

Abstract
Adenine phosphoribosyltransferase (APRT) deficiency is a hereditary disorder that leads to excessive urinary excretion of 2,8-dihydroxyadenine (DHA), causing nephrolithiasis and chronic kidney disease. Treatment with allopurinol or febuxostat reduces DHA production and attenuates the renal manifestations. Assessment of DHA crystalluria by urine microscopy is used for therapeutic monitoring, but lacks sensitivity. We report a high-throughput assay based on ultra-performance liquid chromatography coupled to tandem mass spectrometry (UPLC–MS/MS) for quantification of urinary DHA.
The UPLC–MS/MS assay was optimized by a chemometric approach for absolute quantification of DHA, utilizing isotopically labeled DHA as an internal standard. Experimental screening was conducted with D-optimal design and optimization of the DHA response was performed with central composite face design and related to the peak area of DHA using partial least square regression. Acceptable precision and accuracy of the DHA concentration were obtained over a calibration range of 100 to 5000 ng/mL on three different days. The intra- and inter-day accuracy and precision coefficients of variation were well within ±15% for quality control samples analyzed in replicates of six at three concentration levels. Absolute quantification of DHA in urine samples from patients with APRT deficiency was achieved wihtin 6.5 min. Measurement of DHA in 24 h urine samples from three patients with APRT deficiency, diluted 1:15 (v/v) with 10 mM ammonium hydroxide (NH4OH), yielded a concentration of 3021, 5860 and 10563 ng/mL and 24 h excretion of 816, 1327 and 1649 mg, respectively. A rapid and robust UPLC–MS/MS assay for absolute quantification of DHA in urine was successfully developed. We believe this method will greatly facilitate diagnosis and management of patients with APRT deficiency.
Keywords: APRT deficiency, Clinical mass spectrometry, Chemometrics, D-optimal design, Therapeutic drug monitoring
1. Introduction
Adenine phosphoribosyltransferase (APRT) deficiency (OMIM 102600) is a rare autosomal recessive disorder of purine metabolism, primarily manifested by nephrolithiasis and chronic kidney disease (CKD) [1–3]. The absence of functional APRT prevents recycling of adenine, which in turn is oxidized by xanthine dehydrogenase to the poorly soluble 2,8-dihydroxyadenine (DHA). Precipitation of DHA in the kidneys leads to progressive crystal-associated tubulointerstitial nephropathy and urinary stone formation [4–8]. Since APRT deficiency can be successfully treated with the xanthine dehydrogenase inhibitors allopurinol and febuxostat, early diagnosis and timely initiation of pharmacotherapy prevents stone formation and irreversible loss of kidney function [4–8]. Although the characteristic round, brown DHA crystals are usually readily detected by urine microscopy, they are frequently missed or misidentified [4,5,9]. Infrared spectrophotometry and x-ray crystallography are the recommended methods for detecting DHA in urinary calculi [1,10]. Nevertheless, the authors have observed several cases of false identification of DHA in kidney stone specimens using these techniques. Thus, the diagnosis of APRT deficiency still relies on the demonstration of absent APRT activity in red cell lysates or biallelic pathogenic variants in the APRT gene [11,12]. Monitoring of pharmacological therapy is currently based on microscopic evaluation of crystalluria, with the absence of DHA crystals considered indicative of adequate therapy [12]. Several methods for analysis of urinary DHA concentration have previously been described in the literature, including high-performance liquid chromatography (HPLC) coupled to a multichannel ultraviolet detector [13], capillary electrophoresis (CE) [14–17] and HPLC coupled to tandem mass spectrometry (HPLC–MS/MS) [18]. None of these methods have been established for use in clinical practice. Hence, a rapid and accurate clinical assay for DHA detection is needed to enhance the diagnosis and therapeutic monitoring of patients with APRT deficiency.
Since HPLC–MS/MS has proved to be a highly reliable method for the identification and quantification of various biomarkers [19–21], we developed and report herein a novel ultra-performance liquid chromatography-tandem mass spectrometry (UPLC–MS/MS) assay for absolute quantification of urinary DHA, employing an isotopically labeled DHA as an internal standard. Optimization of the UPLC–MS/MS quantification method was carried out using a chemometric approach [22–25]. The assay was pre-validated in terms of selectivity, sensitivity, concentration function, accuracy, precision, carryover, stability and matrix effect for diluted urine samples [26,27].
2. Experimental
2.1. Chemicals and reagents
DHA (95% pure), manufactured by Sigma-Aldrich (Taufkirchen, Germany), but no longer commercially available, was kindly provided by L. Fairbanks, Purine Research Laboratory, St Thomas' Hospital, London, UK. Adenine (99.6% pure), adenosine (100% pure), 2-deoxyadenosine (99.9% pure), hypoxanthine (99.6% pure), ino-sine (99.2% pure), 2-deoxyinosine (99.8% pure), uric acid (99.3% pure), acetonitrile (gradient grade), methanol (gradient grade), ammonia (25%), ammonium acetate (reagent grade), ammonium hydroxide (NH4OH, reagent grade), formic acid (reagent grade) and dimethyl sulfoxide (DMSO, Chromasolv grade) were purchased from Sigma-Aldrich (Taufkirchen, Germany). Deionized water was produced by the Millipore® technique. 2-Deoxyguanosine-13C (15N2) (98% pure) was obtained from Santa Cruz (Dallas, TX, USA) and applied as an internal standard for purines during method development.
2.2. Internal standard for DHA
In order to perform absolute quantification of DHA in urine samples, an isotopically labeled internal standard was required. 2,8-Dihydroxyadenine-2-13C-1,3-15N2 (DHA-2-13C-1,3-15N2) was synthesized by our group, following a previously reported protocol [28,29], using thiourea-13C,15N2 as a starting material.
