

Abstract
Tofacitinib is an oral Janus kinase inhibitor. An integrated analysis was conducted to evaluate dosage optimality for tofacitinib in patients with moderate‐to‐severe plaque psoriasis and the impact of body weight on optimality in this patient population. Data were pooled from one phase IIb trial (2, 5, and 15 mg twice daily (b.i.d.)) and four phase III trials (5 and 10 mg b.i.d.). A longitudinal exposure–response model for Psoriasis Area and Severity Index (PASI) improvement (percent change from baseline) was established. Body weight influenced potency; heavier subjects require higher doses to achieve comparable benefit to lighter subjects. Disease severity, sex, and prior biologic usage were also predictive of response. The 10 and 5 mg doses were predicted to achieve 81% and 65%, respectively, of the maximum effect based on a 75% improvement in PASI. The greater efficacy of 10 mg over 5 mg was clinically meaningful.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Tofacitinib is an oral Janus kinase inhibitor. Based on efficacy results from a phase IIb study in psoriasis patients, a model‐based dose–response model successfully predicted appropriate doses for investigation in phase III confirmatory trials; however, the effect of subject body weight was not clearly defined.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ A longitudinal dose–response model was developed to improve understanding of how subject‐specific factors, including body weight, might influence the dose–response relationship.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The model predicted that the magnitude of effect of tofacitinib 5 and 10 mg b.i.d. depended on previous biologic usage (difference in PASI75 response: 12% biologic‐naïve; 16% biologic‐experienced), and body weight (10% 64 kg; 14% 116 kg). Heavier subjects and those with prior biologic experience were predicted to require a higher dose to achieve benefit comparable to that in lighter subjects.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This analysis demonstrates the benefit that modeling and simulation methods can provide to optimize treatment of individuals within a patient population.
Chronic plaque psoriasis is a complex disease that predominantly affects the skin, yet has nondermatological comorbidities, including psoriatic arthritis, cardiovascular disease, and metabolic syndrome.1, 2, 3 Psoriasis markedly degrades patients' quality of life and can be associated with shortened life expectancy.4 Continuous, long‐term therapy is necessary to control psoriasis and prevent incidence or progression of comorbidities.
Current systemic treatments for moderate‐to‐severe psoriasis include oral systemic and biologic agents. While these agents can be highly effective in some patients, a large proportion of patients do not achieve the desired improvement and no single agent is effective in all patients.5, 6 These agents have been associated with significant short‐term and long‐term organ toxicities, which limit long‐term usage.5, 6 Biologic agents can yield greater therapeutic results, yet not all patients respond to or tolerate these treatments,7, 8 and loss of effectiveness is frequently observed over time, which can limit long‐term treatment success.9 Such issues with current therapies manifest in low patient satisfaction and treatment persistence.10
Although pharmacokinetic–pharmacodynamic models have been well utilized in the psoriasis disease area, dosage optimization to achieve target effectiveness has only been performed for the biologics treatments ustekinumab11 and secukinumab.12 Dose selection using exposure–response modeling of phase II data for another recently approved biologic agent, ixekizumab, has also been reported.13 However, the effects of intrinsic patient characteristics on efficacy in psoriasis that could suggest modification of the dosage are typically only evaluated using subgroup analyses.
The issue of weight as a pharmacodynamic (PD) factor has been discussed previously.14 A population model for etanercept, relating subject‐specific predicted cumulative area under the curve (AUC) to the 75% improvement from baseline in Psoriasis Area and Severity Index (PASI) score (PASI75),15 estimated a 130% increase in half‐maximal effective concentration (EC50) for a 2‐fold weight change. PASI75 is a response threshold commonly used to evaluate the efficacy of treatment for psoriasis and as a primary endpoint in clinical trials.19 Despite the selection of the weight effect on EC50, closer inspection suggested difficulty in assigning the effect to EC50 or delay in onset of drug action.
Tofacitinib is an oral Janus kinase (JAK) inhibitor. The main focus of this analysis was to evaluate the exposure–response (ER) for tofacitinib in the treatment of psoriasis, specifically evaluating the optimality of the selected dosages in the presence of the effect of body weight and other patient factors. A large integrated database constructed from pooling data from one phase IIb study and four phase III studies in psoriasis (all available data from phase IIb and phase III studies of tofacitinib at the time of analysis) was used to support the analysis—the largest analysis of a JAK inhibitor to our knowledge (>3,400 subjects with >17,000 observations). Subject factors were investigated for their influence on the ER relationship.
METHODS
Study design
PASI data were pooled from five studies for the ER analysis. Data prior to forced withdrawal due to lack of efficacy or study discontinuation were included (Table 1), while data from the active comparator arm in OPT Compare (subjects administered etanercept) were not included. All studies were conducted according to the International Conference on Harmonisation Good Clinical Practice Guidelines. Prior to the start of each study, written consent was provided by all subjects and the protocols were approved by appropriate Institutional Review Boards or Ethics Committees.
Table 1.
