

Abstract
Herein we have shown that electrodeposited NiSe2 can be used as a bifunctional electrocatalyst under alkaline conditions to split water at very low potential by catalyzing both oxygen evolution and hydrogen evolution reactions at anode and cathode, respectively, achieving a very high electrolysis energy efficiency exceeding 80% at considerably high current densities (100 mA cm−2). The OER catalytic activity as well as electrolysis energy efficiency surpasses any previously reported OER electrocatalyst in alkaline medium and energy efficiency of an electrolyzer using state-of-the-art Pt and RuO2 as the HER and OER catalyst, respectively. Through detailed electrochemical and structural characterization, we have shown that the enhanced catalytic activity is attributed to directional growth of the electrodeposited film that exposes a Ni-rich lattice plane as the terminating plane, as well as increased covalency of the selenide lattice which decreases the Ni(II) to Ni(III) oxidation potential. Thereby, the high efficiency along with extended stability makes NiSe2 as the most efficient water electrolyzer known to-date.
Introduction
Water splitting reactions producing readily useable clean fuel H2, has been the focus of major research activities in alternative energy since they are benign in terms of impact on the environment and human health. The oxygen and hydrogen evolution reactions (OER and HER, respectively), an intricate part in water oxidation/reduction, respectively, also plays a crucial role in other alternative energy devices including fuel cells, metal-oxygen batteries and solar water splitting devices1–3. Among these the oxygen evolution reaction (OER) occurring at the anode is a major hurdle since it is a kinetically sluggish process that involves 4 electron transfer associated with the formation of dioxygen molecule from water, and requires a large anodic potential4. Therefore, this OER half reaction is regarded as the most limiting step for water splitting, and electrocatalysts/photocatalysts are typically used to reduce the applied potentials for O2 evolution. The most commonly used high-efficiency catalysts are the precious metal oxides (IrOx, RuOx) which exhibit some of the lowest overpotential for practical current densities5–12. In addition significant advances have been recently made in identifying OER electrocatalysts13 based on transition metal compounds including alloys (often containing significant amounts of Ni, Co or Fe)5, oxide/hydroxide6–8, phosphide9, spinels5, 10, 11 and perovskite oxides5, 12. Among these, Ni-based oxides, oxyhydroxides, and hydroxides, along with Ni-Fe based oxyhydroxides have shown the most potent catalytic efficiencies with performance closer to that of precious metal oxides14–17. Very recently transition metal chalcogenides has been receiving increasing interest as OER and HER catalysts18–21. Compared to the oxides, the chalcogenides are expected to have more covalency, smaller bandgaps, and better band alignment with water redox levels for efficient charge transfer, thus promoting better catalytic efficiency. In particular Ni- and Co-based chalcogenides have shown great promise for water electrolysis in alkaline medium18–25. Indeed we have identified a nickel selenide, Ni3Se2 which shows a very low onset potential and overpotential at 10 mA cm−2 for oxygen evolution in alkaline medium20. Other groups have also reported chalcogenides such as NiSx as a bifunctional HER and OER active catalysts18. The bifunctional electrocatalysts that can be applied both at the cathode and anode and produce hydrogen and oxygen, respectively, are highly appealing technologically for water electrolysis22.
In this article we report the highly efficient OER and HER catalytic activity of electrodeposited pure NiSe2 films and emphasize the importance of film orientation and growth conditions on the catalytic activity. The OER catalytic activity in these films with the onset potential for O2 evolution at 1.36 V (vs RHE) and overpotential at 10 mA cm−2 at 140 mV in alkaline medium, was observed to be superior to any other OER electrocatalysts reported till date including state-of-the-art precious metal oxides, transition metal oxides, and other nickel chalcogenides. There has been another report of the OER catalytic activity of NiSe2 (coated with native oxide layer) albeit with much higher overpotential at 10 mA cm−2 (~290 mV)18, 23. This difference can be attributed to several factors including the surface chemistry, phase purity and the preferential growth direction which influences exposure of the catalytic sites to the electrolyte as discussed below. The later proposition has been conclusively proven in this manuscript whereby, we have grown randomly oriented NiSe2 grains through hydrothermal methods and have shown their catalytic activity for OER to be less efficient than the electrodeposited oriented films, thereby providing a very valuable insight for the OER electrocatalysts. The onset potential for H2 evolution was also very low as compared to other non-Pt based HER electrocatalyst. The NiSe2 bifunctional electrocatalyst reported here could effectively split water at 1.43 V and achieve an electrolysis energy efficiency of 83% producing current density as high as 100 mA cm−2, which is the highest that has been obtained so far for a bifunctional water electrolyzer. Electrodeposition being a facile, direct method to grow large area binder-free catalytic films, and the experimental proof that films with preferred orientation will show enhanced catalytic activity indeed highlights the novelty of this report.
Results and Discussion
Structural and Morphological Characterization
As mentioned above and described in the experimental section in Supporting Information, NiSe2 electrocatalysts were grown by two methods – electrodeposition on Au-coated glass and by hydrothermal techniques. The powder X-ray diffraction patterns (PXRD) of as-synthesized NiSe2 films electrodeposited on Au-coated glass substrates as well as NiSe2 powder synthesized hydrothermally showed peaks that matched well with standard NiSe2 (PDF # 00-041-1495) (Fig. 1). The electrodeposited film also showed peaks characteristic of Au arising from the underlying substrate. The peaks were considerably broader indicating that the films were composed of nanostructured grains. Using the Scherrer formula (See Supplementary Information), average particle size in the electrodeposited film was estimated as 30.0 nm.
Figure 1.

PXRD pattern of electrodeposited NiSe2 @ Au (blue) and NiSe2 powder (red) showing the presence of NiSe2 (PDF # 00-041-1495).