2.3. Preparation of solutions and samples for UPLC–MS/MSanalysis
Standard stock solutions were prepared in 100 mM NH4OH at a concentration of 100 μg/mL for DHA, adenine and 2-deoxyadenosine, 200 μg/mL for adenosine, hypoxanthine, inosine, 2-deoxyinosine, and uric acid, and 1 mg/mL for DHA-2-13C-1,3-15N2 and 2-deoxyguanosine13C,15N2. Urine samples donated by healthy volunteers, diluted 1:15 (v/v) with 10 mM NH4OH, were used as a blank matrix. An intermediate working solution was prepared in 10 mM NH4OH at the concentration of 10 μg/mL for DHA, adenine, adenosine, 2-deoxyadenosine, hypoxanthine, inosine, 2-deoxyinosine, uric acid and 2-deoxyguanosine13C,15N2.This intermediate working solution was diluted 1:100 (v/v) with the blank matrix, producing a 1000 ng/mL solution of each analyte for use during the design of experiments (DoE). A working solution of the internal standard, DHA-2-13C-1,3-15N2 (8 μg/mL) was prepared in 10 mM NH4OH. All solutions were stored at −20°C. For the quantification of DHA, a calibration standard and QC samples were prepared fresh each day by adding the intermediate working solutions prepared in 10 mM NH4OH at an analyte concentration of 0.04, 0.08, 0.16, 0.2, 0.3, 0.5, 0.6, 0.8, 1.2, 2.0, 4.0, 6.0 and 10.0 μg/mL as well as by adding the internal standard working solution by proper dilution with the blank matrix. The calibration points were selected at the DHA concentrations of 100, 150, 250, 400, 600, 1000, 3000, 5000 ng/mL and 3 QC samples at the concentrations of 300, 800, 2000 ng/mL for DHA and 8 μg/mL for the internal standard.
2.4. Multivariate experimental design for the UPLC–MS/MS assay
Waters ACQUITY UPLC device coupled to a Quattro Premier™ XE triple quadrupole mass spectrometer (Waters Corporation, Milford, MA) was utilized for the analysis. Multiple reaction monitoring (MRM) in both negative and positive electrospray ionization (ESI) modes was employed and the fragmentation behavior of each compound characterized using collision-induced dissociation (CID).The MRM transition of each analyte is presented in Table 1. Nitrogen was used as a desolvation and cone gas and high purity argon as a collision gas. Source temperature and desolvation gas temperature were set at 130°C and 400°C, respectively. The analytical column, Acquity HSS T3 (1.8 μm, 100 × 2.1 mm) (Waters Corporation, Wexford, Ireland), was maintained at 35 °C. The injection volume was 5 or 10 μL. D-optimal design with interaction model (MODDE Pro 11, Data Analytics Solutions, MKS Instruments AB, Umea, Sweden), consisting of 3 quantitative and 3 qualitative factors, was generated to determine significant variables for a UPLC–MS/MS screening assay of DHA in human urine. The assay was designed to include simultaneous measurement of DHA, adenine, adenosine, 2-deoxyadenosine, hypoxanthine, inosine, 2-deoxyinosine, and uric acid to ensure adequate separation between all analytes. Capillary voltage, gradient steepness, type of gradient slope, flow rate, and different composition of the mobile phase were optimized and related to the UPLC–MS/MS responses, utilizing partial least squares (PLS) regression. Mobile phase A consisted of MQ water containing 0.1% (v/v) formic acid (pH 2.8) or MQ water with 2 mM ammonium acetate (pH 6.7) and mobile phase B was either methanol (MeOH) or acetonitrile (ACN). The gradient elution program was designed as follows: 0–1.0 min 0.5% B; 1.5, 2.5 or 3.5 min, respectively, to 20% B; 2.5, 3.5 or 4.5–5.0 min to 80% B; 5.0–5.5 min, hold at 80% B; 5.5–5.7 min to 0.5% B; and 5.7–6.5 min, hold at 0.5% B. The levels of each experimental factor are presented in Table 2. The experimental design consisted of 39 experiments, including 3 center point runs performed in 2 different domains, ESI+ and ESI-. The retention time and peak area for each of the purine analytes were studied. A central composite face (CCF) design with a quadratic model was generated to identify the optimal settings for the mass spectrometer at an optimal flow rate for the UPLC for quantification of DHA in human urine. Three factors, capillary voltage, cone voltage and flow rate, were investigated and related to the peak area of DHA, using PLS regression in 12 experiments plus central point replications.
Table 1.
MRM transitions for the analytes in both positive and negative ESI.
| Analyte | MW [g/mol] | Negative ESI [m/z] | Positive ESI [m/z] | ||
|---|---|---|---|---|---|
|
|
|
||||
| Precursor ion | Product ion | Precursor ion | Product ion | ||
| 2,8-Dihydroxyadenine | 167.1 | 165.6 | 122.7 | 168.1 | 125.0 |
| Adenine | 135.1 | 133.9 | 106.9 | 136.0 | 119.0 |
| Adenosine | 267.2 | 311.9 | 133.7 | 268.2 | 136.0 |
| 2-Deoxyadenosine | 251.2 | 250.2 | 133.9 | 252.3 | 136.0 |
| Hypoxanthine | 136.1 | 134.5 | 91.6 | 137.0 | 110.0 |
| Inosine | 268.2 | 266.9 | 134.7 | 269.3 | 137.0 |
| 2-Deoxyinosine | 252.2 | 251.0 | 134.8 | 253.3 | 137.0 |
| Uric acid | 168.1 | 166.6 | 123.8 | 169.0 | 141.0 |
| 2,8-Dihydroxyadenine-2-13C-1,3-15N2 | 170.1 | 170.7 | 152.9 | ||
| 2-Deoxyguanosine-13C,15N2 | 270.2 | 271.3 | 155.0 | ||
ESI, electrospray ionization.
