells: time to rethink combination therapy in chronic hepatitis B? J Hepatol. 2013;58:205–9. doi: 10.1016/j.jhep.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Dienstag JL. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology. 2009;49:S112–21. doi: 10.1002/hep.22920. [DOI] [PubMed] [Google Scholar]
- 7.Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49:S103–11. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 8.Rehermann B, Ferrari C, Pasquinelli C, et al. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T- lymphocyte response. Nat Med. 1996;2:1104–8. doi: 10.1038/nm1096-1104. [DOI] [PubMed] [Google Scholar]
- 9.Block TM, Gish R, Guo H, et al. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res. 2013;98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 11.Wu TT, Coates L, Aldrich CE, et al. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175:255–61. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 12.Newbold JE, Xin H, Tencza M, et al. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. Journal of Virology. 1995;69:3350–7. doi: 10.1128/jvi.69.6.3350-3357.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Y, Yamamoto T, Cullen J, et al. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75:311–22. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokosuka O, Omata M, Imazeki F, et al. Changes of hepatitis B virus DNA in liver and serum caused by recombinant leukocyte interferon treatment: Analysis of intrahepatic replicative hepatitis B virus DNA. Hepatololgy. 1985;5:707–713. doi: 10.1002/hep.1840050505. [DOI] [PubMed] [Google Scholar]
- 15.Chang J, Guo F, Zhao X, et al. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B. 2014;4:248–57. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang J, Guo JT. Treatment of chronic hepatitis B with pattern recognition receptor agonists: Current status and potential for a cure. Antiviral Res. 2015;121:152–9. doi: 10.1016/j.antiviral.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang E, Kosinska A, Lu M, et al. Current status of immunomodulatory therapy in chronic hepatitis B, fifty years after discovery of the virus: Search for the “magic bullet” to kill cccDNA. Antiviral Res. 2015;123:193–203. doi: 10.1016/j.antiviral.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology. 2007;46:1759–68. doi: 10.1002/hep.21896. [DOI] [PubMed] [Google Scholar]
- 19•.Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. This landmark paper identifies sodium taurocholate cotransporting polypeptide (NTCP) as the bona fide receptor of HBV and HDV. This discovery not only helps to improve HBV infection cell culture system, but also identifies a therapeutic target for inhibition of HBV and HDV infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni Y, Lempp FA, Mehrle S, et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology. 2014;146:1070–83. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Macovei A, Radulescu C, Lazar C, et al. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J Virol. 2010;84:243–53. doi: 10.1128/JVI.01207-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verrier ER, Colpitts CC, Bach C, et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology. 2016;63:35–48. doi: 10.1002/hep.28013. [DOI] [PubMed] [Google Scholar]
- 23.Petersen J, Dandri M, Mier W, et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol. 2008;26:335–41. doi: 10.1038/nbt1389. [DOI] [PubMed] [Google Scholar]
- 24.Volz T, Allweiss L, Ben MM, et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58:861–7. doi: 10.1016/j.jhep.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Blank A, Markert C, Hohmann N, et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor Myrcludex B. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 26.Bogomolov P, Alexandrov A, Voronkova N, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 27•.Nkongolo S, Ni Y, Lempp FA, et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol. 2014;60:723–31. doi: 10.1016/j.jhep.2013.11.022. This paper shows that myrcludex B, a first-in-class HBV/HDV entry inhibitor, significantly reduces HDV load and normalizes liver function as well as demonstrates a synergistic effect with pegylated IFN-alpha in 26 weeks of mono- and combinatory therapies of chronic hepatitis D. [DOI] [PubMed] [Google Scholar]