For the electrodeposited films, the catalyst loading was measured by weighing the electrode before and after electrodeposition, while the thickness of the catalytic film was estimated from 3-dimensional profiling (Supplementary Fig. 1). Table 1 lists the typical catalyst loading and average thickness of the electrodeposited film. The morphology of the as-synthesized films was investigated through detailed SEM and TEM studies (Fig. 2a and b) which showed that they were indeed composed of nanostructured grains. The TEM and HRTEM images, Fig. 2b and c, respectively, suggests that the film contained mainly nanocrystallites where individual grains were single crystalline showing lattice fringes corresponding to 〈211〉 planes of NiSe2. Selected area electron diffraction (SAED) collected from these nanocrystallites showed diffraction spots corresponding to 〈200〉 and 〈023〉 lattice planes of NiSe2 (Fig. 2d). The NiSe2 granules were free of any amorphous or heterogeneous coating on the surface as apparent from the TEM images, also confirming the purity of these nanograins. The composition of the electrodeposited films were confirmed through EDS (Supplementary Fig. 2) which confirmed the presence of Ni and Se in the film with an approximate ratio of 1:2 and validated the composition as NiSe2. The XPS binding energy observed at 853.3 and 870.4 eV as shown in Fig. 2e, corresponds well to the Ni 2p3/2 and Ni 2p1/2 respectively, similar to that obtained from NiSe2 26, 27, while, binding energies of Se 3d5/2 and Se 3d3/2 observed at 54.0 eV and 54.6 eV (Fig. 2f), respectively were similar to the reported values for nickel selenides20, 23. It should be noted that the peaks corresponding to metal oxide and/or selenium oxide were not observed in the XPS spectrum, indicating that the electrodeposited films were devoid of oxidic impurities.
Table 1.
Electrochemical parameters of the NiSe2 catalysts measured in 1.0 M KOH.
| Catalyst | Loading/mg/cm2 | ECSA/cm2 | RF | Onset potential/Va | η to 10 mAcm−2/Va | Tafel slope/mV dec−1 |
|---|---|---|---|---|---|---|
| NiSe2/Au | 0.13 | 4.7 | 16.6 | 1.36 | 0.14 | 48.7 |
| NiSe2 (powder)/CFP | 1.40 | — | — | 1.38 | 0.22 | 56.6 |
| RuO2/Au | — | — | — | 1.43 | 0.32 | 121.1 |
aPotential reported with respect to RHE.
Figure 2.
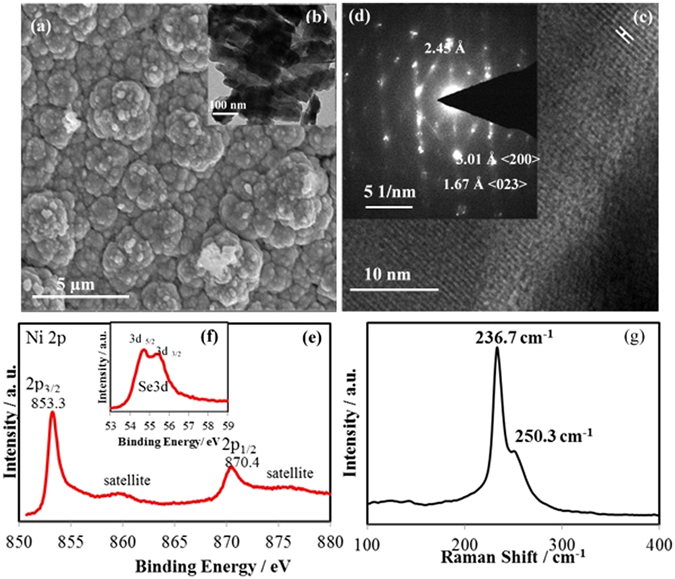
SEM (a) and TEM (b) images of electrodeposited NiSe2, (c) HRTEM image of the catalyst showing lattice spacing of 2.45 A corresponding to <211> lattice planes, (d) Typical SAED pattern showing characteristic diffraction spots, (e) XPS spectra of the catalyst showing the Ni 2p peaks, (f) XPS peaks corresponding to Se 3d. (g) Raman spectrum of the catalyst.
The electrodeposited films were also studied with Raman spectroscopy which showed peaks at 236.7 and 250.3 cm−1 characteristic of NiSe2 as shown in Fig. 2g 28, 29. The spectrum shows no other major Raman lines attributed to the Se phase and/or Ni–O modes20. All of these characterization indicates that the electrodeposited film was purely NiSe2 with no discernible impurity phases.
Electrochemical Performance, OER and HER Catalytic Activities
Electrochemical characterization of the electrodeposited NiSe2 films was conducted in N2-saturated 1 M KOH (pH 13.6) with a scan rate at 10 mV s−1 using a 3-electrode setup. The actual catalytically active region of the material was estimated from the electrochemical surface area (ECSA) measurement (see Supplementary Information, Supplementary Fig. 3) and provided in Table 1.
The activity of the catalyst for OER was determined from LSVs and CVs (Fig. 3a and b) by measuring the onset potential and overpotential at 10 mA cm−2 (vs RHE). All the potentials have been reported after iR compensation. Current density with bare Au-glass electrode and RuOx coated electrodes were also measured for comparison. As expected bare Au-coated glass slide showed very minimal catalytic activity for OER, while RuO2 coated slide showed an onset potential of 1.43 V (vs. RHE) and overpotential of 320 mV at 10 mA cm−2, which matches perfectly with the reported values8. It was observed that NiSe2 on Au-coated glass electrode exhibits a very low onset potential of about 1.36 V, reaching current density 100 mA cm−2 at overpotential of about 200 mV. It must be mentioned here that these value of onset and overpotentials were very reproducible and different batches of NiSe2 films on Au-glass showed similar values. The CV for NiSe2 showed a large pre-oxidation peak before the increase of current density due to oxygen evolution. For Ni-based OER electrocatalysts, the pre-oxidation peak typically attributed to the Ni2+ to Ni3+ oxidation has been observed previously. The intensity of the pre-oxidation peak was considerably large, possibly due to exposure of large number of catalytically active sites and high surface roughness that may lead to a high ECSA of the catalyst. Typically, activities of the OER electrocatalysts are benchmarked by comparing the overpotential at current densities of 10 mA cm−2 (per geometric surface area), which is considered to be equivalent to 10% solar water-splitting device under 1 sun illumination30. In the present case, however, the onset of OER activity as well as the overpotential at 10 mA cm−2 was heavily masked by the large pre-oxidation peak and the overpotential at 10 mA cm−2 was obtained from the cyclic voltammogram (CV) by analyzing the current density of the reverse scan which corresponds to the OER current only (Fig. 3b). Accordingly, it was observed that a current density of 10 mA cm−2 was obtained at an overpotential of 140 mV. This value is much better than the RuO2 (320 mV overpotential required to get 10 mA cm−2 current density) and is the lowest value reported till date amongst all the known OER electrocatalysts as well as amongst the recently reported chalcogenides (see Supplementary Table 1)15. To the best of our knowledge, this is the first time that pure NiSe2 has been reported as an active electrocatalyst for OER with such a low onset potential as well as overpotential at 10 mA cm−2. To understand the role of catalyst growth conditions, we have also measured the catalytic activity of NiSe2 grown through hydrothermal methods. For this purpose, the NiSe2 powder was mixed with Nafion and drop-casted on carbon fiber paper (CFP) and used as an anode in the OER set-up (see Supporting Information for details). The CV plot showed that NiSe2 powder on CFP was still catalytically active for O2 evolution with a low onset potential (1.38 V) and overpotential at 10 mA cm−2 (220 mV) (Fig. 3b) indicating that this is indeed an intrinsic property of this material. Noticeably, the catalytic activity of hydrothermally grown pure NiSe2 powder was better than previous reports of NiSe2 23, possibly due to the nanostructured morphology of the NiSe2 grains as has been frequently obtained with hydrothermal synthesis. Recently we have also shown that nanostructuring increases catalytic activity31. However the catalytic activity was inferior to the electrodeposited NiSe2 film, thereby, emphasizing the fact that growth conditions indeed play a vital role in determining the catalytic activity of these electrocatalysts (vide infra). A small value of Tafel slope of 48.7 mV dec−1 for NiSe2 compared to RuO2 (121.2 mV dec−1) confirmed that the catalytic process was very facile. All the parameters defining the electrocatalytic activity of NiSe2 has been listed in Table 1.