Table 2.
Experimental factors and settings for the multivariate designs of experiments.
| Design | Qualitative factor | Settings | |
|---|---|---|---|
| D-optimal design | pH of mobile phase | pH 2.8; pH 6.7 | |
| Organic solvent | Methanol; Acetonitrile | ||
| Type of gradient slopea | 4; 6; 8 | ||
| Quantitative factor | Lower limit | Upper limit | |
| Capillary voltage (kV) | 0.5 | 3.5 | |
| Gradient steepness (min) | 1.5 | 3.5 | |
| Flow rate (mL/min) | 0.4 | 0.6 | |
| CCF-design | Capillary voltage (kV) | 0.4 | 2.0 |
| Cone voltage (V) | 20 | 35 | |
| Flow rate (mL/min) | 0.35 | 0.45 | |
CCF, central composite face design.
Slope 4 is concave downwards; slope 6 is linear; slope 8 is concave upwards.
2.5. Pre-validation of the UPLC–MS/MS assay
The following parameters were assessed during pre-validation of the UPLC–MS/MS assay: selectivity, concentration function, limit of detection (LOD) and lower limit of quantification (LLOQ), intra-and inter-day assay precision and accuracy, auto-sampler stability, carryover, dilution integrity and matrix effect of the corresponding matrix. The selectivity was assessed by analyzing blank urine samples, with and without internal standard, from six healthy individuals, as well as blank urine samples spiked with DHA at the LLOQ concentration level (100 ng/mL). The concentration function of the method was evaluated at concentrations ranging from 100 to 5000 ng/mL for DHA. Calibration curves were generated by plotting the corresponding peak area ratio under the specific MRM chromatogram for the analyte or internal standard versus the corresponding analyte concentrations using weighted (1/x) least squares quadratic regression. The LOD and LLOQ were evaluated by replicate analysis (n = 6) and defined as the lowest concentrations that gave a signal-to-noise (S/N) ratio of at least 3-fold and 20-fold, respectively. Intra- and inter-assay precision (the relative standard deviation, RSD) and accuracy (the relative error) were determined by replicate analysis (n = 6) of the LLOQ and QC samples, conducted over three different days. The stability of the processed samples, which were kept in the autosampler at 22 °C for at least 18 h, was tested by replicate analysis (n = 6) of each QC concentration level and compared to QC samples measured immediately after sample preparation. Carryover in the UPLC–MS/MS system was tested by analyzing a blank urine sample spiked with internal standard, following analysis of urine samples at the upper limit of quantification. Matrix effect was assessed by comparing the peak area for DHA in spiked blank urine samples from six healthy individuals with MQ water spiked with DHA. In both cases, the samples were diluted with NH4OH 1:15 (v/v).
2.6. Measurement of DHA in urine samples from patients with APRT deficiency
This part of the study was approved by the National Bioethics Committee of Iceland (NBC 09-072) and the Icelandic Data Protection Authority. Informed consent was obtained from all living participants. Twenty-four hour urine samples were collected from 3 adult patients with APRT deficiency, who were not receiving xanthine dehydrogenase inhibitor (allopurinol or febuxostat) treatment and 6 healthy individuals. All 3 patients had an estimated glomerular filtration rate (eGFR) ≥60 mL/min/1.73 m2. Within 2 h of arrival at the laboratory, the 24 h collection bottle was inverted 3, 13, 18 and 28 times and subsequently aliquoted. After the last inversion, the remaining urine was aliquoted and stored for up to 72 h at 20 °C, 4°C and at −20 °C (pH 8.6). Finally, the pH of the urine samples, which were kept at room temperature for 72 h, was raised to pH 10 by adding 25% NH4OH during continuous stirring with a magnetic mixer. Prior to UPLC–MS/MS analysis, urine samples were diluted 1:15 (v/v) with 10 mM NH4OH. Then, 50 μL of the diluted samples were pipetted into a 96-well plate, followed by addition of 100 μL of 10 mM NH4OH and subsequently 50 μL of the internal standard working solution. All samples were subsequently mixed in the plate for 3 min and centrifuged at 3100 rpm for 10 min at 4°C before injection into the UPLC–MS/MS system.
2.7. Data processing
All data were acquired using the MassLynx 4.1 software program and processed with TargetLynx™ Application Manager (Waters Corp., Milford, MA, USA). GraphPad prism (GraphPad software Inc, La Jolla, LA, USA) was used for statistical analysis, including a one-way ANOVA with Tukey Multiple Comparison test. Data shown represent the results of 6 separate analyses.
All multivariate experimental design data analyses and calculations were carried out using the MODDE Pro 11 software (Data Analytics Solutions, MKS Instruments AB, Umea, Sweden). This included generation of response surface models, analysis of regression coefficients at 95% confidence intervals and prediction of optimum conditions of the experimental design, based on criteria for the highest DHA peak area at the minimum retention time. All responses were centered and scaled to unit variance and log scale transformation was applied for the peak area of all analytes.