- 28.Watashi K, Sluder A, Daito T, et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP) Hepatology. 2014;59:1726–37. doi: 10.1002/hep.26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urban S, Bartenschlager R, Kubitz R, et al. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology. 2014;147:48–64. doi: 10.1053/j.gastro.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 30.Yan H, Liu Y, Sui J, et al. NTCP opens the door for hepatitis B virus infection. Antiviral Res. 2015;121:24–30. doi: 10.1016/j.antiviral.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology. 2015;479–480:672–86. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang TJ, Block TM, McMahon BJ, et al. Present and future therapies of hepatitis B: From discovery to cure. Hepatology. 2015;62:1893–908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Liu F, Guo F, et al. Characterization of novel hepadnaviral RNA species accumulated in hepatoma cells treated with viral DNA polymerase inhibitors. Antiviral Res. 2016;131:40–8. doi: 10.1016/j.antiviral.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark DN, Hu J. Hepatitis B virus reverse transcriptase – Target of current antiviral therapy and future drug development. Antiviral Res. 2015;123:132–7. doi: 10.1016/j.antiviral.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tavis JE, Cheng X, Hu Y, et al. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog. 2013;9:e1003125. doi: 10.1371/journal.ppat.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Cheng X, Cao F, et al. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res. 2013;99:221–9. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Cai CW, Lomonosova E, Moran EA, et al. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res. 2014;108:48–55. doi: 10.1016/j.antiviral.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu G, Lomonosova E, Cheng X, et al. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity. Antimicrob Agents Chemother. 2015;59:1070–9. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tavis JE, Lomonosova E. The hepatitis B virus ribonuclease H as a drug target. Antiviral Res. 2015;118:132–8. doi: 10.1016/j.antiviral.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu J, Flores D, Toft D, et al. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol. 2004;78:13122–31. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stray SJ, Bourne CR, Punna S, et al. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stray SJ, Zlotnick A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit. 2006;19:542–8. doi: 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- 43.Bourne CR, Finn MG, Zlotnick A. Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. Journal of Virology. 2006;80:11055–11061. doi: 10.1128/JVI.00933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu G, Liu B, Zhang Y, et al. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother. 2013;57:5344–54. doi: 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campagna MR, Liu F, Mao R, et al. Sulfamoylbenzamide Derivatives Inhibit the Assembly of Hepatitis B Virus Nucleocapsids. J Virol. 2013 doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King RW, Ladner SK, Miller TJ, et al. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-L- 2′,3′-dideoxy-3′-thiacytidine [published erratum appears in Antimicrob Agents Chemother 1999 Mar;43(3):726] Antimicrob Agents Chemother. 1998;42:3179–86. doi: 10.1128/aac.42.12.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang L, Wang YJ, Chen HJ, et al. Effect of a hepatitis B virus inhibitor, NZ-4, on capsid formation. Antiviral Res. 2016;125:25–33. doi: 10.1016/j.antiviral.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Wang YJ, Lu D, Xu YB, et al. A novel pyridazinone derivative inhibits hepatitis B virus replication by inducing genome-free capsid formation. Antimicrob Agents Chemother. 2015;59:7061–72. doi: 10.1128/AAC.01558-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katen SP, Tan Z, Chirapu SR, et al. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure. 2013;21:1406–16. doi: 10.1016/j.str.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Klumpp K, Lam AM, Lukacs C, et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A. 2015;112:15196–201. doi: 10.1073/pnas.1513803112. This paper reports a high resolution crystal structure of a HAP derivative-bound HBV capsid. The structure reveals that improvement in antiviral potency of HAP derivatives can be achieved by improving in their stabilization of core protein dimer interaction and thus provides clear avenues for compound optimization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan Z, Pionek K, Unchwaniwala N, et al. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J Virol. 2015;89:3275–84. doi: 10.1128/JVI.03545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu Z, Lin X, Zhou M, et al. Design and Synthesis of Orally Bioavailable 4-Methyl Heteroaryldihydropyrimidine Based Hepatitis B Virus (HBV) Capsid Inhibitors. J Med Chem. 2016 doi: 10.1021/acs.jmedchem.6b00879. [DOI] [PubMed] [Google Scholar]
- 53.Guo JT, Guo H. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res. 2015;122:91–100. doi: 10.1016/j.antiviral.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–8. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 55.Sung JJ, Wong ML, Bowden S, et al. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology. 2005;128:1890–7. doi: 10.1053/j.gastro.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Wursthorn K, Lutgehetmann M, Dandri M, et al. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology. 2006;44:675–84. doi: 10.1002/hep.21282. [DOI] [PubMed] [Google Scholar]