Figure 3.

(a) LSVs measured at NiSe2 (Electrodeposited) @ Au, NiSe2 (Hydrothermal) @ CFP, RuO2 @ Au and bare Au at in N2 saturated 1.0 M KOH solution at a scan rate of 10 mV s−1. (b) CVs measured under identical condition to estimate the overpotential at 10 mA cm−2. (c) Tafel plots of catalysts.
A common concern with the Ni-based OER electrocatalysts in the alkaline medium is that the catalytic activity is obtained from the oxide-hydroxide phases formed in situ. To address this issue, we have intentionally electrodeposited Ni(OH)2 films following a reported procedure on Au-coated glass. The electrocatalytic activity of the Ni(OH)2 was measured in the same way as described above. It was observed that the onset potential for O2 evolution as well as overpotential at 10 mA cm−2 was much higher at 1.49 V vs RHE and 330 mV, respectively (Supplementary Fig. 4). This proves that the highly efficient catalytic activity obtained with the electrodeposited NiSe2 is definitely an intrinsic property of the selenide itself and not merely arising from the oxide-hydroxide. This claim has been supported by other electrochemical studies (vide infra).
The composition of the evolved gas was confirmed to be O2 through further electrochemical studies using rotating ring disk electrodes (see Supplementary Information and Supplementary Fig. 5a) and the Faradaic efficiency was observed at about 99.5% at the applied disk potential of 1.4 V (vs. RHE) (see Supplementary Information and Supplementary Fig. 5b), that corresponds to about 1.0 mA cm−2 disk current density. As the disk voltage increased to 1.46 V (vs. RHE), the Faradaic efficiency decayed to 50.2%. This decrease could be attributed to large amounts of oxygen being produced at the disk electrode that cannot be efficiently collected by the Pt ring electrode20.
Stability of the catalysts under conditions of continuous O2 evolution (in 1.0 M KOH) was investigated using constant potential electrolysis (chronoamperometry) for 24 h where the potential was held at a constant value of (1.37 V vs RHE) to deliver 10 mA cm−2 per geometric area (Fig. 4a). As can be seen from the chronoamperometry data, there was no degradation of the current density with time indicating extended catalyst stability. It should be noted here that the constant potential of 1.37 V was chosen based on the reverse scan of the CV plot which provided a better estimate of the potential needed for achieving a 10 mA cm−2 (Fig. 3b). A separate set of stability studies was conducted by holding the potential constant in several stages of the OER reaction (from onset to rapid O2 evolution stage) and the current density was measured at each of these steps for extended period. As can be seen from Fig. 4c, the current density at 1.28 V was almost 0, which can be expected from the LSV curve since it falls below the onset potential. As the potential step was increased to 1.34 and 1.37 V, the current density showed a steady step-wise increase to ~10 mA cm−2 where it stayed stable for the duration of the potential step. As the potential step was increased further to 1.39 V and 1.41 V respectively, the current density showed a much steeper increase indicating that OER was indeed the major process in these potential ranges. The current density at 1.41 V was noisy possibly due to evolution of large amounts of O2 which led to accumulation of bubbles near the electrode surface and had to be dislodged with mechanical agitation, thereby producing a noisy data. The composition of the film after chronoamperometry and extended periods of continuous O2 evolution was analyzed through PXRD (Fig. 4d) and XPS (Fig. 4e) studies which revealed that the films were still predominantly NiSe2 even after 24 h of continuous O2 evolution. However, a small intensity peak was also observed in the pxrd pattern that corresponds to Ni0.85 Se, indicating that there might be some Se loss from the films during extended period of O2 evolution leading to formation of a selenium deficient phase. The formation of selenium deficiency on the surface after OER activity was also evidenced further by EDS (Supplementary Fig. 6) as the ratio of Ni to Se increased slightly with progress of reaction.
Figure 4.
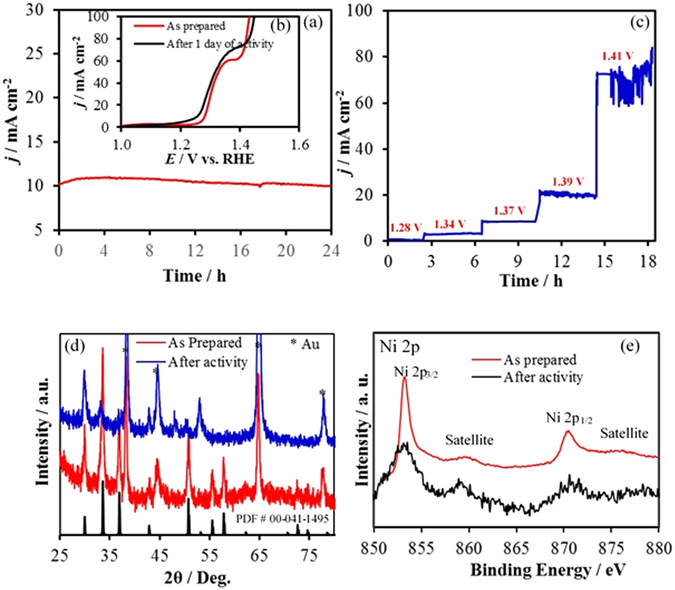
(a) Stability study of catalyst under continuous O2 evolution at a constant potential to achieve 10 mA cm−2 for 24 h, (b) LSVs of catalyst in N2 saturated 1.0 M KOH before and after chronoamperometry for 24 h, (c) Stability of catalyst at different potential by using potential step method. (d) XRD patterns before and after current transient experiment for 24 h and (e) XPS spectra before and after of stability.