3. Results and discussion
3.1. Chemometric optimization of the UPLC–MS/MS assay
D-optimal design with an interaction model was selected for screening of important experimental variables and their appropriate ranges for the UPLC–MS/MS assay, as this model allows for simultaneous evaluation of both multilevel qualitative factors and quantitative factors. The retention time of all purines studied and the peak area of DHA were selected as responses, since our primary goal was to develop a sensitive and accurate method for quantification of DHA in human urine samples, without any interference from early eluting matrix components or other purines. PLS regression analyses of the peak area of all purines studied, both in ESI+ and ESI- modes, revealed two distinguishable groups (Fig. 1). Group A, corresponding to all experimental values in the ESI+ mode, demonstrated a much higher response (peak area) compared with group B, which represented experiments carried out in ESI- mode. Therefore, the ESI+ mode was utilized for the remainder of the work as it resulted in much higher sensitivity for each purine. The fraction of variance (R2), explained by the PLS regression analysis of the responses in the ESI+ mode, was > 91% for all responses with an acceptable prediction ability of the model (Q2 > 80%). The plot from the PLS regression modeling of the purines showed that flow rate, pH and the type of organic modifier of the mobile phase had the most significant effect on retention time, with a low flow rate, pH of 6.7 and methanol as mobile phase B resulting in a higher retention time (data not shown). Adequate separation of the purines was achieved for all conditions studied during the experimental screening in the ESI+ mode as is illustrated in Fig. 2. This is important as spontaneous fragmentation of adenosine and 2-deoxyadenosine into the same ion fragment as adenine and of inosine and 2-deoxyinosine into the same ion fragment as hypoxanthine may occur.
Fig. 1.
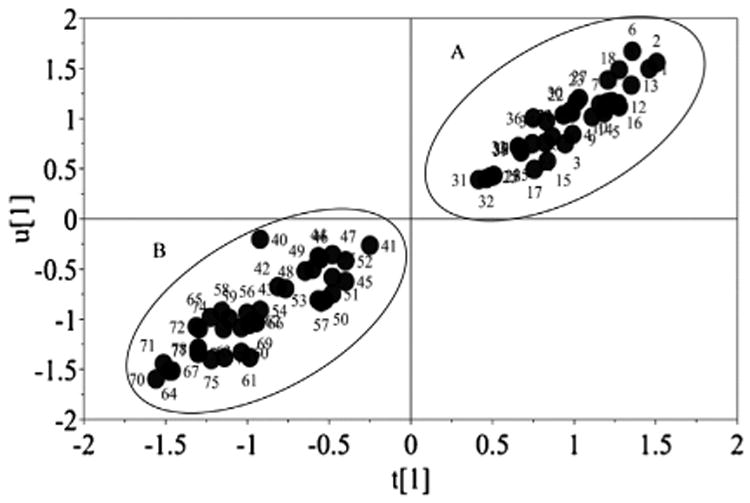
Partial least squares (PLS) scores plot for area of 2,8-dihydroxyadenine, adenine, adenosine, 2-deoxyadenosine, hypoxanthine, inosine, 2-deoxyinosine and uric acid. Positive ionization mode (A) and negative ionization mode (B) are shown. u [1] is the Y scores of component 1 and t [1] is the X scores of PLS component 1.
Fig. 2.

MRM chromatogram for positive ionization mode of adenine (A), adenosine (B), 2-deoxyadenosine (C), hypoxanthine (D), inosine (E) and 2-deoxyinosine (F). Adenine m/z 136.0→ 119.0, retention time 1.97 min. Adenosine m/z 268.2→ 136.0, retention time 2.66 min. 2-Deoxyadenosine m/z 252.3→ 136.0, retention time 2.81 min. Hypoxanthine m/z 137.0→ 110.0, retention time 1.41 min. Inosine m/z 269.3→ 137.0, retention time 1.96 min. 2-Deoxyinosine m/z 253.3→ 137.0, retention time 2.09 min.
The regression coefficient plot of the scaled and centered peak area of DHA showed that capillary voltage, flow rate and mobile phase A at pH 2.8 had a significant negative effect, while nonlinear gradient (slope 4) and mobile phase A at pH 6.7 had a significant positive effect on the peak area (Fig. 3). Several interaction effects between capillary voltage and flow rate, capillary voltage and pH of the mobile phase, and type of gradient slope and organic modifier in the mobile phase, were significant so these variables cannot be independently controlled to obtain maximum sensitivity. This is further illustrated in Fig. 4, showing a counterplot of the DHA peak area as a function of flow rate and capillary voltage at different mobile phase conditions. Mobile phase A at pH 6.7, as opposed to pH 2.8, yielded a much higher peak area of DHA and the same was true for acetonitrile as an organic modifier in mobile phase B. Moreover, the sensitivity of the method could be improved by the decreasing capillary voltage and flow rate.
Fig. 3.
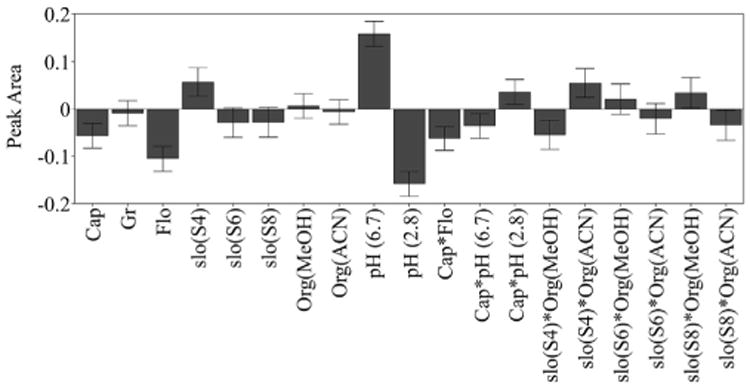
Regression coefficients plot scaled and centered for the peak area of 2,8-dihydroxyadenine. X-axis displays experimental factors and factor interactions that have significant effect on the peak area of 2,8-dihydroxyadenine and y-axis peak area. Error bars represent the 95% confidence interval. Cap, capillary voltage (kV); Gr, gradient steepness (min); Flo, flow rate (ml/min); Slo, slope; Org, organic mobile phase; MeOH, methanol; ACN, acetonitrile.