- 57.Ramanan V, Shlomai A, Cox DB, et al. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep. 2015;5:10833. doi: 10.1038/srep10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X, Hao R, Chen S, et al. Inhibition of Hepatitis B Virus by CRISPR/Cas9 System via Targeting the Conserved Regions of Viral Genome. J Gen Virol. 2015 doi: 10.1099/vir.0.000159. [DOI] [PubMed] [Google Scholar]
- 59.Dong C, Qu L, Wang H, et al. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110–7. doi: 10.1016/j.antiviral.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 60.Ren Q, Li C, Yuan P, et al. A Dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci Rep. 2015;5:8865. doi: 10.1038/srep08865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin SR, Yang HC, Kuo YT, et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Mol Ther Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seeger C, Sohn JA. Targeting Hepatitis B Virus With CRISPR/Cas9. Mol Ther Nucleic Acids. 2014;3:e216. doi: 10.1038/mtna.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63•.Seeger C, Sohn JA. Complete Spectrum of CRISPR/Cas9-induced Mutations on HBV cccDNA. Mol Ther. 2016;24:1258–66. doi: 10.1038/mt.2016.94. This paper reports the complete spectrum of CRISPR/Cas9 editing of cccDNA in HBV infected hepatoma cells by next generation DNA sequence technology, which provides a basis for further development of the gene-editing technology to inactivate cccDNA in infected hepatocytes in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang J, Xu ZW, Liu S, et al. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J Gastroenterol. 2015;21:9554–65. doi: 10.3748/wjg.v21.i32.9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pollicino T, Belloni L, Raffa G, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130:823–37. doi: 10.1053/j.gastro.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 66.Tropberger P, Mercier A, Robinson M, et al. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc Natl Acad Sci U S A. 2015;112:E5715–24. doi: 10.1073/pnas.1518090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bock CT, Schwinn S, Locarnini S, et al. Structural organization of the hepatitis B virus minichromosome. Journal of molecular biology. 2001;307:183–196. doi: 10.1006/jmbi.2000.4481. [DOI] [PubMed] [Google Scholar]
- 68.Guo Y-H, Li Y-N, Zhao J-R, et al. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics. 2011;6:720–726. doi: 10.4161/epi.6.6.15815. [DOI] [PubMed] [Google Scholar]
- 69•.Lucifora J, Xia Y, Reisinger F, et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science. 2014 doi: 10.1126/science.1243462. This paper reveals an novel mechanism by which host antiviral immune response non-cytopathically eliminates cccDNA from the nuclei of hepatocytes through APOBEC3A or APOBEC3Bmediated cytosine deamination of cccDNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lucifora J, Arzberger S, Durantel D, et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55:996–1003. doi: 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 71.Tsuge M, Hiraga N, Akiyama R, et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J Gen Virol. 2010;91:1854–64. doi: 10.1099/vir.0.019224-0. [DOI] [PubMed] [Google Scholar]
- 72.Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–8. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- 73.Kouwaki T, Okamoto T, Ito A, et al. Hepatocyte Factor JMJD5 Regulates Hepatitis B Virus Replication through Interaction with HBx. J Virol. 2016;90:3530–42. doi: 10.1128/JVI.02776-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gearhart TL, Bouchard MJ. Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology. 2010;407:14–25. doi: 10.1016/j.virol.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Melegari M, Wolf SK, Schneider RJ. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J Virol. 2005;79:9810–20. doi: 10.1128/JVI.79.15.9810-9820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rawat S, Bouchard MJ. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J Virol. 2015;89:999–1012. doi: 10.1128/JVI.02440-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Riviere L, Gerossier L, Ducroux A, et al. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol. 2015;63:1093–102. doi: 10.1016/j.jhep.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 78.van Breugel PC, Robert EI, Mueller H, et al. Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology. 2012;56:2116–24. doi: 10.1002/hep.25928. [DOI] [PubMed] [Google Scholar]