XPS studies on the other hand, revealed that the Ni and Se (Supplementary Fig. 7) XPS signals were unaltered after the chronoamperometric studies indicating that the catalyst did not undergo a drastic decomposition. Also oxidic peaks were notably absent in the XPS spectra, demonstrating that the catalytic film did not undergo conversion to the oxide, as has been reported for some selenides18, 23. The O 1 s signal was also monitored through XPS before and after chronoamperometry (Supplementary Fig. 8) which revealed that there was no evidence of Ni-oxides. In the as-synthesized sample, the O 1 s peak showed only surface adsorbed oxygen. The deconvoluted O 1 s peak after chronoamperometry showed evidence of surface-absorbed oxygen along with physisorbed H2O and traces of Se-oxide.
Mechanism
As was observed with the OER catalytic process, the Ni2+ → Ni3+ oxidation preceded the oxygen evolution reaction indicating that Ni3+ may be the actual catalytic center as has been reported by other researchers for Ni-based OER electrocatalysts6, 7, 31, 32. However, in NiSe2, the Ni2+/Ni+3 oxidation potential shows a shift towards lower value as compared to that observed in the Ni-based oxides or oxide-hydroxides as seen from the LSVs. We have probed this point further by collecting cyclic voltammograms (CVs) with electrodes coated with pure NiSe2 and Ni(OH)2 in 1.0 M KOH with a three electrode set-up. It should be noted here that to check the oxidation peak, the CVs were measured on electrodes prepared with less NiSe2 deposition time (5 min), such that the Ni2+ → Ni3+ oxidation peak was not masked by the stronger OER peak. It was observed that the Ni2+ to Ni3+ oxidation indeed occurs at much lower potential (1.345 V vs. RHE) compared to that in Ni(OH)2 (1.395 V vs. RHE), indicating that the catalytically active species (Ni3+) is generated at a lower potential in the NiSe2 electrode (Fig. 5). Such kind of downward shift of the oxidation peak potential can be expected by considering the effect of surrounding ligands on the oxidation potential of the central metal atom. In fact, the electronegativity values of Ni (1.91) and Se (2.55) are very close to each other indicating that as the ligands change from oxo-based to seleno-based, the covalent nature of the metal-chalcogen bond increases. The observation of a lower Ni2+ to Ni3+ oxidation potential in seleno-based coordination environment is also supported by our recent studies with a seleno-based molecular Ni coordination complex containing tetrahedral NiSe4 core, similar to that found in most Ni-selenides33. Single crystals of this complex showed that Ni2+ to Ni3+ oxidation occurred at 1.34 V (vs Ag|AgCl) in 1.0 M KOH, confirming that the low Ni2+ → Ni3+ oxidation potential observed in NiSe2 is indeed due to the selenide coordination. The increasing covalency in the selenides compared to oxides is also supported from Fajan’s rule34. This increase in covalency leads to several important consequences including a decreased bandgap of the selenides as compared to that of oxides (3.5 eV) and an upward shift of the valence band edge. These changes in the orbital energy levels will directly influence their alignment with respect to the water oxidation and reduction levels, which in turn will affect the charge transfer between the catalyst and water. For the water splitting catalysts, one of the most influential factors in light of the electronic band structure is that water oxidation-reduction levels are bracketed within the valence and conduction band edges of the catalyst. In these electrocatalytic systems charge transfer occurs at the semiconductor (catalyst)-electrolyte interface which will be influenced by the relative energy levels of the semiconductor and aqueous electrolyte, and an efficient charge transfer will occur when these two levels are closer in energy, thereby facilitating the catalysis mechanism as proposed above.
Figure 5.

CVs measured at NiSe2 and Ni(OH)2 catalysts in N2 saturated 1.0 M KOH at 10 mV s−1. The Ni(OH)2 CV has been scaled by a factor of 8.