Fig. 4.
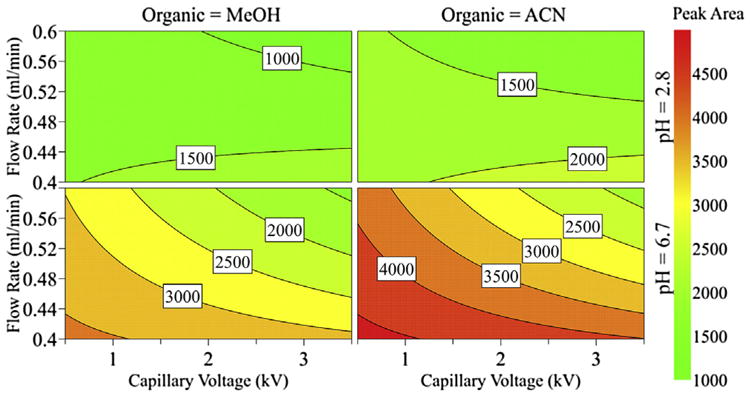
Counterplot for the peak area of 2,8-dihydroxyadenine, according to flow rate, capillary voltage, pH of the mobile phase and the organic modifiers, methanol (MeOH) and acetonitrile (ACN).
The results of the screening studies were used to select significant variables for final optimization of the quantification method. A CCF design was chosen and correlated to the sensitivity of the analyte employing PLS regression. The experimental domain was redefined and the experimental factors studied included capillary voltage, cone voltage and flow rate (Table 2). A nonlinear gradient slope was used for mobile phase A (pH 6.7) and mobile phase B (acetonitrile) with the gradient steepness set at 1.5 min. A response surface model was fitted utilizing PLS regression analysis of the peak area of DHA. Two PLS components were significant, the explained variance > 96% and the acceptable predictability of the model (Q2 >85%). The optimal experimental conditions observed and subsequently used for the analysis of urine samples were: mobile phase A at pH 6.7; acetonitrile as mobile phase B; flow rate of 0.35 mL/min with a nonlinear gradient slope, raising the concentration of mobile phase B from 0.5 to 20% in 1.5 min; capillary voltage of 0.4 kV; and cone voltage for DHA set at 35 V. The final experimental conditions selected for the quantification method are summarized in Table 3.
Table 3.
Experimental conditions of the UPLC–MS/MS assay for quantification of DHA.
| Instrument/conditions | Details | ||
|---|---|---|---|
| UPLC–MS/MS | Waters ACQUITY – Quattro Premier™ XE | ||
| Analytical column | Waters Acquity HSS T3 2,1 × 100 mm, 1.8 μm | ||
| Column temperature | 35°C | ||
| Autosampler temperature | 22°C | ||
| Flow rate | 0.35 mL/min | ||
| Mobile phase | A: 2 mM Ammonium acetate at pH 6.7; B: Acetonitrile | ||
| Gradient slope | Time (min) | %A | %B |
| 0.0 | 99.5 | 0.50 | |
| 1.0 | 99.5 | 0.50 | |
| 2.5 | 80.0 | 20.0 | |
| 5.0 | 20.0 | 80.0 | |
| 5.5 | 20.0 | 80.0 | |
| 5.7 | 99.5 | 0.50 | |
| 6.5 | 99.5 | 0.50 | |
| Gradient slope | Concave downwards | ||
| Sample injection | 5 μLof urine sample diluted 1:15 (v/v) with 10 mM NH4OH | ||
| Ionization mode | Positive electrospray ionization | ||
| Capillary voltage | 0.4 kV | ||
| Cone voltage | 35 V | ||
3.2. Analytical performance of the optimized UPLC–MS/MS assay
We report for the first time an absolute quantification of DHA in human urine samples by UPLC–MS/MS, using the isotopically labeled reference compound, DHA-2-13C-1,3-15N2, as an internal standard. The results from the pre-validation study of the assay showed acceptable selectivity without any interfering peaks in the human urine samples from the 6 individuals tested. There were no interfering peaks detected in the blank matrix at the same retention time as for DHA, as demonstrated by the MRM chromatogram of blank urine from a healthy individual (Fig. 5a) compared with the blank matrix spiked with DHA at LLOQ concentration (Fig. 5b). The results also indicate that urine from healthy individuals does not contain significant endogenous levels of DHA, as DHA was not detected in any of the six urine matrixes analyzed (data not shown). The back calculated concentration of all calibration standards, including the LLOQ, were within acceptable limits of ±15% of the nominal value (data only shown for LLOQ), in the concentration range of 100 to 5000 ng/mL (Table 4). The limit of detection for DHA was 20 ng/mL and the LLOQ of the method was set at 100 ng/mL. The analytical performance of the assay was tested for LLOQ and QC samples using blank matrix spiked with DHA at 3 different concentrations, 300, 800 and 2000 ng/mL, on three different days. Intraday coefficients of variation (CVs) were 4.5–5.7% with accuracy of −2.0–8.5% for all QC samples. Intraday CVs for LLOQ were 8.3% with accuracy 3.3%. The interday accuracy and precision CVs for QC samples and LLOQ were well within the ±15% limits (Table 4). The processed samples (n = 6) were stable up to 18 h at 22°C in the autosampler with CV for all QC samples < 6% and with a mean bias of the back calculated concentration <11%. To evaluate carryover, a blank urine sample was injected immediately following analysis of the highest DHA standard at 5000 ng/mL but no DHA peak was detected. Dilution integrity was evaluated by spiking DHA into a blank urine sample at a concentration of 10.000 ng/mL and diluting the sample with 10 mM NH4OH down to the range of the concentration curve. The DHA concentration of the diluted samples (n = 3) was within ±15% of nominal values, demonstrating good sample integrity. The matrix effect was evaluated by comparing the response for DHA spiked at the same concentration in blank urine and in neat solution. The response for DHA was the same in blank urine samples from 6 different biological sources as in the neat solution (data not shown), indicating no significant matrix effect.