- 79.Belloni L, Pollicino T, De Nicola F, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A. 2009;106:19975–9. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alarcon V, Hernandez S, Rubio L, et al. The enzymes LSD1 and Set1A cooperate with the viral protein HBx to establish an active hepatitis B viral chromatin state. Sci Rep. 2016;6:25901. doi: 10.1038/srep25901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ducroux A, Benhenda S, Riviere L, et al. The Tudor domain protein Spindlin1 is involved in intrinsic antiviral defense against incoming hepatitis B Virus and herpes simplex virus type 1. PLoS Pathog. 2014;10:e1004343. doi: 10.1371/journal.ppat.1004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cougot D, Allemand E, Riviere L, et al. Inhibition of PP1 phosphatase activity by HBx: a mechanism for the activation of hepatitis B virus transcription. Sci Signal. 2012;5:ra1. doi: 10.1126/scisignal.2001906. [DOI] [PubMed] [Google Scholar]
- 83•.Decorsiere A, Mueller H, van Breugel PC, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386–9. doi: 10.1038/nature17170. This landmark paper demonstrates that cellular “structural maintenance of chromosomes” (Smc) complex Smc5/6 suppressed the transcription of cccDNA minichromosome and HBx activates the transcription by hijacking DDB1-containing E3 ubiquitin ligase to target the Smc5/6 for degradation. [DOI] [PubMed] [Google Scholar]
- 84.van de Klundert MA, Zaaijer HL, Kootstra NA. Identification of FDA-approved drugs that target hepatitis B virus transcription. J Viral Hepat. 2016;23:191–201. doi: 10.1111/jvh.12479. [DOI] [PubMed] [Google Scholar]
- 85.Cai D, Wang X, Yan R, et al. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res. 2016;132:26–37. doi: 10.1016/j.antiviral.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bai W, Cui X, Chen R, et al. Re-Designed Recombinant Hepatitis B Virus Vectors Enable Efficient Delivery of Versatile Cargo Genes to Hepatocytes with Improved Safety. Viruses. 2016;8 doi: 10.3390/v8050129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. 2014;40:13–24. doi: 10.1016/j.immuni.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chu CM, Liaw YF. HBsAg seroclearance in asymptomatic carriers of high endemic areas: appreciably high rates during a long-term follow-up. Hepatology. 2007;45:1187–92. doi: 10.1002/hep.21612. [DOI] [PubMed] [Google Scholar]
- 89.Marcellin P, Ahn SH, Ma X, et al. Combination of Tenofovir Disoproxil Fumarate and Peginterferon alpha-2a Increases Loss of Hepatitis B Surface Antigen in Patients With Chronic Hepatitis B. Gastroenterology. 2016;150:134–144 e10. doi: 10.1053/j.gastro.2015.09.043. [DOI] [PubMed] [Google Scholar]
- 90.Chu CM, Liaw YF. Hepatitis B surface antigen seroclearance during chronic HBV infection. Antivir Ther. 2010;15:133–43. doi: 10.3851/IMP1497. [DOI] [PubMed] [Google Scholar]
- 91•.Zhu D, Liu L, Yang D, et al. Clearing Persistent Extracellular Antigen of Hepatitis B Virus: An Immunomodulatory Strategy To Reverse Tolerance for an Effective Therapeutic Vaccination. J Immunol. 2016;196:3079–87. doi: 10.4049/jimmunol.1502061. This paper demonstrates the important role of circulating HBsAg in the induction and maintenance of immune tolerance to HBV in a mice model and requirement of the depletion of circulating HBsAg in the activation of antiviral immune response against HBV by immunization and immunotherapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wooddell CI, Rozema DB, Hossbach M, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–85. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu YB, Yang L, Wang GF, et al. Benzimidazole derivative, BM601, a novel inhibitor of hepatitis B virus and HBsAg secretion. Antiviral Res. 2014;107:6–15. doi: 10.1016/j.antiviral.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 94.Dougherty AM, Guo H, Westby G, et al. A substituted tetrahydro-tetrazolo-pyrimidine is a specific and novel inhibitor of hepatitis B virus surface antigen secretion. Antimicrob Agents Chemother. 2007;51:4427–37. doi: 10.1128/AAC.00541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Noordeen F, Scougall CA, Grosse A, et al. Therapeutic Antiviral Effect of the Nucleic Acid Polymer REP 2055 against Persistent Duck Hepatitis B Virus Infection. PLoS One. 2015;10:e0140909. doi: 10.1371/journal.pone.0140909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vaillant A. Nucleic acid polymers: Broad spectrum antiviral activity, antiviral mechanisms and optimization for the treatment of hepatitis B and hepatitis D infection. Antiviral Res. 2016;133:32–40. doi: 10.1016/j.antiviral.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 97.Drug watch: compounds in development for chronic hepatitis B. Hepatitis B Foundation. 2016 [cited 2016 Oct 17]. Available from: http://www.hepb.org/treatment-and-management/drug-watch/
- 98.Lanford RE, Guerra B, Chavez D, et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology. 2013;144:1508–17. 1517 e1–10. doi: 10.1053/j.gastro.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]