Nickel selenides are an interesting family of compounds and have been researched by solid state chemists for a long time owing to their novel electronic as well as magnetic properties35. Several stoichiometries of the nickel selenides has been known including NiSe, NiSe2, Ni3Se2, Ni3Se4, and Ni7Se8 36. Among these, NiSe2 has a pyrite structure (space group Th 6(Pa3)) with dumbbell-shaped Se2 units between two Ni atoms36. Most of these nickel selenides are reported to be narrow bandgap semiconductors or semimetals. Previous electronic band structure calculations reported zero bandgap for NiSe2 as obtained using GGA, exact exchange hybrid functional, and GW approximation37. Another factor that might enhance the catalytic activity is easy access to the active site, which in turn can be influenced by growth direction of the film. Hence, the preferential growth direction of the film was analyzed by studying the texture of the film from PXRD data. Specifically, to perform this kind of texturing study, a symmetric diffraction was utilized, where incident and detector angles were always equal. Under these conditions, diffraction from planes that are oriented parallel to the sample surface was only observed. This is the same general method used for powder samples. However, for a powder, all expected reflections are observed as the powder itself consists of randomly oriented grains. On the other hand, if texturing is present in the sample, it implies that there is a preferential growth direction, and the plane normal to that growth direction will predominate in the diffraction pattern. In this case, the pattern obtained was consistent with (311)-textured NiSe2 (Fig. 6) which crystallizes with a primitive cubic structure, penroseite25. Substantial (311) texture was evident by the enhanced intensity of the (311) reflection at 2θ~50.5° when compared to the intensity of the (210) reflection at 2θ~33.5° while the underlying Au substrate showed a 〈111〉 orientation as shown in Fig. 6a. In a randomly oriented film, based on the theoretical PXRD generated from the atomic coordinates, one would expect the (311) reflection to be roughly one-third the intensity of the (210) as has been observed in Fig. 1. Our results suggested that (311) planes are preferentially oriented parallel to the growth direction of the film. In other words, the average terminating planes of the film were the 〈311〉 lattice planes of NiSe2. Similar texturing studies on the hydrothermally grown NiSe2 did not show any preferred orientation. The Au surface with a preferred orientation along the 〈111〉 direction may play a substantial role in determining the growth direction of the film. Similar results has been recently reported by Switzer et al. by growing preferentially oriented Cu2O grains on Au-coated silicon substrates38. Figure 6b shows the (311) plane of the NiSe2 lattice and interestingly it can be seen that this crystallographic plane is Ni-rich, and can be considered as predominantly Ni-terminated plane. Since, Ni is the catalytically active center for OER, the anion deficient terminating lattice plane will facilitate the attachment of the hydroxyl anions to the metal center leading to a low onset potential and low Tafel slope. The Ni-rich terminating lattice plane may also reduce Se loss with prolonged OER catalytic activity thereby reinforcing catalyst’s stability. In fact, recently, another group has reported OER electrocatalytic activities of NiSe2 in alkaline medium albeit with much higher overpotential (250 mV)23. They have also shown through in situ Raman measurements that the catalytic activity proceeds with the immobilization of OH− on the surface. However, they have also mentioned that their as-prepared catalyst had a surface coating of oxide-hydroxide. That may be the reason the overpotential showed a higher value than the pure NiSe2 reported in this study. As we have shown above, the generation of the catalytically active center (Ni3+) is much more facile in the chalcogenide lattice than the oxide matrix. The higher conductivity of the inner NiSe2 lattice will aid in faster electron transfer leading to higher current densities even at low potentials. Hence, the enhanced intrinsic OER catalytic activity in NiSe2 can be explained from the increased covalency in the lattice which affects the electronic band structure of NiSe2, band positioning, zero band gap of the material, while the highly efficient catalytic activity of the electrodeposited film can be explained from the preferential growth during electrodeposition. It should be also noted that the catalyst loading in this report was very comparable and in fact lower than that reported for other OER electrocatalysts (Supplementary Table 1). Nevertheless, to rule out any effect of catalyst loading on the OER activity, we have compared the catalytic activity between the electrodeposited 〈311〉 -textured NiSe2 film and hydrothermally synthesized NiSe2 powder containing identical loading. As can be seen from the CV plot (Supplementary Fig. 9), there was no change of overpotential with variation in catalyst loading, however, the current density increased with increased loading. More importantly, the overpotential for the textured NiSe2 film showed the same difference with that obtained for the NiSe2 powder, indicating that this difference in activity is intrinsic.
Figure 6.

(a) Pxrd pattern of the (311) textured NiSe2 film. Inset shows the crystal structure with the marked (311) plane. (b) An illustration of the NiSe2 lattice terminated with the (311) plane showing excess Ni atoms on the surface.
Electrocatalytic performance for HER and Overall Water Splitting
Electrodeposited NiSe2 showed considerable catalytic activity for HER in 1 M KOH measured using a three-electrode system with a scan rate of 10 mV s−1 and was compared with commercially available Pt electrocatalyst. As shown in the polarization curve (j s vs. E), NiSe2 required an overpotential [η(HER)] of 170 mV to achieve 10 mA cm−2 (Fig. 7a), which is slightly higher than that observed with Pt39. A Tafel slope of 107 mV dec−1 was obtained for the HER process (Fig. 7b). Non-precious HER catalysts often exhibit Tafel slopes ranging from 40 to 120 mV dec−1 40–49.
Figure 7.

(a) LSVs measured at NiSe2/Au and Pt in N2 saturated 1.0 M KOH solution at a scan rate of 0.01 V s−1. Inset shows the HER stability of NiSe2. (b) Tafel plots of catalysts.
Remarkably, an early onset overpotential (50 mV vs. RHE), indicated that the negative current increased rapidly under more cathodic potentials. Such low onset potential places NiSe2 amongst the most active non-precious HER catalysts in an alkaline medium50–52. Previous report of HER activity with NiSe2 corroborates very well with our report signifying the accuracy of the results. Chronoamperometric studies for continuous H2 evolution for 6 h showed that the catalyst was also stable for HER activities in 1.0 M KOH for extended time (inset of Fig. 7a). To confirm that the composition of the evolved gas was indeed H2 we designed an electrochemical experiment where the evolved gas was oxidized at Pt electrode. Specifically, a series of HER experiments were carried out in 1.0 M KOH, where H2 was generated for different time intervals (0.5, 1 and 3 h) at NiSe2/Au. After periods of continuous gas evolution for certain periods of time, the evolved gas was oxidized at Pt electrode. As has been shown previously, Pt can electrochemically oxidize H2 at 0.0 V53. Hence monitoring the H2 oxidation potential and oxidation current could confirm the composition of the gas was indeed H2. Initially the electrolyte was purged with N2 and as expected the current density for H2 oxidation at Pt electrode was very close to 0. After the HER reaction was done for 0.5, 1 and 3 h, the oxidation current density increased indicating the increasing concentration of dissolved H2 in the electrolyte (Fig. 8). This was further confirmed by measuring the oxidation current after purging the electrolyte with N2 after 3 h of HER to get rid of any dissolved gas. As expected, the oxidation current reduced back to the initial value after purging with N2. This simple experiment proved conclusively that H2 was indeed produced at the NiSe2 electrode. The composition of the evolved gas was further confirmed as H2 following the well-established catalytic hydrogenation of para-nitrophenol (PNP) to para-aminophenol (PAP). The hydrogenation reaction was performed as described in supporting information, and the progress of reaction was monitored through UV-Vis spectroscopy since PNP and PAP shows distinctly different λmax values (Supplementary Fig. 10). When the electrolyte was maintained at conditions for HER, it was clearly observed that the absorbance peak due to PNP showed a gradual decrease accompanied by an increase of the PAP absorbance (Fig. 8b) indicating conversion of PNP to PAP. After 5 h, the conversion of PNP to PAP was complete and the solution turned nearly colorless. Two separate control experiments involved passing H2 through the reaction mixture in the absence of applied voltage and maintaining a blank electrode (Au-glass) under the HER conditions mentioned above. In both of these control experiments, absorbance of PNP did not show any detectable change even after 5 h. This indicates that not only H2 is being produced by NiSe2, but it is actually forming catalytically active hydrogen capable of performing liquid phase hydrogenation under ambient conditions. This itself is a significant achievement with long-term technological impact.