Descriptive summary of studies
| Study | Ph |
Duration (weeks) |
Planned sample size |
Dose cohorts (allocation) |
PASI schedule (weeks) |
Description and notes |
|---|---|---|---|---|---|---|
|
A3921047 NCT0067821020 |
IIb | 12 | 176 |
Placebo Tofacitinib: 2 mg b.i.d. 5 mg b.i.d. 15 mg b.i.d. (1:1:1) |
Used: 0, 2, 4, 8, 12 Not used: 14, 16 |
Double‐blinded and placebo‐controlled |
|
OPT Pivotal 1 A3921078 NCT0127663916 |
3 | 52 | 825 |
Placebo Tofacitinib 5 mg b.i.d. 10 mg b.i.d. (1:2:2) |
Used: 0, 2, 4, 8, 12, 16 (all) 20, 28 (tofacitinib) Not used: 20, 28 (placebo) 40, 52 (all) |
Double‐blinded and placebo‐controlled Re‐randomization of placebo subjects at Week 16 to tofacitinib 5 or 10 mg Discontinuation of subjects who did not achieve PASI75 or PGA response at Week 28, with option to enroll in LTE study |
|
OPT Pivotal 2 A3921079 NCT0130973716 |
3 | 52 | 825 |
Placebo Tofacitinib: 5 mg b.i.d. 10 mg b.i.d. (1:2:2) |
Used: 0, 2, 4, 8, 12, 16 (all) 20, 28 (tofacitinib) Not used: 20, 28 (placebo) 40, 52 (all) |
Double‐blinded and placebo‐controlled Re‐randomization of placebo subjects at Week 16 to tofacitinib 5 or 10 mg Discontinuation of subjects who did not achieve PASI75 or PGA response at Week 28, with option to enroll in LTE study |
|
OPT Compare A3921080 NCT0124159117 |
3 | 12 | 1,100 |
Placebo Tofacitinib: 5 mg b.i.d. 10 mg b.i.d. Etanercept: 50 mg b.i.w. (1:3:3:3) |
Used: 0, 2, 4, 8, 12 |
Double‐blinded, placebo‐controlled Stratification based on previous failed experience with systemic therapies Patients previously exposed to etanercept or efalizumab were not eligible for study participation |
|
OPT Re‐Treatment A3921111 NCT0118674418 |
3 | 56 | 660 |
Tofacitinib: 5 mg b.i.d. 10 mg b.i.d. (1:1) |
Used: 0, 4, 8, 16, 24 (Period A) Not used: 28, 32, 36 (Period B) +4, +8, +16 (Period C) |
Period A was dose‐blinded tofacitinib treatment (initial treatment), Period B was double‐blinded tofacitinib or placebo (treatment withdrawal), and Period C was dose‐blinded tofacitinib (re‐treatment) Discontinuation at Week 24 based on efficacy or re‐randomization (entry to Period B) to same treatment (as in Period A) or placebo Loss of 50% of Week 24 PASI response in Period B triggers enrollment into Period C and re‐treatment based on dose administered in Period A |
b.i.d., twice daily; b.i.w., twice weekly; LTE, long‐term extension; NCT, National Clinical Trial identifier (ClinicalTrials.gov); PASI, Psoriasis Area and Severity Index; PASI75, 75% improvement from baseline in PASI score; PGA response, Physician Global Assessment of “clear” or “almost clear”; Ph, phase.
PASI is a subjective continuous bounded outcome score (BOS) with possible values ranging from 0 to 72, inclusive. Such data are not normally distributed due to skewness and the frequency of PASI = 0. A pharmacometric methodology was used throughout the analysis to mitigate these issues.22
A primary objective of this analysis was to quantify the influence of body weight on the PASI PD profile. It was of key interest to understand if heavier subjects have longer time to onset of drug action, or a different steady‐state dose–effect profile, as these have a direct bearing on long‐term dosing. To increase the rigor in evaluating weight, a validation was performed using the pooled data. Subject data were stratified by study and treatment arm. Data from 65% of the subjects from each strata were selected for the training dataset (TD). The remaining data were used in the internal validation dataset (VD). Base and covariate model development was performed using the TD dataset. The VD dataset was reserved for model evaluation through predictive assessment.
Base model development
The dependent variable used in the model was based on a percentage change from baseline of PASI. This strategy was used to improve the accuracy of predictions of PASI responder measures (e.g., PASI50 and PASI75) while also being able to predict the general PASI distribution. Direct modeling of the PASI did not yield accurate predictions of these responder rates.
An inhibition of kin indirect (IDR)27 model was selected as the structural model and is described by Eq. 1:
| (1) |
where t is the time after the first dose of study medication; Y is the response variable (percentage change from baseline on the transformed PASI scale); PASB is the baseline PASI; λ(t) represents the time‐dependent transformation of PASI and PASB; BASE is the baseline parameter or intercept (anticipated to be close to 0); PMAX is the slope and also the maximum nondrug effect achieved at time Tc (Ix represents an indicator function that = 1 when the condition x is true or = 0 otherwise); EMAX is the maximum drug effect; E represents drug exposure; E50 is the exposure at which half of the EMAX is achieved; T½ is the half‐life of onset of drug effect; g(t) is a residual error function that changes with time; and ε is a residual random variable, assumed to be normally distributed with mean = 0 and variance = σ 2 (i.e., ε ∼ N(0,σ 2)). The transformation function λ(t) has a slope parameter (λ2) and an intercept parameter (λ1) to adjust for skewness that changed with time.
The vectors of subject‐specific random effects were assumed to be distributed as multivariate normal: η ∼ N(0,Ω), with mean = 0 and covariance matrix Ω. Parameters that are inherently positive (e.g., rate constants or potency parameters) were modeled using a multiplicative or lognormal structure, e.g., . Additive effects (e.g., maximum placebo or drug effects) were modeled using an additive or normal structure, e.g., .