Fig. 5.

MRM chromatogram of 2,8-dihydroxyadenine (DHA). a. Urine sample from a healthy individual; b. Urine sample from a healthy individual spiked with 2,8-dihydroxyadenine at a concentration of 100 ng/mL; c-e. Urine samples from patients with APRT deficiency.
Table 4.
Intra-assay and inter-assay accuracy and precision of DHA measurement in urine samples.
| QC Level | Concentration (ng/mL) | Intraday (n = 6) | Interday (n = 3) | ||
|---|---|---|---|---|---|
|
|
|
||||
| %CVa | %Biasb | %CVa | %Biasb | ||
| LLOQ | 100 | 8.3 | 3.3 | 7.8 | 0.5 |
| QC-low | 300 | 5.7 | −2.0 | 5.4 | −4.6 |
| QC-medium | 800 | 4.7 | 6.5 | 7.4 | 3.1 |
| QC-high | 2000 | 4.5 | 8.5 | 3.4 | 4.4 |
Calibration range 100-5000 ng/mL, R2 = 0.9989 with weighing factor 1/X
QC, quality control; LLOQ, lower limit of quantification; CV, coefficient of variation.
% RSD = (standard deviation/mean) × 100.
%Bias = (mean calculated concentration – nominal concentration)/(nominal concentration) × 100.
Hartmann et al. [18] reported relative quantification of urinary DHA and other key purine metabolites in a total analysis time of 20 min, whereas the analytical run time was only 6.5 min with our method. The limit of detection for DHA was much higher than in our assay, 167 ng/mL vs. 20 ng/mL and the interday CV for DHA was quite high or 25% [18]. Urinary DHA has also been measured using HPLC coupled to a multichannel ultraviolet detector, but this method required computer-aided data analysis and a 30 min analytical run time [13]. In addition, CE has been used for relative quantification of DHA in urine samples with a markedly shorter analytical run time than can be achieved with the HPLC-based methods [14–17]. However, reproducibility has been a challenge when CE methods are used in conjunction with UV detection as the peak identification is based on detection time which can vary greatly amongst different matrices. None of the previously reported techniques for urinary DHA determination has been fully developed for clinical application. In fact, only relative quantification of DHA has been accomplished due to lack of isotopically labeled reference standards.
3.3. Measurement of DHA concentration in urine samples from patients with APRT deficiency
Due to the poor solubility of DHA, storage and handling of urine specimens from patients with APRT deficiency may influence the measurement of the DHA concentration. Since 24 h urine collection is almost exclusively conducted by patients at home, we investigated the effect of storage temperature on the measurement of DHA in urine specimens obtained from 3 patients. No significant difference was observed in the peak area of DHA at +20°C, −4°Cor at −20°C for up to at least 72 h at pH 8.6, indicating no significant effect of storage temperature on the DHA signal response (Fig. 6a). Accordingly, urine samples may be kept at room temperature up to 72 h prior to analysis without affecting the results. Raising the pH of the urine samples to pH 8.6 improved the response of DHA, but additional increase of the pH to 10 with 25% NH4OH resulted in a lower response (Fig. 6b). These results are consistent with those reported by Hartmann et al. [18], who observed no change in urinary DHA concentration at pH values ranging from 3 to 9. Although Peck et al. [30] also reported that the solubility of DHA can be increased by raising the pH beyond baseline values, this resulted in very unstable solutions. Increased sample mixing did not have significant effect on the peak area of DHA (data not shown) and, therefore, it was decided to invert the collection bottle only 3 times during further experiments.
Fig. 6.
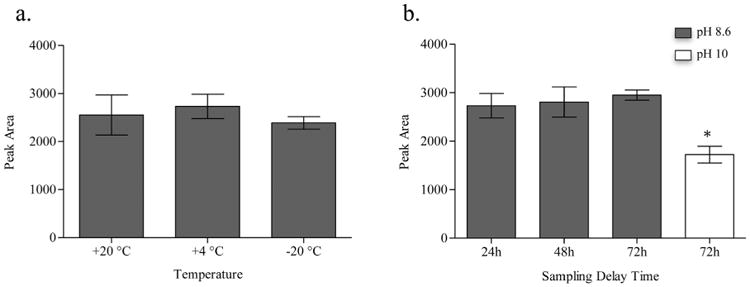
Effect of different storage conditions on the peak response of 2,8-dihydroxyadenine (DHA) in a urine sample from patient with APRT deficiency. a. Effect of storage temperature on the peak area of DHA. b. Effect of storage time on the peak area of DHA at pH 8.6 and 10. *Indicates a significant decrease of the peak area (P < 0.001).
Twenty-four hour urine samples from 3 patients with APRT deficiency, who were not receiving pharmacological treatment and from 6 healthy individuals were analyzed using the UPLC–MS/MS assay. MRM chromatogram illustrated a peak corresponding to DHA in the patients' samples, which was not detected in the sample from the healthy individuals (Fig. 5a, c, d and e). An unidentified peak was observed in the MRM chromatogram of all the urine samples from the patients and the control individuals (data shown for 1 healthy individual) that was well separated from the DHA peak and did not interfere with the quantification of DHA. The clinical applicability of the assay was demonstrated by determining the concentration of DHA in 24 h urine samples from patients with APRT deficiency, diluted 1:15 (v/v) with 10 mM NH4OH. The urinary DHA concentration in the three patients was 3021, 5860 and 10563 ng/mL and the 24 h urinary DHA excretion 816, 1327 and 1649 mg, respectively, suggesting a highly variable excretion rate between patients (Fig. 7).