Figure 8.
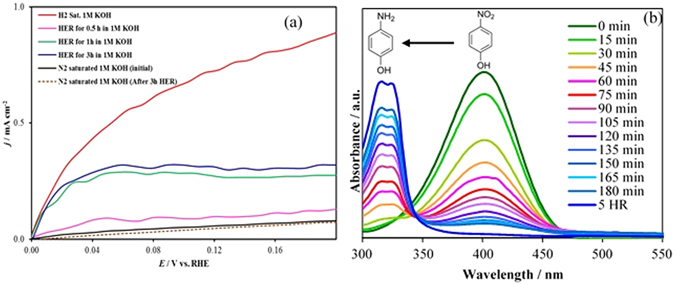
(a) Evidence of H2 evolution was confirmed by electrochemical oxidation of hydrogen in a Pt electrode. Initially HER was performed at NiSe2/Au at a constant potential of −0.2 V vs RHE for different periods of time (0.5, 1 and 3 h) under N2 saturated and blanketed 1.0 M KOH. The evolved gas was then oxidized with Pt electrode, whereby the oxidation potential and current density confirmed the presence of H2. In the absence of H2 [before HER, (black solid line) and after purging with N2 (reddish dashed line)] the current density was very close to zero. (b) Catalytic hydrogenation of p-nitrophenol to p-aminophenol monitored by the change in UV-visible absorbance spectra, confirming the evolution of active hydrogen.
The Faradic efficiency for HER process was calculated by quantifying the amount of H2 produced and comparing it with the theoretical yield (details has been provided supplementary information). A Faradaic efficiency close to 100% was obtained for HER (Supplementary Fig. 11).
Since NiSe2 shows high catalytic activity for both OER and HER processes in alkaline medium, we have designed a full water splitting system using NiSe2 for both anode and cathode. This bifunctional catalyst exhibits high performance towards overall water splitting and could deliver a current density of ~6 mA cm−2 with an applied cell voltage of 1.5 V in 1 M KOH (Fig. 9) during which bubbles of O2 and H2 were steadily released at both electrodes. It should be noted here that although the thermodynamic water splitting voltage is 1.23 V, it is reckoned that an ideal electrolyzer should be able to split water at 1.48 V for maximum efficiency without any heat exchange with the surrounding, also referred to as the thermoneutral potential54, 55. In the present case, water electrolysis was carried out in a closed electrochemical cell containing NiSe2 as the anodic and cathodic catalyst without any external source of heat. Electrolysis energy efficiency is calculated as the ratio between 1.48 V and the water splitting cell voltage. Electrolysis energy efficiency of the NiSe2 catalyst was calculated to be ~100% (Supplementary Table 2) at the onset potential of electrolysis (1.43 V), while at higher current densities (100 mA cm−2) the efficiency was 83%. This was compared with the energy efficiency obtained from an electrolyzer containing Pt as the cathode catalyst for HER and RuO2 as the anodic catalyst for OER, which gave a cell voltage of 1.83 V at 100 mA cm−2 (81% efficiency). In comparison with the reported literature, as a bifunctional catalyst, NiSe2 exhibits the highest electrolysis energy efficiency obtained so far under ambient conditions and is closest to the elusive most efficient and green water electrolyzer.
Figure 9.

Demonstration of water-splitting device driven by a DC power supply at a cell voltage of 1.5 V to deliver a current density of ~6 mA cm−2.
Conclusions
In summary, we have electrodeposited nanostructured NiSe2 films as an efficient bifunctional electrocatalyst for overall water splitting with energy efficiency exceeding 83% at high current density. As-deposited NiSe2 reported here shows the lowest onset potential and overpotential at 10 mA cm−2 amongst all the Ni-based OER electrocatalysts (Supplementary Table 3). Through electrochemical measurements and structural characterization, we have proven that this enhancement in primarily due to lowering of the oxidation potential of Ni2+ to Ni3+ that in turn is a consequence of changing the oxide lattice to the more covalent selenide lattice, as well as the preferential growth direction of the film which exposes a Ni-rich surface as the terminating lattice plane.
Electronic supplementary material
Author Contributions
M.N. wrote the manuscript text with some contributions from A.T.S. and J.M. A.T.S. did most of the experiments. J.M. interpreted the data. S.U. performed the hydrothermal reaction and provided data for Figures 1, 3a and b. W.P.L. designed and collected data for Figure 8b. E.B. designed the texturing pxrd experiment and analyzed data presented in Figure 6a. Figures were prepared by A.T.S. (Figures 1, 2a, 3a,b, 4a,b,c,d, 5, 7a and 9), J.M. (Figures 2c, d,e,f,g, 3c, 4e, 7b and 8), W.P.L. (Figure 6, Figure 8b). J.E.M. helped with understanding the band structure calculations and its dependence on directionality.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02285-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chen G, Bare SR, Mallouk TE. Development of supported bifunctional electrocatalysts for unitized regenerative fuel cells. J. Electrochem. Soc. 2002;149:A1092–A1099. doi: 10.1149/1.1491237. [DOI] [Google Scholar]
- 2.Gorlin Y, Jaramillo TF. A bifunctional nonprecious metal catalyst for oxygen reduction and water oxidation. J. Am. Chem. Soc. 2010;132:13612–13614. doi: 10.1021/ja104587v. [DOI] [PubMed] [Google Scholar]
- 3.Cook TR, et al. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 2010;110:6474–6502. doi: 10.1021/cr100246c. [DOI] [PubMed] [Google Scholar]
- 4.Walter MG, et al. Solar water splitting cells. Chem. Rev. 2010;110:6446–6473. doi: 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- 5.Lewis NS, Nocera DG. Powering the planet: Chemical challenges in solar energy utilization. Proc Natl Acad Sci USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong M, Dai H. A mini review of NiFe-based materials as highly active oxygen evolution reaction electrocatalysts. Nano Res. 2015;8:23–39. doi: 10.1007/s12274-014-0591-z. [DOI] [Google Scholar]
- 7.Fabbri E, Habereder, Waltar AK, Kotz R, Schmidt TJ. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2014;4:3800–3821. doi: 10.1039/C4CY00669K. [DOI] [Google Scholar]
- 8.Lee Y, Suntivich J, May KJ, Perry EE, Horn YS. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions. J. Phys. Chem. Lett. 2012;3:399–404. doi: 10.1021/jz2016507. [DOI] [PubMed] [Google Scholar]
- 9.Masud J, Umapathi S, Ashokan N, Nath M. Iron phosphide nanoparticles as an efficient electrocatalyst for the OER in alkaline solution. J. Mater. Chem. A. 2016;4:9750–9754. doi: 10.1039/C6TA04025J. [DOI] [Google Scholar]
- 10.Kinoshita, K. Electrochemical Oxygen Technology (Wiley-Interscience, New York 1992).