Maximum likelihood estimation, implemented in NONMEM (Version 7.3, ICON Development Solutions, Ellicott City, MD), was used to fit the posited models and estimate the parameters. Rounding errors and convergence issues occurred during model development using the Laplacian approximation to the likelihood. As a result, the iterative two‐stage method followed by stochastic approximation expectation maximization estimation was used to mitigate these issues. Importance sampling was used to calculate the objective function and drive approximate standard errors of the estimates.
Study effects were added to the base model fitted to the TD to assess heterogeneity between studies and to potentially adjust for the lack of randomization between these. The number of study‐specific effects was systematically reduced to avoid overparameterization and to yield a stable model for the covariate and weight‐effect determinations. The study‐effect model components are provided in the Supplementary Material.
Covariate model development
A full covariate model was fitted to the TD as part of the covariate selection procedure. This full model was systematically reduced to a working full model to achieve model stability by eliminating covariates that had weak and irrelevant predictive power. This working full model was subjected to the covariate selection procedure.28 Weight was not assessed at this point and was considered a structural model. The covariate model equations are provided in the Supplementary Information.
Assessment of model adequacy (goodness of fit)
Conditional population‐weighted residuals29 and individual‐weighted residuals were computed. Given the BOS nature of the PASI data, ½ × 0.1 was used to impute PASI for PASI = 0 when calculating residuals. Note that 0.1 was the lowest observed value not equal to 0 (the value was used as the censoring thresholds). No PASI values of 72, the upper bound of the PASI scale, were observed, so imputation was not necessary to calculate residuals. Traditional diagnostic plots were then constructed.
Predictive performance
Visual predictive checks (VPCs)30 (graphical posterior predictive checks)21 were performed using the final model. The model was also used to make predictions of the VD dataset. Population mean predictions on the transformed scale (based on 1,000 Monte Carlo replicates) for each individual at each timepoint were computed using his/her observed trial design and covariates. The absolute deviation between these predictions and the observed data were computed (i.e., |Y – E(Y)|) while the sample median of these was calculated to estimate the median absolute deviation (MAD). MAD was selected as the measure of prediction error because of the nonnormal nature of the data. These VPCs are provided in Supplementary Figure S2 online. After the adequacy of the model was established, all the data were pooled and the final model estimates were updated (Table 2).
Table 2.
Final model parameter estimates based on the training dataset and the complete pooled dataset
| Parameter |
Estimate ASE (TD) |
Estimate ASE (pooled) | Trans. estimate (TD) |
Transformed 90% CI (TD) |
Trans. estimate (pooled) |
Transformed 90% CI (pooled) |
Trans. units | |
|---|---|---|---|---|---|---|---|---|
| OFV = −26,441.365 | OFV = −39,867.620 | N = 2,233, n = 11,204 | N = 3,431, n = 17,221 | |||||
| BASE | ||||||||
| OPT 2 | θ1 | 0.0798 (0.00762) | 0.0917 (0.00668) | 0.0798 | (0.0649, 0.0947) | 0.0917 | (0.0786, 0.105) | tp |
| Pbo | θ11 | −0.128 (0.00939) | −0.143 (0.00811) | −0.128 | (−0.147, −0.110) | −0.143 | (−0.159, −0.127) | tp |
| OPT 1 | θ14 | −0.00570 (0.00917) | −0.00995 (0.0079) | −0.00570 | (−0.0237, 0.0123) | −0.00995 | (−0.0254, 0.00555) | tp |
| OPT Com | θ15 | −0.0191 (0.0127) | −0.0114 (0.0109) | −0.0191 | (−0.0440, 0.00589) | −0.0114 | (−0.0329, 0.00999) | tp |
| PMAX | ||||||||
| OPT 2 | θ2 | 0.00276 (0.00113) | 0.00174 (0.000999) | 0.00276 | (0.000545, 0.00498) | 0.00174 | (−0.000219, 0.00370) | tp/w |
| 1047 | θ17 | 0.00471 (0.00364) | 0.00619 (0.00292) | 0.00471 | (−0.00243, 0.0118) | 0.00619 | (0.000466, 0.0119) | tp/w |
| OPT 1 | θ18 | −0.00508 (0.00166) | −0.00379 (0.00139) | −0.00508 | (−0.00833, −0.00184) | −0.00379 | (−0.00652, −0.00106) | tp/w |
| OPT ReT | θ20 | −0.00300 (0.00225) | −0.00314 (0.00192) | −0.00300 | (−0.00742, 0.00141) | −0.00314 | (−0.00691, 6.35E‐4) | tp/w |
| EMAX | ||||||||
| OPT 2 | θ3 | 1.10 (0.0355) | 1.09 (0.0503) | 1.10 | (1.03, 1.17) | 1.09 | (0.988, 1.18) | tp |
| 1047 | θ21 | −0.208 (0.0838) | −0.196 (0.0686) | −0.208 | (−0.372, −0.0438) | −0.196 | (−0.331, −0.0617) | tp |