Fig. 7.

24 h urinary 2,8-dihydroxyadenine (DHA) excretion in 3 patients with APRT deficiency who were not receiving treatment with a xanthine dehydrogenase inhibitor. Patient 1, 2 and 3 correspond to c, d and e in Fig. 5, respectively. Data represents mean values and SD of six measurements by the UPLC–MS/MS assay.
4. Conclusions
We have successfully developed a rapid and robust UPLC-MS/MS assay with minimal sample preparation for absolute quantification of DHA in urine, utilizing isotopically labeled DHA as an internal standard. The chemometric approach (DOE), for optimization of the UPLC–MS/MS method was demonstrated to be highly effective, requiring only a fraction of the number of experiments that would have been needed when changing only one separate factor at a time.
We believe this UPLC–MS/MS assay will greatly facilitate clinical diagnosis and monitoring of pharmacotherapy in patients with APRT deficiency. Preliminary data from patients with APRT deficiency demonstrate that urinary DHA is easily detected by the assay and that absolute concentration of DHA can be determined in patient's samples at a broad concentration range.
Future work will focus on evaluation of the UPLC–MS/MS urinary assay in individuals with APRT deficiency, aiming for full validation for use in clinical practice.
Acknowledgments
Part of this work was presented in an abstract form at the American Society of Nephrology Kidney Week in Atlanta, GA, November 2013 and at the American Society for Mass Spectrometry in St. Louis, MO, May 2015. The study was supported by the Rare Kidney Stone Consortium (U54DK083908), a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). This consortium is funded through a collaboration between NCATS, and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The authors would like to thank L. Fairbanks, Purine Research Laboratory, St Thomas' Hospital, London, UK, who kindly provided our research team with a sample of 2,8-dihydroxyadenine (manufactured by Sigma-Aldrich, Germany, but no longer commercially available) for the initial assay development.
Abbreviations
- APRT
adenine phosphoribosyltransferase
- CCF
central composite face
- CE
capillary electrophoresis
- CID
collision induced dissociation
- CKD
chronic kidney disease
- CV
coefficient of variation
- DHA
2,8-dihydroxyadenine
- DMSO
dimethyl sulfoxide
- DoE
design of experiments
- eGFR
estimated glomerular filtration rate
- ESI
electrospray ionization
- HPLC–MS/MS
high-performance liquid chromatography-tandem mass spectrometry
- LLOQ
lower limit of quantification
- LOD
limit of detection
- MRM
multiple reaction monitoring
- NH4OH
ammonium hydroxide
- PLS
partial least squares
- QC
quality control
- RSD
relative standard deviation
- S/N
signal-to-noise ratio
- UPLC–MS/MS
ultra-performance liquid chromatography-tandem mass spectrometry
- UV
ultraviolet
References
- 1.Simmonds HA, Van Acler KJ, Cameron JS, Snedden W. The identification of 2,8-dihydroxyadenine, a new component of urinary stones. Biochem J. 1976;157:485–487. doi: 10.1042/bj1570485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeuchi H, Kaneko Y, Fujita J, Yoshid O. A case of compound heterozygote for adenine phosphoribosyltransferase deficiency (APRT*J/APRT*Q0) leading to 2,8-dihydroxyadenine urolithiasis: review of the reported cases with 2,8-dihydroxyadenine stones in Japan. J Urol. 1993;149:824–826. doi: 10.1016/s0022-5347(17)36222-5. [DOI] [PubMed] [Google Scholar]
- 3.Sahota A, Tischfield J, Kamatani N, Simmonds HA. Adenine phosphoribosyltransferase deficiency and 2,8-dihydroxyadenine lithiasis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Volgelstein B, Childs B, editors. The Metabolic and Molecular Bases of Inherited Disease. eighth. McGraw-Hill; New York: 2001. pp. 2571–2584. [Google Scholar]
- 4.Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in Iceland. Am J Kidney Dis. 2001;38:473–480. doi: 10.1053/ajkd.2001.26826. [DOI] [PubMed] [Google Scholar]
- 5.Bollée G, Dollinger C, Boutaud L, Guillemot D, Bensman A, Harambat J, Deteix P, Daudon M, Knebelmann B, Ceballos-Picot I. Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J Am Soc Nephrol. 2010;21:679–688. doi: 10.1681/ASN.2009080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harambat J, Bollée G, Daudon M, Ceballos-Picot I, Bensman A. APRT study group, adenine phosphoribosyltransferase deficiency in children. Pediatr Nephrol. 2012;27:571–579. doi: 10.1007/s00467-011-2037-0. [DOI] [PubMed] [Google Scholar]
- 7.Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, Palsson R. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28:1923–1942. doi: 10.1007/s00467-012-2329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Runolfsdottir HL, Palsson R, Agustdottir IM, Indridason OS, Edvardsson VO. Kidney disease in adenine phosphoribosyltransferase deficiency. Am J Kidney Dis. 2015;67:431–438. doi: 10.1053/j.ajkd.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollée G, Harambat J, Bensman A, Knebelmann B, Daudon M, Ceballos-Picot I. Adenine phosphoribosyltransferase deficiency. Clin J Am Soc Nephrol. 2012;7:1521–1527. doi: 10.2215/CJN.02320312. [DOI] [PubMed] [Google Scholar]