- 11.Hall DE. Alkaline water electrolysis anode materials. J. Electrochem. Soc. 1985;132:41C–48C. doi: 10.1149/1.2113856. [DOI] [Google Scholar]
- 12.Hall DE. Ni(OH)2-impregnated anodes for alkaline water electrolysis. J. Electrochem. Soc. 1983;130:317–321. doi: 10.1149/1.2119702. [DOI] [Google Scholar]
- 13.Jasem SM, Tseung ACC. A potentiostatic pulse study of oxygen evolution on teflon‐bonded nickel‐cobalt oxide electrodes. J. Electrochem. Soc. 1979;126:1353–1360. doi: 10.1149/1.2129276. [DOI] [Google Scholar]
- 14.Yu X, et al. Nickel-based thin film on multiwalled carbon nanotubes as an efficient bifunctional electrocatalyst for water splitting. ACS Appl. Mater. Interfaces. 2014;6:15395–15402. doi: 10.1021/am503938c. [DOI] [PubMed] [Google Scholar]
- 15.Zhou WJ, et al. Ni3S2 nanorods/Ni foam composite electrode with low overpotential for electrocatalytic oxygen evolution. Energy Environ. Sci. 2013;6:2921–2924. doi: 10.1039/c3ee41572d. [DOI] [Google Scholar]
- 16.Merrill MD, Dougherty RC. Metal oxide catalysts for the evolution of O2 from H2O. J. Phys. Chem. C. 2008;112:3655–3666. doi: 10.1021/jp710675m. [DOI] [Google Scholar]
- 17.Gewirth AA, Thorum MS. Electroreduction of dioxygen for fuel-cell applications: materials and challenges. Inorg. Chem. 2010;49:3557–3566. doi: 10.1021/ic9022486. [DOI] [PubMed] [Google Scholar]
- 18.Pu Z, Luo Y, Asiri AM, Sun X. Efficient electrochemical water splitting catalyzed by electrodeposited nickel diselenide nanoparticles based film. ACS Appl. Mater. Interfaces. 2016;8:4718–4723. doi: 10.1021/acsami.5b12143. [DOI] [PubMed] [Google Scholar]
- 19.Xu X, Song F, Hu X. A nickel iron diselenide-derived efficient oxygen-evolution catalyst. Nat. Commun. 2016;7:12324. doi: 10.1038/ncomms12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swesi AT, Masud J, Nath M. Nickel selenide as a high-efficiency catalyst for oxygen evolution reaction. Energy Environ. Sci. 2016;9:1771–1782. doi: 10.1039/C5EE02463C. [DOI] [Google Scholar]
- 21.Tang C, Cheng N, Pu Z, Xing W, Sun X. NiSe nanowire film supported on nickel foam: an efficient and stable 3D bifunctional electrode for full water splitting. Angew. Chem. Int. Ed. 2015;54:9351–9355. doi: 10.1002/anie.201503407. [DOI] [PubMed] [Google Scholar]
- 22.Zhu W, et al. Nickel sulfide microsphere film on Ni foam as an efficient bifunctional electrocatalyst for overall water splitting. Chem. Commun. 2016;52:1486–1489. doi: 10.1039/C5CC08064A. [DOI] [PubMed] [Google Scholar]
- 23.Kwak IH, et al. CoSe2 and NiSe2 nanocrystals as superior bifunctional catalysts for electrochemical and photoelectrochemical water splitting. ACS Appl. Mater. Interfaces. 2016;8:5327–5334. doi: 10.1021/acsami.5b12093. [DOI] [PubMed] [Google Scholar]
- 24.Masud J, Swesi AT, Liyanage WPR, Nath M. Cobalt Selenide Nanostructures: An Efficient Bifunctional Catalyst with High Current Density at Low Coverage. ACS Appl.Mater. Interfaces. 2016;8:17292–17302. doi: 10.1021/acsami.6b04862. [DOI] [PubMed] [Google Scholar]
- 25.Kong D, Cha JJ, Wang H, Lee HR, Cui Y. First-row transition metal dichalcogenide catalysts for hydrogen evolution reaction. Energy Environ. Sci. 2013;6:3553–3558. doi: 10.1039/c3ee42413h. [DOI] [Google Scholar]
- 26.Heide HVD, Hemmel R, Bruggen FV, Haas C. X-Ray photodectron spectra of 3d transition metal pyrites. J. Solid State Chem. 1980;33:17–25. doi: 10.1016/0022-4596(80)90543-5. [DOI] [Google Scholar]
- 27.Wagner, C. D. Handbook of X-ray photoelectron Spectroscopy (Perkin-Elmer Corporation: Minnesota 1979).
- 28.Heras CDL, Agulló-Rueda F. Raman spectroscopy of NiSe2 and NiS2−xSex (0 < × < 1) thin films. J. Phys.: Condens. Matter. 2000;12:5317–5324. [Google Scholar]
- 29.Fan H, Zhang M, Zhang X, Qian Y. Hydrothermal growth of NiSe2 tubular microcrystals assisted by PVA. J. Cryst. Growth. 2009;311:4530–4534. doi: 10.1016/j.jcrysgro.2009.08.005. [DOI] [Google Scholar]
- 30.Louie MW, Bell AT. An investigation of thin-film Ni–Fe oxide catalysts for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 2013;135:12329–12337. doi: 10.1021/ja405351s. [DOI] [PubMed] [Google Scholar]
- 31.Swesi AT, Masud J, Nath M. Enhancing electrocatalytic activity of bifunctional Ni3Se2 for overall water splitting through etching-induced surface nanostructuring. J. Mater. Res. 2016;31:2888–2896. doi: 10.1557/jmr.2016.301. [DOI] [Google Scholar]
- 32.Trotochaud L, Ranney JK, Williams KN, Boettcher SW. Solution-cast metal oxide thin film electrocatalysts for oxygen evolution. J. Am. Chem. Soc. 2012;134:17253–17261. doi: 10.1021/ja307507a. [DOI] [PubMed] [Google Scholar]
- 33.Masud, J., Ioannou, P. C., Levesanos, N., Kyritsis, P. & Nath, M. A Molecular Ni-complex Containing Tetrahedral Nickel Selenide Core as Highly Efficient Electrocatalyst for Water Oxidation. ChemSusChem, doi:10.1002/cssc.201601054 (2016). [DOI] [PubMed]
- 34.Douglas, B. E., McDaniel, D. H. & Alexander, J. J. Concepts and Models of Inorganic Chemistry (John Wiley & Sons, Inc. New York 1983).