| OPT ReT | θ24 | −0.0352 (0.0513) | 0.0557 (0.0647) | −0.0352 | (−0.136, 0.0653) | 0.0557 | (−0.0712, 0.183) | tp |
| ED50 | ||||||||
| OPT 2 | θ4 | 0.919 (0.134) | 0.907 (0.163) | 2.51 | (2.01, 3.13) | 2.48 | (1.89, 3.24) | mg |
| OPT 1 | θ26 | −0.334 (0.133) | −0.200 (0.130) | 0.716 | (0.575, 0.892) | 0.819 | (0.661, 1.01) | frac |
| OPT ReT | θ28 | −0.188 (0.194) | 0.226 (0.181) | 0.828 | (0.602, 1.14) | 1.25 | (0.93, 1.69) | frac |
| Sex = F | θ64 | −0.472 (0.101) | −0.400 (0.0911) | 0.624 | (0.528, 0.736) | 0.670 | (0.577, 0.778) | frac |
| PASB | θ68 | −0.473 (0.124) | −0.287 (0.100) | −0.473 | (−0.716, −0.230) | −0.287 | (−0.483, −0.0906) | frac |
| BWT | θ69 | 1.04 (0.191) | 0.844 (0.155) | 1.04 | (0.669, 1.42) | 0.844 | (0.541, 1.15) | frac |
| PRBL = Y | θ71 | 0.841 (0.0948) | 0.638 (0.0703) | 2.32 | (1.98, 2.71) | 1.89 | (1.69, 2.12) | frac |
| T½ | ||||||||
| OPT 2 | θ5 | 1.58 (0.0375) | 1.49 (0.0330) | 4.87 | (4.58, 5.18) | 4.42 | (4.19, 4.67) | w |
| 1047 | θ29 | −0.548 (0.156) | −0.631 (0.135) | 0.578 | (0.447, 0.747) | 0.532 | (0.426, 0.665) | frac |
| OPT Com | θ31 | −0.0221 (0.0775) | −0.118 (0.0655) | 0.978 | (0.861, 1.11) | 0.889 | (0.798, 0.990) | frac |
| OPT ReT | θ32 | −0.274 (0.0682) | −0.358 (0.0608) | 0.760 | (0.679, 0.850) | 0.699 | (0.632, 0.773) | frac |
| PASB | θ77 | 0.265 (0.0676) | 0.236 (0.0562) | 0.265 | (0.133, 0.398) | 0.236 | (0.126, 0.347) | frac |
| Ω:Var(η) | ||||||||
| BASE | ηBASE | 0.00861 (0.000875) | 0.00889 (0.000786) | 0.0928 | (0.0835, 0.102) | 0.0943 | (0.0861, 0.102) | SD |
| PMAX | ηPMAX | 0.000470 (2.16E‐5) | 0.000430 (1.89E‐5) | 0.0217 | (0.0207, 0.0227) | 0.0207 | (0.0198, 0.0216) | SD |
| EMAX | ηEMAX | 0.00330 (0.00200) | 0.0273 (0.00878) | 0.0574 | (0.0233, 0.0915) | 0.165 | (0.113, 0.217) | SD |
| T½ | ηT½ | 0.553 (0.0384) | 0.565 (0.0321) | 0.744 | (0.693, 0.794) | 0.752 | (0.71, 0.793) | SD |
| g | ||||||||
| OPT 2 | θ7 | −2.44 (0.0176) | −2.41 (0.0141) | 0.0875 | (0.0850, 0.0901) | 0.0900 | (0.0879, 0.0921) | SD |
| 1047 | θ33 | −0.0782 (0.0504) | −0.0596 (0.0409) | 0.925 | (0.851, 1.00) | 0.942 | (0.881, 1.01) | frac |
| TIME | θ8 | 0.0149 (0.00129) | 0.0146 (0.00103) | 1.01 | (1.01, 1.02) | 1.01 | (1.01, 1.02) | frac/w |
ASE, approximate standard error; BASE, baseline; BWT, body weight; CI, confidence interval; ED50, dose at which half of the EMAX is achieved; EMAX, maximum drug effect; F, female; frac, fraction; mg, milligram; N, number of subjects; n, number of observations; OFV, objective function value; OPT 1, OPT Pivotal 1 study; OPT 2, OPT Pivotal 2 study; OPT Com, OPT Compare study; OPT ReT, OPT Re‐Treatment study; PASB, baseline PASI score; PASI, Psoriasis Area and Severity Index; PMAX, maximum non‐drug effect; Pbo, placebo (dose = 0); PRBL, prior biologic therapy; T1/2, half‐life of onset of drug effect; TD, training dataset; tp, transformed PASI; Trans, transformed; SD, standard deviation; Var, variance; w, week; Y, yes; 1047, Phase IIb study A3921047.
Note: study effects are relative to those for OPT 2.
Effects of body weight
The effects of weight were evaluated (after the covariate selection described above) by finding the covariate model of weight that predicted a VD dataset (dataset independent of model building) with the least MAD (or prediction) error. VPCs were also performed on the validation dataset to assess the adequacy of the model and covariate selection.
RESULTS
Data
Data from one phase IIb study (A3921047; NCT00678210)20 and four phase III studies (OPT Pivotal 1 (A3921078; NCT01276639),16 OPT Pivotal 2 (A3921079, NCT01309737),16 OPT Compare (A3921080, NCT01241591),17 and OPT Re‐Treatment (A3921111; NCT01186744)18), conducted to assess the efficacy and safety of oral tofacitinib in subjects with moderate‐to‐severe psoriasis, were pooled to support the integrated ER analysis. Table 1 provides pertinent summary information of these studies.
A total of 3,431 subjects with 17,221 observations were used in the ER analysis. The dataset included ∼85% White, 7% Asian, and 2.5% Black subjects, and 70% were males (Supplementary Table S1 online). The median age was 45 years and the median weight was 86 kg. The median disease severity (measured by baseline PASI) was 19.8, median disease duration was 16 years, and 79% of subjects were naïve to previous biologic usage. The covariate distributions for weight, disease severity, and disease duration were skewed (Supplementary Figure S1 online), but were generally similar across the studies. OPT Compare had the greatest percentage of subjects naïve to biologic treatment, with 91.4% compared with 73–78% across the other studies.