- 10.Simmonds HA, Potter CF, Sahota A, Cameron JS, Rose GA, Barratt TM, Williams DI, Arkell DG, Van Acker KJ. Adenine phosphoribosyltransferase deficiency presenting with supposed uric acid stones: pitfall of diagnosis. J R Soc Med. 1978;71:791–795. doi: 10.1177/014107687807101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassidy MJ, McCulloch T, Fairbanks LD, Simmonds HA. Diagnosis of adenine phosphoribosyltransferase deficiency as the underlying cause of renal failure in renal transplant. Nephrol Dial Transplant. 2004;19:736–738. doi: 10.1093/ndt/gfg562. [DOI] [PubMed] [Google Scholar]
- 12.Edvardsson VO, Palsson R, Sahota A, Adenine phosphoribosyltransferase deficiency. Pagron RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K. GeneReviews®. University of Washington; Seattle: [accessed 15.02.16]. pp. 1993–2016. Internet. 30.08.12 (updated 18.06.15), http://www.genetests.org. [PubMed] [Google Scholar]
- 13.Kojima T, Nishina T, Kitamura M, Kamatani N, Nishloka K. Liquid chromatography with multichannel ultraviolet detection used for studying disorders of purine metabolism. Clin Chem. 1987;33:2052–2056. [PubMed] [Google Scholar]
- 14.Sevcik J, Adam T, Mazacova H. A fast and simple screening method for detection of 2,8-dihydroxyadenine urolithiasis by capillary zone electrophoresis. Clin Chim Acta. 1996;245:85–92. doi: 10.1016/0009-8981(95)06175-4. [DOI] [PubMed] [Google Scholar]
- 15.Wessel T, Lanvers C, Fruend S, Hempel G. Determination of purines including 2,8-dihydroxyadenine in urine using capillary electrophoresis. J Chromatogr A. 2000;894:157–164. doi: 10.1016/s0021-9673(00)00439-8. [DOI] [PubMed] [Google Scholar]
- 16.Adam T, Friedecky D, Fairbanks LD, Sevcik J, Bartak P. Capillary electrophoresis for detection of inherited disorders of purine and pyrimidine metabolism. Clin Chem. 1999;45:2086–2093. [PubMed] [Google Scholar]
- 17.Adam T, Lochman P, Friedecky D. Screening method for inherited disorders of purine and pyrimidine metabolism by capillary electrophoresis with reversed electroosmotic flow. J Chromatogr B. 2002;767:333–340. doi: 10.1016/s1570-0232(01)00591-8. [DOI] [PubMed] [Google Scholar]
- 18.Hartmann S, Okun JG, Schmidt C, Langhans CD, Garbade SF, Burgard P, Haas D, Sass JO, Nyhan WL, Hoffmann GF. Comprehensive detection of disorders of purine and pyrimidine metabolism by HPLC with electrospray ionization tandem mass spectrometry. Clin Chem. 2006;52:1127–1137. doi: 10.1373/clinchem.2005.058842. [DOI] [PubMed] [Google Scholar]
- 19.Dooley KC. Tandem mass spectrometry in the clinical chemistry laboratory. Clin Biochem. 2003;36:471–481. doi: 10.1016/s0009-9120(03)00105-x. [DOI] [PubMed] [Google Scholar]
- 20.Himmelsbach M. 10 years of MS instrumental developments –impact on LC-MS/MS in clinical chemistry. J Chromatogr B Anal Technol Biomed Life Sci. 2012;883-884:3–17. doi: 10.1016/j.jchromb.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 21.van den Ouweland JM, Kema IP. The role of liquid chromatography-tandem mass spectrometry in the clinical laboratory. J Chromatogr B Anal Technol Biomed Life Sci. 2012;883-884:18–32. doi: 10.1016/j.jchromb.2011.11.044. [DOI] [PubMed] [Google Scholar]
- 22.Box GEP, Hunter WG, Hunter JS. Statistics for Experimenters: an Introduction to Design Data Analysis and Model Building. John Wiley and Sons Inc; New York: 1978. [Google Scholar]
- 23.Stahle L, Wold S. Multivariate data analysis and experimental design in biomedical research. Prog Med Chem. 1988;25:291–338. doi: 10.1016/s0079-6468(08)70281-9. [DOI] [PubMed] [Google Scholar]
- 24.Hibbert DB. Experimental design in chromatography: a tutorial review. J Chromatogr B Anal Technol Biomed Life Sci. 2012;910:2–13. doi: 10.1016/j.jchromb.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Thorsteinsdottir M, SigurĐsson BB, Bragason G. In: Determination of pharmacokinetic parameters by HPLC-MS/MS and UPLC-MS/MS. Lee MS, editor. Mass Spectrometry Handbook, John Wiley and Sons, Inc; Hoboken, NJ: 2012. pp. 191–208. [Google Scholar]
- 26.Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2001. [Google Scholar]
- 27.Liquid Chromatography-Mass Spectrometry Methods. Approved Guideline, CLSI document C62-A. Clinical and Laboratory Standards Institute; Wayne PA: 2014. [Google Scholar]
- 28.Bendich A, Tinker JF, Brown GB. A synthesis of isoguanine labeled with isotopic nitrogen. J Am Chem Soc. 1948;70:3109–3113. doi: 10.1021/ja01189a081. [DOI] [PubMed] [Google Scholar]
- 29.Liebe F, Cavalieri F, Bendich A. The ultraviolet absorption spectra of pyrimidines and purines. J Am Chem Soc. 1950;72:2587–2594. doi: 10.1021/ja01191a102. [DOI] [PubMed] [Google Scholar]
- 30.Peck CC, Bailey J, Moore GL. Enhanced solubility of 2,8 dihydroxyadenine (DOA) in human urine. Transfusion. 1977;17:383–390. doi: 10.1046/j.1537-2995.1977.17477216867.x. [DOI] [PubMed] [Google Scholar]