- 35.Noue I, Yasuoka H, Ogawa S. Paramagnetism in NiSe2. J. Phys. Soc. Jpn. 1980;48:850–856. doi: 10.1143/JPSJ.48.850. [DOI] [Google Scholar]
- 36.Baker, H. ASM Handbook vol. 3: Phase Diagrams (New York 1992).
- 37.Schuster C, Gatti M, Rubio A. Electronic and magnetic properties of NiS2, NiSSe and NiSe2 by a combination of theoretical methods. Eur. Phys. J. B. 2012;85:325. doi: 10.1140/epjb/e2012-30384-7. [DOI] [Google Scholar]
- 38.Switzer JA, Hill JC, Mahenderkar NK, Liu YC. Nanometer-Thick Gold on Silicon as a Proxy for Single-Crystal Gold for the Electrodeposition of Epitaxial Cuprous Oxide Thin Films. ACS Appl. Mater. Interfaces. 2016;8:15828–15837. doi: 10.1021/acsami.6b04552. [DOI] [PubMed] [Google Scholar]
- 39.Luo J, et al. Water photolysis at 12.3% efficiency via perovskite photovoltaics and earth-abundant catalysts. Science. 2014;345:1593–1596. doi: 10.1126/science.1258307. [DOI] [PubMed] [Google Scholar]
- 40.Jaramillo TF, et al. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science. 2007;317:100–102. doi: 10.1126/science.1141483. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, et al. MoS2 nanoparticles grown on graphene: an advanced catalyst for the hydrogen evolution reaction. J. Am.Chem. Soc. 2011;133:7296–7299. doi: 10.1021/ja201269b. [DOI] [PubMed] [Google Scholar]
- 42.Chen Z, et al. Core–shell MoO3–MoS2 nanowires for hydrogen evolution: a functional design for electrocatalytic materials. Nano Lett. 2011;11:4168–4175. doi: 10.1021/nl2020476. [DOI] [PubMed] [Google Scholar]
- 43.Kibsgaard J, Chen Z, Reinecke BN, Jaramillo TF. Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis. Nat. Mater. 2012;11:963–969. doi: 10.1038/nmat3439. [DOI] [PubMed] [Google Scholar]
- 44.Lukowski MA, et al. Enhanced hydrogen evolution catalysis from chemically exfoliated metallic MoS2 nanosheets. J. Am. Chem. Soc. 2013;135:10274–10277. doi: 10.1021/ja404523s. [DOI] [PubMed] [Google Scholar]
- 45.Laursen AB, Kegnaes S, Dahl S, Chorkendorff I. Molybdenum sulfides-efficient and viable materials for electro- and photoelectrocatalytic hydrogen evolution. Energy Environ. Sci. 2011;5:5577–5591. doi: 10.1039/c2ee02618j. [DOI] [Google Scholar]
- 46.Merki D, Hu X. Recent developments of molybdenum and tungsten sulfides as hydrogen evolution catalysts. Energy Environ. Sci. 2011;4:3878–3888. doi: 10.1039/c1ee01970h. [DOI] [Google Scholar]
- 47.Chen WF, et al. Highly active and durable nanostructured molybdenum carbide electrocatalysts for hydrogen production. Energy Environ. Sci. 2013;6:943–951. doi: 10.1039/c2ee23891h. [DOI] [Google Scholar]
- 48.Popczun EJ, et al. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2013;135:9267–9270. doi: 10.1021/ja403440e. [DOI] [PubMed] [Google Scholar]
- 49.Brown DE, Mahmood MN, Man MCM, Turner AK. Preparation and characterization of low overvoltage transition metal alloy electrocatalysts for hydrogen evolution in alkaline solutions. Electrochim. Acta. 1984;29:1551–1556. doi: 10.1016/0013-4686(84)85008-2. [DOI] [Google Scholar]
- 50.Tang C, Pu Z, Liu Q, Asiri AM, Sun X. NiS2 nanosheets array grown on carbon cloth as an efficient 3D hydrogen evolution cathode. Electrochim. Acta. 2015;153:508–514. doi: 10.1016/j.electacta.2014.12.043. [DOI] [Google Scholar]
- 51.Tang C, et al. Ni3S2 nanosheets array supported on Ni foam: A novel efficient three-dimensional hydrogen-evolving electrocatalyst in both neutral and basic solutions. Int. J. Hydrogen Energy. 2015;40:4727–4732. doi: 10.1016/j.ijhydene.2015.02.038. [DOI] [Google Scholar]
- 52.Gao MR, et al. An efficient molybdenum disulfide/cobalt diselenide hybrid catalyst for electrochemical hydrogen generation. Nat. Commun. 2015;6:5982. doi: 10.1038/ncomms6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar SMS, Hidyatai N, Herrero JS, Irusta S, Scott K. Efficient tuning of the Pt nano-particle mono-dispersion on Vulcan XC-72R by selective pre-treatment and electrochemical evaluation of hydrogen oxidation and oxygen reduction reactions. Int. J. Hydrogen Energy. 2011;36:5453–5465. doi: 10.1016/j.ijhydene.2011.01.124. [DOI] [Google Scholar]
- 54.Kreuter W, Hofmann H. Electrolysis: the important energy transformer in a world of sustainable energy. Int. J. Hydrogen Energy. 1998;23:661–666. doi: 10.1016/S0360-3199(97)00109-2. [DOI] [Google Scholar]
- 55.Wang X, Li W, Xiong D, Liu L. Fast fabrication of self-supported porous nickel phosphide foam for efficient, durable oxygen evolution and overall water splitting. J. Mater. Chem. A. 2016;4:5639–5646. doi: 10.1039/C5TA10317G. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.