ER model
An inhibition of Kin IDR drug‐effect model was applied to a percentage change from baseline endpoint computed from transformed PASI scores. The percentage change from baseline endpoint was used to account for the skewed baseline distribution of PASI scores, and the transformation was used to normalize the change from baseline scores due to the skewness of the post‐baseline scores. The use of the percentage change from baseline endpoint also improved prediction of PASI responder rates relative to predictions achieved using models of PASI. A piecewise linear model was used for the placebo/nondrug model component. The slope of the second linear piece was 0 after Week 14, when the maximum placebo effect was obtained. Study effects were evaluated on the drug‐related parameters of maximum drug effect, potency, and drug onset, as well as baseline and placebo/nondrug components to evaluate heterogeneity between studies. Differences in baseline and maximum drug effect were detected; however, the potency was similar across the studies. Steady‐state average plasma concentration (Cavg; derived from the population pharmacokinetics (PK) model23) was evaluated as a subject‐specific measure of exposure and was not found to be a better predictor than dose. It is important to note that tofacitinib achieved steady‐state PK on Day 2 due to short half‐life (∼3 hours) relative to the dosing interval.24 Dose was retained as the measure of exposure in the model because the individual predictions of exposure did not improve model predictions.
Age, disease severity, disease duration, sex, race, and previous biologic usage were evaluated as covariates on the model parameters for baseline, maximum placebo effect or slope, maximum drug, potency, and drug effect onset rate (mediated through the IDR). Effects of weight were included but not evaluated during the covariate selection procedure. The procedure selected: sex, disease severity, and previous biologic usage as covariate effects for potency; and disease severity for drug effect onset.
Prediction onto a VD (the full dataset was divided, 65% as a TD and 35% as a VD) was used to evaluate the effect of weight specifically with respect to the prediction of PASI change. Weight on potency was the only effect retained based on prediction errors (not included on the drug onset rate parameter). The final model thus consisted of the effects of sex, disease severity, previous biologic usage, and weight on potency, and disease severity on drug effect onset. The parameter estimates for the final model fitted to the entire pooled datasets (training and validation) are provided in Table 2. A doubling of body weight increased the dose achieving half the maximum effect (ED50) estimate by 1.8‐fold (90% confidence interval (1.45, 2.20)). The typical half‐life of drug effect onset was ∼4 weeks.
Model predictions
The selection of the 5 and 10 mg doses for the pivotal trials was based on a Bayesian analysis of the phase II study (A3921047) data.25 To evaluate the optimality of the selection of these dose levels, the percentage of maximum drug effect (EMAX) based on PASI75 for the pivotal trials (Table 1) was predicted at Week 16 as a function of dose using the current ER model and is displayed in Figure 1. The mean predictions are based on typical values of the subject covariates (male; body weight, 86 kg; baseline PASI, 20; biologic agent‐naïve). The 10 mg dose is just on the cusp of the plateau of the ER curve at 81% of the maximum effect, a value that can be judged to be the optimum when considering benefit/risk (i.e., in general with unspecified risk). The 5 mg dose was predicted to be at 65% of EMAX on the ER curve. The predicted median values for maximum effect on the PASI percentage change from baseline scale were 77% and 88% for the 5 and 10 mg doses, respectively (data not shown). The overall absolute predicted PASI75 rates for 5 and 10 mg twice daily (b.i.d.) were 49% and 61%.
Figure 1.
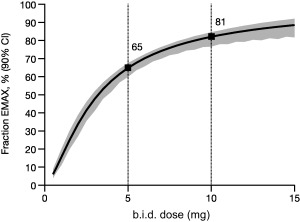
Fraction of EMAX based on PASI75. Model‐predicted relationship vs. dose at Week 16 for a reference subject (median or typical covariate values) based on the pivotal studies. Solid line is the mean prediction and shading represents 90% CI bounds. b.i.d., twice daily; CI, confidence interval; EMAX, maximum drug effect; PASI75, 75% improvement from baseline in Psoriasis Area and Severity Index.
PASI75 responder rates were also predicted for the 5 and 10 mg b.i.d. doses vs. a range of covariate values. The effects of weight, previous biologic usage, and sex on ED50, which affects the long‐term (or steady‐state) dose–response profile, were prominent. The effect of 10 mg b.i.d. yielded increased response rates for covariates that increased ED50, suggesting higher doses are necessary to adjust for those covariate effects. For example, Figure 2 displays PASI75 responses and the differences between 5 and 10 mg b.i.d. over a body weight range of 64–116 kg (corresponding to the 10th and 90th percentiles of the observed weight distribution, respectively). Over this weight range, PASI75 rates decreased with increasing weight and this trend was seen for both doses. The predicted differences in PASI75 responses between 5 and 10 mg b.i.d. at Week 16 increased with increasing weight, ranging from 10% for 64 kg to 14% for 116 kg. Differences in PASI75 responses between 5 and 10 mg b.i.d. at Week 16 were 12% for biologic‐naïve vs. 16% for biologic‐experienced subjects, and 9% for females vs. 12% for males (Figure 2). The effect of sex on potency may be due to a poor approximation of body weight to body composition. Differences in PASI75 responses between 5 and 10 mg b.i.d. were 14% for a baseline PASI of 14 and 10% for a baseline PASI of 35 (Figure 2). Overall, 10 mg b.i.d. provided greater response rates and mostly offset these covariate effects relative to 5 mg b.i.d.
Figure 2.
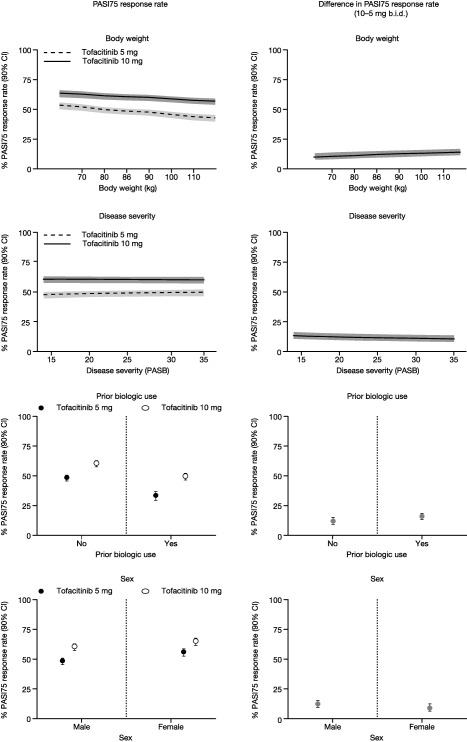
Predicted PASI75 response rates and differences between tofacitinib 10 and 5 mg b.i.d. vs. covariate value at Week 16. The solid lines represent population mean predictions while the gray lines or shaded regions represent 90% CIs. Covariates not represented in the particular graph were fixed at their median or reference (no previous biologic usage, male) value. Predictions are based on OPT Pivotal 1 and 2. b.i.d., twice daily; CI, confidence interval; PASB, baseline Psoriasis Area and Severity Index score; PASI75, 75% improvement from baseline in Psoriasis Area and Severity Index score.
Body weight
Given the influence of body weight on potency, the relative locations of the 5 and 10 mg doses on the dose–response curve were assessed vs. weight. Prior biologic usage also affected potency with a noticeable magnitude, so it was important to look at body weight and prior biologic usage simultaneously. The effects are illustrated in Figure 3 for the 10th and 90th percentiles of body weight in biologic‐naïve or ‐experienced subjects. The results indicated that even though PASI75 response rates decreased with increasing body weight, dose–response was evident for heavy weight (116 kg) and light weight (64 kg) subjects in both biologic‐naïve and ‐experienced subjects. For subjects weighing 116 kg, the predicted difference between 5 and 10 mg b.i.d. was 14% for biologic‐naïve subjects and 17% for biologic‐experienced subjects. For subjects weighing 64 kg, the difference was 10% and 15% for biologic‐naïve and ‐experienced subjects, respectively. Figure 3 also demonstrates higher response rates for both heavy and light weight subjects in the biologic‐naïve compared to biologic‐experienced subjects. Differences in efficacy with weight could not be explained by differences in PK with weight, as shown by the weak relationship between predicted Cavg and body weight (Figure 4).
Figure 3.
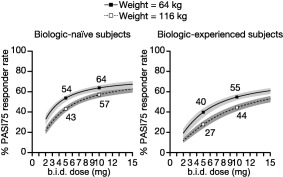
PASI75 responder rate vs. dose in heavy vs. light weight subjects by prior biologic usage. The lines represent population mean PASI75 responder rate predictions, while the shaded regions represent 90% confidence intervals. The predictions are for baseline PASI of 20, males, with groups defined by covariates as shown in the legend and title. Predictions are based on OPT Pivotal 1 and 2. b.i.d., twice daily; PASI75, 75% improvement from baseline in Psoriasis Area and Severity Index score.
Figure 4.
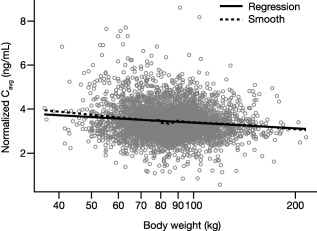
The relationship between dose‐normalized Cavg vs. body weight. Gray open circles are individual predictions from the PK model. Cavg, average plasma concentration; PK, pharmacokinetics.
DISCUSSION
The ER analysis discussed herein was performed to evaluate the optimality of the 5 and 10 mg dosage levels. Additionally, the effect of body weight, an issue noted by practicing clinicians, as well as other important patient factors, were evaluated.
The importance of the issue of weight was demonstrated during the dosage optimization for ustekinumab. During regulatory review, two‐tiered, three‐tiered, and five‐tiered dosing regimens based on weight categories were presented to the Dermatologic and Ophthalmic Drugs Advisory Committee due to the suboptimality, in terms of benefit to heavier subjects, of a single fixed‐dose regimen.26 A two‐tiered fixed‐dose regimen of 45 mg for a patient weighing ≤100 kg or 90 mg for patients weighing >100 kg was approved for ustekinumab.11 The basis for this regimen was the difference in PK as a function of weight. Another recent example for dose optimization using modeling and simulation comes from the Advisory Committee for secukinumab, which recommended a weight‐based dosing regimen (as opposed to the sponsor's proposal of a flat dosing regimen) for maximizing efficacy in patients ≥90 kg.12 These examples illustrate the acceptance of pharmacometrics methodologies for quantifying the impact of body weight and thereby optimizing dosing for psoriasis therapies.
Weight was found to influence PASI response following administration of tofacitinib—specifically ED50, the determinate of the long‐term dose–response relationship. The following unique properties of tofacitinib facilitated a focused assessment of weight. Tofacitinib achieves PK steady state on Day 2 of administration, while the PD of PASI does not achieve steady state until after Week 16. In addition, body weight is not a meaningful determinant of Cavg of a subject; as demonstrated by the weak relationship between predicted Cavg and body weight. Therefore, the effect of weight on ED50 is an induced PD phenomenon and is not confounded by using dose instead of Cavg.
The results presented are not dissimilar to those reported for etanercept,14 even with regard to the parameter estimate linking potency to weight. This is despite differences in the modeling strategy (PASI PCFB for tofacitinib vs. PASI75 for etanercept), and different mechanisms of drug action (JAK inhibitor vs. tumor necrosis factor inhibitor). However, it is unknown if this weight‐potency relationship would hold for treatments with other mechanisms of action. ER model‐dependent methods of assessment would be necessary because of the typically strong relationship of body weight to exposure and the longer time frame used to achieve steady‐state exposure. In this regard, this tofacitinib ER model provides an advancement in understanding the effects of body weight on dose–response and also suggests that trials should be of sufficient duration to achieve and evaluate PD steady state such that predictors of onset of effect and steady‐state dose–response are not confounded.
The model predicted the magnitude of effect at clinical doses of 5 and 10 mg b.i.d. depended on sex, previous biologic usage, disease severity, and body weight. Because weight influenced ED50 and hence the level of steady‐state efficacy, heavier subjects would require higher dosages to achieve comparable benefit to lighter subjects. Overall, across the covariate and weight distributions, the predictions suggest that tofacitinib 10 mg b.i.d. demonstrates consistently higher response levels relative to 5 mg b.i.d. and that it offsets to some extent the effect of the covariates; subjects in these subpopulations can achieve a similar response with tofacitinib 10 mg b.i.d. compared with subjects in the subpopulation counterpart who receive 5 mg b.i.d.
In conclusion, moderate‐to‐severe plaque psoriasis is a serious chronic systemic disease that, while predominantly affecting the skin, also profoundly affects patients' quality of life.4 Many patients with psoriasis are undertreated, often due to suboptimal dosing regimes and reduced efficacy in heavier patients. Despite the introduction of new therapies, such as biologic agents, little progress has been made in the past decade in reducing the undertreatment of moderate‐to‐severe psoriasis.10 This analysis highlights the importance of considering the PD effects of body weight, in addition to possible PK effects, when making treatment and dosage decisions, to help optimize dosing in order to reduce undertreatment of patients with psoriasis.
Supporting information
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
Supporting Information Table 5.
Acknowledgments
The studies included in this analysis were sponsored by Pfizer Inc. Editorial support, under direction from the authors, was provided by Alice Palmer, PhD, of Complete Medical Communications and funded by Pfizer Inc.
Disclosures
MM Hutmacher is a consultant for Pfizer Inc. K Papp has received grant/research support from Abbott, Amgen, Anacor, Astellas, Celgene, Celtic, Dow Pharma, Eli Lilly, Galderma, Janssen, Janssen Biotech (Centocor), Merck, Novartis, and Pfizer Inc.; has acted as a consultant for 3M, Abbott, Akesis, Akros, Alza, Amgen, Astellas, Baxter, Boehringer Ingelheim, Celgene, Cipher, Eli Lilly, Forward Pharma, Funxional Therapeutics, Galderma, Genentech, Isotechnika, Janssen, Janssen Biotech (Centocor), J&J, Kataka, Kirin, Kyowa Lypanosys, Meiji Seika Pharma Co., Ltd., Merck, Mitsubishi Pharma, Mylan, Novartis, Pfizer Inc., Regeneron Pharmaceuticals Inc., Sanofi Aventis, Serono, Stiefel, Takeda, UCB Pharma, and Vertex; and has participated in speakers' bureaus for 3M, Abbott, Amgen, Astellas, Boehringer Ingelheim, Celgene, Eli Lilly, Galderma, Janssen, Merck, Novartis, and Pfizer Inc. H Tan, K Ito, R Wolk, C Mebus, H Valdez, S Krishnaswami, and P Gupta are employees and shareholders of Pfizer Inc. ST Rottinghaus was an employee of Pfizer Inc at the time of the analysis.
Author Contributions
M.M.H. and P.G. wrote the article; M.M.H., S.K., K.I., H.T., R.W., H.V., C.M., S.T.R., and P.G. designed the research; M.M.H., K.P., and P.G. performed the research; M.M.H. and P.G. analyzed the data.
References
- 1. Neimann, A.L. , Shin, D.B. , Wang, X. , Margolis, D.J. , Troxel, A.B. & Gelfand, J.M. Prevalence of cardiovascular risk factors in patients with psoriasis. J. Am. Acad. Dermatol. 55, 829–835 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Miller, I.M. , Skaaby, T. , Ellervik, C. & Jemec, G.B. Quantifying cardiovascular disease risk factors in patients with psoriasis: a meta‐analysis. Br. J. Dermatol. 169, 1180–1187 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Onumah, N. & Kircik, L.H. Psoriasis and its comorbidities. J. Drugs Dermatol. 11, s5–10 (2012). [PubMed] [Google Scholar]
- 4. Augustin, M. & Radtke, M.A. Quality of life in psoriasis patients. Expert Rev. Pharmacoecon. Outcomes Res. 14, 559–568 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Pathirana, D. et al European S3‐guidelines on the systemic treatment of psoriasis vulgaris. J. Eur. Acad. Dermatol. Venereol. 23(suppl. 2), 1–70 (2009). [DOI] [PubMed] [Google Scholar]
- 6. Menter, A. et al Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J. Am. Acad. Dermatol. 58, 826–850 (2008). [DOI] [PubMed] [Google Scholar]
- 7. Wadhwa, M. , Knezevic, I. , Kang, H.N. & Thorpe, R. Immunogenicity assessment of biotherapeutic products: an overview of assays and their utility. Biologicals 43, 298–306 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Mansouri, Y. & Goldenberg, G. Biologic safety in psoriasis: review of long‐term safety data. J. Clin. Aesthet. Dermatol. 8, 30–42 (2015). [PMC free article] [PubMed] [Google Scholar]
- 9. Levin, E.C. , Gupta, R. , Brown, G. , Malakouti, M. & Koo, J. Biologic fatigue in psoriasis. J. Dermatolog. Treat. 25, 78–82 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Armstrong, A.W. , Robertson, A.D. , Wu, J. , Schupp, C. & Lebwohl, M.G. Undertreatment, treatment trends, and treatment dissatisfaction among patients with psoriasis and psoriatic arthritis in the United States: findings from the National Psoriasis Foundation surveys, 2003‐2011. JAMA Dermatol. 149, 1180–1185 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Lebwohl, M. et al Impact of weight on the efficacy and safety of ustekinumab in patients with moderate to severe psoriasis: rationale for dosing recommendations. J. Am. Acad. Dermatol. 63, 571–579 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Oussova, T. Dermatologic and Ophthalmic Drugs Advisory Committee Meeting; FDA Introductory Remarks. <http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DermatologicandOphthalmicDrugsAdvisoryCommittee/UCM420461.pdf>, Last updated 20 Oct 2014; Accessed 21 Nov 2016.
- 13. Tham, L.S. , Tang, C.C. , Choi, S.L. , Satterwhite, J.H. , Cameron, G.S. & Banerjee, S. Population exposure‐response model to support dosing evaluation of ixekizumab in patients with chronic plaque psoriasis. J. Clin. Pharmacol. 54, 1117–1124 (2014). [DOI] [PubMed] [Google Scholar]
- 14. Hutmacher, M.M. , Nestorov, I. , Ludden, T. , Zitnik, R. & Banfield, C. Modeling the exposure‐response relationship of etanercept in the treatment of patients with chronic moderate to severe plaque psoriasis. J. Clin. Pharmacol. 47, 238–248 (2007). [DOI] [PubMed] [Google Scholar]
- 15. Fredriksson, T. & Pettersson, U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica 157, 238–244 (1978). [DOI] [PubMed] [Google Scholar]
- 16. Papp, K.A. et al Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo‐controlled dose‐ranging study. Br. J. Dermatol. 167, 668–677 (2012). [DOI] [PubMed] [Google Scholar]
- 17. Papp, K.A. et al Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two, randomized, placebo‐controlled, phase III trials. Br. J. Dermatol. 173, 949–961 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Bachelez, H. et al Tofacitinib versus etanercept or placebo in moderate‐to‐severe chronic plaque psoriasis: a phase 3 randomised non‐inferiority trial. Lancet 386, 552–561 (2015). [DOI] [PubMed] [Google Scholar]
- 19. Bissonnette, R. et al Tofacitinib withdrawal and retreatment in moderate‐to‐severe chronic plaque psoriasis: a randomized controlled trial. Br. J. Dermatol. 172, 1395–1406 (2015). [DOI] [PubMed] [Google Scholar]
- 20. Feldman, S.R. & Krueger, G.G. Psoriasis assessment tools in clinical trials. Ann Rheum Dis 64(suppl. 2), ii65–ii68 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hutmacher, M.M. , French, J.L. , Krishnaswami, S. & Menon, S. Estimating transformations for repeated measures modeling of continuous bounded outcome data. Stat. Med. 30, 935–949 (2011). [DOI] [PubMed] [Google Scholar]
- 22. Dayneka, N.L. , Garg, V. & Jusko, W.J. Comparison of four basic models of indirect pharmacodynamic responses. J. Pharmacokinet. Biopharm. 21, 457–478 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kowalski, K.G. & Hutmacher, M.M. Efficient screening of covariates in population models using Wald's approximation to the likelihood ratio test. J. Pharmacokinet. Pharmacodyn. 28, 253–275 (2001). [DOI] [PubMed] [Google Scholar]
- 24. Hooker, A.C. , Staatz, C.E. & Karlsson, M.O. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm. Res. 24, 2187–2197 (2007). [DOI] [PubMed] [Google Scholar]
- 25. Karlsson, M.O. & Holford, N. A tutorial on visual predictive checks. Abstract presented at the 17th Annual Meeting of the Population Approach Group in Europe 2008.
- 26. Yano, Y. , Beal, S.L. & Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 28, 171–192 (2001). [DOI] [PubMed] [Google Scholar]
- 27. Ma, G.L. et al Population pharmacokinetics of tofacitinib in adult patients with moderate to severe chronic plaque psoriasis. Abstract presented at the World Congress of Dermatology 2015, 2015.
- 28. Dowty, M.E. et al The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a Janus kinase inhibitor, in humans. Drug Metab. Dispos. 42, 759–773 (2014). [DOI] [PubMed] [Google Scholar]
- 29. Tan, H. et al Dose‐response and pharmacokinetics of tofacitinib (CP‐690,550), an oral Janus kinase inhibitor, in the treatment of chronic plaque psoriasis. CPT Pharmacometrics Syst. Pharmacol. 2, e44 (2013). [Google Scholar]
- 30. Food and Drug Administration . Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research Dermatologic and Ophthalmic Drugs Advisory Committee, June 17, 2008. <http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4361b1-01-FDA%20.pdf>. Last updated 2008; Accessed 12 May 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
Supporting Information Table 5.